
Abstract
This study investigates the suitability of heat conduction calorimeters for determining the specific heat capacity of solid and liquid substances. Accurate and precise measurements were obtained for various substances, including water, ethylene glycol, the ionic liquid [EMIM][TCM], and copper, with relative standard deviations averaging less than 1%. Measurements on [EMIM][DCA] indicated a systematic deviation from the literature values. The study highlights the repeatability of the measurement method, which worked well for both temperature increases and decreases, as well as for the mean of the two. The influence of sample size on the results when it came to liquids was also investigated, revealing that large sample sizes led to underpredictions; while, small sizes yielded the opposite effect. The best results were obtained with half-filled vials; a similar filling level as was used in the electrical calibrations of the calorimeters with heaters in the vials. Additionally, no significant differences were observed among the eight calorimeters of the I-Cal Flex instrument, and different baseline calculation methods had negligible impact on the results. Overall, this study illustrates how a heat conduction calorimeter can be utilized for accurate and precise heat capacity measurements for both solid and liquid substances.
Similar content being viewed by others

Introduction
Heat capacity is a fundamental property of all matter and many method have been developed to determine heat capacities, for example differential scanning calorimetry (DSC) [1], the transient plane source (hot disk) technique [2], adiabatic calorimetry [3], and drop calorimetry [4] (references given are only examples).
With the advent of commercial differential scanning calorimeters (DSCs) in the 1960s, simple techniques to use DSC to determine specific heat capacities of small samples (in the order of 10 mg) were developed. The first methods were “scanning methods” [5] [6] where the heat capacity was calculated from the heat flow measured by the DSC. Later an “enthalpy method” in which the heat capacity was determined from an integral of the heat flow was developed [7] [1], as well as methods based on modulated DSC [8].
The present paper shows that the “enthalpy method” can successfully be used also with heat conduction calorimeters normally used for isothermal measurements. The sample sizes for isothermal calorimeters are typically in the range 1–100 g, i.e., much larger than DSC samples of 5–20 mg. Two advantages with larger samples are that their masses can be accurately determined with less expensive balances, and that measurements on larger samples of inhomogeneous materials will yield more representative results.
The DSC enthalpy method shows similarities to drop calorimetry, but while the sample is exposed to a ramp change in temperature in a DSC, in a drop calorimeter the sample is dropped from one temperature into an isothermal heat conduction calorimeter that is at another temperature. In the method proposed in the present paper the temperature of an “isothermal heat conduction calorimeter” is changed from one temperature to another. This temperature change is not made in a controlled way, but the heat measured is still the heat required to calculate the heat capacity by the enthalpy method.
The method in this study is similar to the method to measure the heat capacity of reactive materials with a heat conduction calorimeter proposed by Bunyan [9]. He measured the heat needed to cool samples in glass vials by 1 K and calculated the heat capacity of the sample from the difference between such measurements with and without a sample. In his study, Bunyan concluded that the method produced accurate results and the specific heat capacity of water was measured with a deviation of only 0.5% from the literature value. Bunyan also used a similar method to calculate the calorimetric calibration coefficients from measurements with samples of sapphire [10], that has a well-known heat capacity [11].
Another similar case is the study performed by Ubelhor et al. in 2015 [12], where an isothermal calorimeter was designed and utilized to measure the heat capacity of whole electrochemical cells (batteries). Silver–zinc cells with a mass slightly over 4 kg were used as samples and measurements were performed with a 10 K increase and decrease in temperature. These measurements determined the heat capacity of these samples with an experimental RSD of approximately 2% and the authors concluded that their heat conduction calorimeter was a useful analytical tool for electrochemical cells.
In this study, the samples were subjected to temperature increases and decreases of 2 K. Both heating and cooling steps were used to calculate the heat capacity of the samples. The substances studied are chemically inert as well as thermally stable to avoid any exothermic or endothermic heat development. The study intends to demonstrate that a heat conduction calorimeter normally used for isothermal measurements also can provide an accurate prediction of the heat capacity of both liquid and solid substances.
Materials
All the experiments presented in this study were performed using the substances listed in Table 1. The list contains both well-studied substances such as copper, sapphire, water and ethylene glycol, as well as the two less studied ionic liquids 1-ethyl-3-methylimidazolium dicyanamide (“DCA”) and 1-ethyl-3-methylimidazolium tricyanomethanide (“TCM”). All the substances are chemically inert and thermally stable. Glycerol was only used in a test of how the sample size influenced the results. The non-aqueous liquids were new, had a low water content, and the bottles were not opened more than necessary to transfer samples to the vials to prevent uptake of water vapor.
Method
The specific heat capacity c (J g−1 K−1) can be determined from the heat transfer Q (J) associated with a temperature change ΔT (K) for a sample of mass m (g):
As c and m are positive quantities, Q and ΔT must have the same sign; in the following we define temperature increase and heat transfer to a sample as positive. Heat capacity measurements are made by changing the temperature, and this can be done either with step changes or by scanning the temperature (in the latter case, Eq. 1 is modified by exchanging Q and ΔT with their rates of change). In the present method, step changes in temperature of the thermostated environment of an eight-channel commercial isothermal (heat conduction) calorimeter (I-Cal Ultra from Calmetrix Inc.) are made and the exchange of heat for each of the eight samples is measured by the calorimeters. Note that this step change of thermostat temperature gives a slow first order (exponential) temperature change of the sample temperature (Newton’s law of cooling). This is a non-standard use of an “isothermal heat conduction calorimeter,” as such calorimeters are made with the aim of keeping the temperature constant, to enable studies of long-term processes like soil respiration [21], degradation of pharmaceutical substances [22], fungal decay [23], and the hydration of cement [24].
The samples were placed in 20 mL glass vials. For liquids, 10 mL samples were used in most cases, i.e., half-filled vials. The sapphire samples filled the lower 30% of the vials; while, the copper ribbons were folded and placed standing up. The vials were sealed with aluminum screw caps with a rubber-Teflon septa.
The aim of the present measurements was to determine the specific heat capacity at 20 °C. For such a measurement, the vials with samples are placed in the calorimeters in the measurement positions (the vial holders) at 19 °C and left there until there is a constant baseline signal. The temperature of the thermostat is then increased to 21 °C, kept there for about 12 h, and then decreased to 19 °C again, and kept there for about another 12 h. Each time that the temperature is changed, the calorimeters—including the samples—need to change their temperature and it is the heat flow rate into or out of the sample that is measured by the calorimeter. This heat flow rate is then integrated to give the heat Q that is needed to heat or cool the sample when it goes between the two temperatures.
A measurement with an empty vial will also give a peak when the temperature is changed. It is therefore needed to do “empty measurements” and subtract the results of them from the “sample measurements.” As the vials and caps were reused in the same calorimeter for all measurements, the results of the empty-runs stayed the same during a measurement series, so it was not needed to do empty measurements in connection to each sample measurement.
Heat conduction calorimeters are twin instruments, i.e., they have both a sample heat flow sensor and a reference heat flow sensor [25]. For isothermal use, the reference side should be charged with a reference sample that has a similar heat capacity as the sample, as this will minimize the noise and make the calorimeter behave in a more ideal way [25]. For the present non-isothermal heat capacity measurements we have instead, on purpose, used a reference heat capacity equal to the heat capacity of 6 g of water. For measurements with 10 g water, the empty peak and the sample peak will have opposite signs. In this way we can cover a large span of thermal powers without going out-of-range. The balancing was made with water in mind, so when measurements were made with ethylene glycol—with a much lower heat capacity than water—the calorimeters were actually almost balanced, which resulted in only small peaks, and for copper, sapphire and the ionic liquids—with even lower heat capacities—the sample and reference peaks had the same sign (Fig. 1A).

A. Example of peaks after temperature increases and decreases. The examples are from different measurements with temperature changes taking place at different times. The double peaks are further discussed in the text. B The evaluated baseline of the first water peak in Fig. 1A using the fraction of heat method (dashed line) and an exponential function with a time constant 0.002 s−1 (dotted line). The start and end points of the baseline functions were calculated from means of the signal in the intervals before and after these points (thick black lines), respectively
In Fig. 1A it is seen that there are double peaks for water and ethylene glycol. We have in all cases integrated both peaks as these double peaks are the result of small non-symmetries in either thermal properties of the calorimeters or in the way that the temperature change takes place between the sample and reference sides of the calorimeters. Similar trends with double peaks have been presented by Mudd [26], where the pre-peak was stated to be caused by a small asymmetry in the chamber heater locations. Computer simulations (see Supplementary material) indicate that if there is heat in a pre-peak to one side, the main peak (in the other direction) will be larger by the same amount of heat, so that these two heats will cancel out. The reason that the pre-peak in the EG measurement in Fig. 1A is comparatively large, is probably that the main peak is so small in this case as the sample and reference sides are almost balanced. For measurements with larger main peaks (like the water measurement in Fig. 1A) the pre-peak is small as it is taken over by the large main peak of opposite sign.
The heat Q in Eq. 1 is calculated from the difference between the integrals of the sample and reference peaks; and in both cases a baseline needs to be subtracted from the measured signal:
Here, ε (W/V) is the calorimetric calibration coefficient, I (V⋅s) is a peak integral (sample or empty), U (V) is the signal during a sample peak after a temperature change, UBL (V) is the corresponding baseline. Note that the integrations are performed on the voltage signals from the calorimeters; the calibration coefficients are later applied on the voltage integrals.
There are small shifts in the baselines between before and after each peak. In the small temperature interval of the present measurements, it is reasonable to assume that the baseline is a linear function of the temperature. The fraction of the heat that has flowed into (or out from) a sample at a certain time is then a measure of how far the change in temperature of the sample has gone. We have therefore constructed baselines (for when the temperature changes) that go from the initial to the final value in proportion to the fraction of the total heat that has passed into or out of the sample. This is shown in Fig. 1B, where it is also shown that this baseline is well approximated by an exponential function with a time constant of 0.002 s−1, that was similar in all evaluations. As the baseline based on the heat fraction described above could not handle the cases with pre-peaks that were large compared to the main peaks, we instead used an exponential function with a time constant of 0.002 s−1 as baseline in all evaluations. Note that the relatively large baseline noise seen in Fig. 1B is a result of the calorimeter being unbalanced; however, the long-term mean value of the signal is correct [25] [27].
All measurements were made with four or six temperature changes (peaks). A mean value of the values of all integrated peaks (with signs corrected for the direction of temperature change) was used as Isample in Eq. 2.
The size of the temperature steps was measured with a calibrated precision thermometer (ASL F250) with two Pt100 sensors placed in the measurement positions of two calorimeters. As all eight calorimeters are placed in the same precision air thermostat, which has a high air velocity, there are no measurable temperature differences between the different calorimeters. Three determinations made at different times agreed well, resulting in that the temperature step was 1.985 K. The mass of the samples was determined on a calibrated balance with a 0.001 g resolution. The calorimetric calibration coefficients were measured with electrical heaters cast into polyurethane resin in glass vials. For each calorimeter, the median value of three calibrations were used. During the period where the presented results were measured, four empty measurements were made. The resulting empty integrals (including the baseline correction) for each calorimeter were similar and we have used the median values in the evaluation. We have used median values as these reduce the likelihood that erroneous outliers influence the results.
Results and discussion
The method was used to measure and determine the specific heat capacity of both liquids and solids. The specific heat of a substance could be determined through either the rise or the fall of the temperature as well as of the mean of the two. It was found that all three options gave similar results. The largest deviation that was found between specific heat results from temperature rise and temperature fall measurements was 0.66%; while, the average deviation was 0.38%. This was similar for both liquids and solids. All heat capacity results presented in this paper are based on the mean of the temperature rise and the temperature fall measurements.
Table 2 gives the heat capacity results and Fig. 2 gives the relative deviations of the results from the literature values given in Table 1 for the first eight measurement series with eight samples in each series. The relative deviation δ is defined by Eq. 4.

Overview of the relative deviations calculated with Eq. 4 and literature data from Table 1 (zero relative deviation is the literature value). Outliers are shown as unfilled circles and starred measurements were made with larger samples. Short horizontal lines indicate plus and minus one percent relative deviation
Two outliers have been identified and removed from the analysis (see Fig. 2). It is apparent that most mean results are within 1% of the corresponding literature values and the RSDs are in most cases about 1%. A notable exception is the values for DCA that are about 2.5% too low, indicating that the literature values may be slightly too high. The sapphire results have significantly larger spread than the other measurements. The significantly lower ethylene glycol results marked with a star in Table 2 are discussed below.
The result of the measurements with different sized samples of glycerol is shown in Fig. 3. There is a trend that larger samples give lower specific heat capacity while smaller samples tend to give a higher measurement result. The results for the sample size (10 g, about half-filled vials) used in most of our measurements agree well—maybe fortuitously—with the literature value. It is worth noting that this trend was detected with glycerol tested at different sample sizes, but when the sample sizes of sapphire and copper was changed no clear trends was observed, as is visible in Fig. 2 when comparing the starred sapphire and copper measurements with the previous ones.

Results of two series of measurements with different sized samples of glycerol. The solid line is a linear regression, and the dashed line is the literature value
As is illustrated in Table 3, the mean specific heat capacity from all the performed measurements correlated well with the reported literature values. Each specific heat capacity presented in the table is the mean of the tests performed of that substance except outliers and the starred data in Fig. 2 and Table 2 (i.e., we only use data from half-filled vials for the liquids). The table illustrates that the overall performance of the method is good. As seen in Table 3, the deviations between measured values and literature values are in most cases low, indicating that the method is both accurate and precise. This is especially the case for the most known and well explored substance, water.
The only substances that lie above one percent deviation from the literature value are the two ionic liquids and sapphire. While the ionic liquid TCM remains just barely outside the 1%-range with a deviation of 1.01%, the remaining two fall a bit further away. The second ionic liquid DCA deviates the most out of all the tested substances with a measured value that is 2.81% lower than its literature value. Sapphire has a measured value 2.34% higher than its reported the literature value.
Uncertainty analysis
With the aim of proposing how the method can be improved, we here make an attempt to understand the origin of the deviations from the literature values and the spread in the measured values. We do this by assessing the spread in the evaluated results, the uncertainty (inaccuracy) in each of the parameters used to calculate the specific heat capacity, and by speculating on different sources of error.
Spread in result
In Fig. 2 it is seen that the RSDs are about 1% for most measurements. Notable exceptions are sapphire that has an RSD of more than 2%, and the final ethylene glycol measurement with a larger sample that has an RSD of about 0.5%.
Uncertainties in parameters
The specific heat capacity is calculated by Eqs. 2–3 and we therefore analyze the uncertainty in each of the parameters in these equations.
The masses were determined on a calibrated balance with a resolution of 1 mg. We assume that the uncertainty in these determinations was 5 mg, so the relative uncertainty of a 10 g sample is 0.05%; a value which we take as the RSD.
The temperature difference was determined by subtracting two measured temperatures of about 19 and 21 °C. The calibration certificate of the instrument and the two Pt100 sensors used in parallel gave values for both a temperature correction (+ 0.011 and −0.036 K) and an extended uncertainty (± 0.015 K). As the evaluation uses the difference between two temperature measurements made with the same sensors in a small temperature interval, we assume that the temperature difference correction is zero and use the extended uncertainty to estimate the uncertainty of the temperature difference. Using a coverage factor of 2 [28] the extended uncertainty can be recalculated to a standard deviation of the temperature difference of 0.0075 K, which is equivalent to an RSD of about 0.4%
The calibration coefficients used were the medians of three determinations; their values were about 7 mW mV-1. The standard deviations were between 0.001 and 0.026 mW mV-1 for the eight calorimeters; the mean standard deviation was 0.013 mW mV-1, which we use as standard deviation for the calibration coefficients, which is equivalent to an RSD of 0.2%.
The baseline correction method using an exponential function was chosen as it gave reasonable baseline functions for all cases. A comparison of this used method and other possible—but clearly less accurate—baseline strategies (for example a straight line between the start and beginning of integration) only gave about 0.5% difference in the evaluated heats. We conclude that the uncertainty of the baseline evaluation method is difficult to assess, but that it probably is negligible compared to some of the other uncertainties as we believe that we use a close-to-optimal method to determine the baselines. We also note that it is possible that errors or uncertainties introduced by the baseline method are reduced if the sample and empty integrals have similar absolute values as two similar baseline corrections will then be subtracted from each other.
Four determinations of the empty integrals were made and for each calorimeter the median value was used. All absolute values of the empty integrals were in the order of 8.5 V⋅s and the standard deviations for the determinations of each of them was about 0.06 V⋅s.
The values of the sample integrals depended on the sample mass and heat capacity. Typical values (in V⋅s) in our measurements were 0 for ethylene glycol, -5 for water, 2 for TCM and DCA, and 5 for copper and sapphire.
It should be noted that the different sources of uncertainty act in different ways on the presented results. It is only the uncertainties in mass and baseline determination that are individual for each measurement, and as discussed above, both these uncertainties are small. The calibration coefficient and the empty integral have the same value for each calorimeter; while, the temperature difference value is common to all evaluations.
An analysis of how the heat capacity values from each calorimeter compared to the mean values from all calorimeters with the same simultaneous measurement of the same substance found no significant differences between the calorimeters, and there is no other systematic trend in these results (see Fig. 4). From visual inspection it appears that all the eight calorimeters operate with a similar level of accuracy.

Relative deviation from the mean of the measurements on the same substance and made at the same time, illustrated for each of the eight calorimeters of the instrument. No clear trends of accuracy differences between the eight calorimeters are visible
Monte Carlo simulations using the standard deviations above (0.0075 K for temperature difference, 0.013 mW mV-1 for calibration coefficients, and 0.06 V⋅s for both sample and empty integrals) gave the following RSDs for the different substances: 1.1% for ethylene glycol, 0.7% for water, 1.4% for TCM and DCA, and 2.5% for copper and sapphire (the differences between these values are only the result of that the sample peaks have different sizes). When the ethylene glycol sample sizes were doubled, the simulated RSD decreased from 1.1 to 0.65%. These values are in reasonable agreement with the RSDs seen in Fig. 2 and also explain the relatively large spread in the sapphire results, albeit they do not explain why copper does not show a similarly large spread.
Other error sources
One potential source of error in the present measurement is that all non-aqueous liquids measured on are hygroscopic and if they take up water, their heat capacity will change. However, the used liquids were new, had a low water content, and the bottles were not opened more than necessary to transfer samples to the vials. Figure 2 also shows that most results were close to the literature values and those liquid that showed deviations of more than 1% gave lower heat capacities than expected, and we assume that the heat capacity of hygroscopic liquids would increase by the uptake of water as water has a significantly higher specific heat capacity than any of the tested non-aqueous liquids.
The present method is based on the assumption that the used “isothermal heat conduction calorimeter” accurately measures the heat that flows into and out of the samples also under non-isothermal conditions (after a change in the thermostat temperature). Under normal “isothermal” use, an isothermal heat conduction calorimeter (of the type used in the present study) will have a maximal temperature difference between the sample and the heat sink of about 0.2 K, and still work in its linear range [25] (i.e., with a heat production/consumption rate proportional to the heat flow sensor output at steady-state conditions); during the transition from one temperature to another in the present type of measurement, the internal temperature differences could be slightly higher, possibly leading to nonlinear behavior. However, we believe that this is not the case.
We can envisage two reasons for a different (nonlinear) behavior of a calorimeter during a temperature change compared to constant temperature conditions. Firstly, a temperature increase could induce convection in the air, which does not occur at isothermal conditions because of the small temperature differences inside the calorimeter. However, the slight (2 K) temperature steps made and the low rate at which they take place does not lead to much higher temperature differences inside the calorimeter than the 0.2 K mentioned above, and significantly higher temperature differences are needed for natural convection to take effect (see Supplementary material). Secondly, that the radiative part of the heat transfer—which has a power-of-four behavior (Stefan–Boltzmann law)—is nonlinear. However, for radiation between two surfaces with a small temperature difference, radiation is also approximately linear (see Supplementary material). So, we believe that the behavior of a heat conduction calorimeter is the same during an “isothermal” measurement and during the 2 K temperature transitions that we have made.
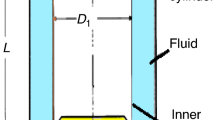
The final ethylene glycol measurement, with 20 mL samples, (Table 2) and the study with different sample sizes of glycerol (Fig. 4) both showed that larger sample sizes tend to result in lower measured specific heat capacity. It is probable that this is related to the fact that the calibration coefficient of a heat conduction calorimeter has a small dependence on where the heater is placed during an electrical calibration; the results from different ways of calibrating can differ by a few percent [10] [25]. This is schematically illustrated in Fig. 5 together with vials with 10- and 20-mL liquid samples. As is seen in Fig. 5A, B, measurements can be made with different sample sizes, and as is seen in Fig. 5C, D, calibrations can be made with heaters placed in different positions. Most commonly a built-in heater (internal/fixed heater, placed in the vial holder of the instrument) is used as such calibrations can be automated in commercial instruments. Sometimes the vial holder is left empty for such calibrations, in other cases empty vials (as is shown in Fig. 5D) or vials with a certain amount of an inert material are used. The alternative to internal is to use calibration heaters placed in the same type of vials as are used for measurements (external/insertion heaters, Fig. 5C). Then heat will be produced in a similar position during calibration and measurement.

Schematic illustrations of a vial in a vial holder in contact with a heat flow sensor placed on a heat sink in the type of calorimeter used in the present study. A With a 10 mL liquid sample (measurement). B With a 20 mL liquid sample (measurement). C With a heater placed in about 8 mL polyurethane rubber (used in the present calibrations) (calibration). D The built-in heater placed in the bottom of the vial holder (calibration)
In the present study, we measured calibration coefficients with both internal and external heaters. From the result (Fig. 6), it is seen that the external (insertion) heaters consistently gave calibration coefficients that were about 1.5% higher than those for the internal heaters. In the present evaluation we used the results from insertion heaters.

Calibration coefficients for the eight calorimeters or the I-Cal Ultra instrument used calculated from three calibrations with external (insertion) heaters and two calibrations with internal (fixed) heaters
The reason that the calibration heater position influences the calibration coefficient is that not all produced heat is conducted out through the heat flow sensor, and the fraction of the heat that leaves through other heat flow paths (heat conduction in air, radiation) is slightly dependent on where the heat is produced. If the heater is closer to the heat flow sensor, less heat will leave through other paths. The same is also true for measurements: if the sample is higher up in the vial, further away from the heat flow sensor, more heat will leave through other parts. These effects are small (a few percent), but we believe that they are clearly seen in the present measurements.
Summary of uncertainty analysis
There were two cases of clear systematic deviations. Firstly, the measured heat capacity of DCA was about 2.5% lower than the literature value. We believe that this discrepancy may be caused by a too high literature value. Secondly, the ethylene glycol measurement with a larger sample also gave about 2.5% lower heat capacity than the literature value. But in this case, the measurements with different masses of glycerol, show that a 20 g sample has approx. 2.5% lower evaluated heat capacity than a 10 g sample, and this effect can be explained by that slightly less heat from the top of a large sample is measured, as discussed above.
There were two cases that deviated significantly from the general 1% RSD: sapphire that has an RSD of more than 2%, and the final ethylene glycol measurement with a larger sample that had an RSD of about 0.5%. We believe that the large spread in the sapphire values is caused by that those samples had significantly lower heat capacities than most other samples and that the low difference between sample and empty integrals then results in an increased spread in the results.
The overall accuracy of the measurements performed using the method proposed in this study is depicted in Fig. 7. In this figure, the results obtained from ethylene glycol (EG) samples exceeding 20 g have been excluded due to the systematic underprediction of oversized samples. Similarly, all measurements of DCA have been excluded as its deviation may stem from a too high literature value. Consequently, the remaining measurements encompass all solid samples, along with water, EG, and TCM. Notably, 62% of these measurements fall within 1% deviation from their respective literature values.

Relative deviation (see text). A total of 62% of the measurements deviated by less than 1% from their corresponding literature values
Conclusions
The results demonstrate the functionality of the employed method, showing that it produces accurate and precise measurements for several types of substances, including water, ethylene glycol, [EMIM][TCM], and copper. The measured values of these substances are close to their respective literature values, with a general RSD of 1%. Notably, water stands out as the most accurately measured substance, with a mean deviation as low as 0.22%. The robustness of the measurement method is evident, as accurate results are obtainable from both temperature increase and temperature decrease measurements.
Furthermore, the study shows that sample size has a significant influence on the results from measurements on liquid samples. This behavior arises from the relationship between sample size and the placement of the calibration heater, which plays a crucial role in achieving a high measurement accuracy.
Lastly, the study highlights the need for further investigations regarding the specific heat capacity measurements of the ionic liquid [EMIM][DCA]. The results obtained consistently fall below its literature value.
Overall, this study shows that isothermal heat conduction calorimeters can be used to determine accurate heat capacity of solid and liquid substances.
Abbreviations
- C :
-
Specific heat capacity (J g−1 K−1)
- I :
-
The peak integral (V s)
- m :
-
Mass (g)
- Q :
-
Heat (J)
- T :
-
Temperature (K)
- t :
-
Time (s)
- U :
-
Voltage (V)
- δ :
-
Relative deviation 1
- ε :
-
Calorimetric calibration coefficient (W V−1)
- λ :
-
Thermal conductivity (W m−1 K−1)
- ρ :
-
Density (g m−3)
- EG:
-
Ethylene Glycol
- DCA:
-
1-Ethyl-3-methylimidazolium Dicyanamide, [EMIM][DCA]
- TCM:
-
1-Ethyl-3-methylimidazolium Tricyanomethanide, [EMIM][TCM]
- RSD:
-
Relative Standard Deviation (standard deviation divided by the mean)
References
Flynn J. Analysis of DSC results by integration. Thermochim Acta. 1993;217:129–49.
Gustafsson S. Transient plane source techniques for thermal conductivity and thermal diffusivity measurements in solid materials. Rev Sci Instr. 1991;62(3):797–804.
Westrum EFJ, Furukawa GT, McCullough JP. In: Experimental thermodynamics. Vol I, London, Butterworths, 1968; 133
Suurkuusk J, Wadsö I. Design and testing of an improved precise drop calorimeter for the measurements of the heat capacity of small samples. J Chem Thermodynam. 1974;6:667–79.
Gaur U, Mehta A, Wunderlich B. Heat capacity measurements by computer-interfaced DSC. J Thermal Anal. 1978;13:71–84.
O’Neill M. Measurements of specific heat functions by differential scanning calorimetry. Anal Chem. 1966;38(10):1331–6.
Mraw S, Naas D. The measurement of accurate heat capacities by differential scanning calorimetry. Comparison of d.s.c. results on pyrite (100 to 800) with literature values from precision adiabatic calorimetry. J Chem Thermodyn. 1979;11:567–84.
Wunderlich B, Boller A, Okazaki I, Ishikiriyama. Heat-capacity determination by temperature-modulated DSC and its separation from transition effects. Thermochim Acta 1997;304–205: 125–136.
Bunyan PF. A technique to measure the specific heat of reactive materials by heat flow calorimetry. Thermochimicu Acta. 1988;130:335–44.
Bunyan P. An absolute calibration method for microcalorimeters. Thermochim Acta. 1999;327:109–16.
Ditmars DA, Ishihara S, Chang S, Bernstein G, West E. Enthalpy and heat-capacity standard reference material: synthetic sapphire (α-Al2O3) from 10 to 2250 K. J Res Natl Bur Stand. 1982;87:159–63.
Ubelhor R, Ellison D, Pierce C. Enhanced thermal property measurement of a silver zinc battery cell using isothermal calorimetry. Thermochim Acta. 2015;606:77–83.
Chase MW. NIST-JANAF themochemical tables fourth edition. J Phys Chem Ref Data 1998;1–1951.
Çengel Y, Cimbala JM, Turner R. Apendix A, Property Tables and Charts. In: Fundamentals of Thermal-Fluid Sciences. 4th ed. New York: McGraw-Hill Education; 2012. p. 998.
Stephens MA, Tamplin WS. Saturated liquid specific heats of ethylene glycol homologues. J Chem Eng Data. 1979;24:81–2.
Popa CV, Nguyyen CT, Gherasim I. New specific heat data for Al2O3 and CuO nanoparticles in suspension in water and Ethylene Glycol. Int J Therm Sci. 2017;111:108–15.
Atalla S, El-Sharkawy A, Gasser F. Measurement of thermal properties of liquids with an AC heated-wire technique. Int J Thermophys 1981;151–162.
Dean JA. Lange's Handbook of Chemistry, McGRAW-HILL, INC., 1999.
Navarro P, Larriba M, García J, Rodríguez F. Thermal stability and specific heats of [emim][DCA] + [emim][TCM] mixed ionic liquids. Thermochim Acta. 2014;588:22–7.
Safarov J, Nachhu P, Hassel E, Müller K. Thermophysical properties of 1-ethyl-3-methylimidazolium dicyanamide in a wide range of temperatures and pressures. J Mol Liq 2021;332.
Barros N, Salgado J, Rodrıigues-Anon JPJ, Villanueva M, Hansen L. Calorimetric approach to metabolic carbon conversion. J Therm Anal Calorim. 2010;99:771–7.
Skaria C, Gaisford S, O’Neill M, Buckton G, Beezer A. Stability assessment of pharmaceuticals by isothermal calorimetry: two component systems. Int J Pharmaceutics. 2005;292:127–35.
Wadsö L, Johansson S, Bardage S. Monitoring of fungal colonization of wood materials using isothermal. Int J Biodeterior Biodegrad. 2017;120:43–51.
Pang X, Sun L, Sun F, Zhang G, Guo S, Bu Y. Cement hydration kinetics study in the temperature range from 15 ◦C to 95 ◦C. Cem Concrete Res. 2021;148: 106552.
Wadsö L. Operational issues in isothermal calorimetry. Cem Concrete Res. 2010;40:1129–37.
Mudd CP, Gershfeld N, Berger R, Tajima K. A differential heat-conduction microcalorimeter for heat-capacity measurements of fluids. J Biochem Biophys Methods. 1993;26:149–71.
Linderoth O, Wadsö L, Jansen D. Long-term cement hydration studies with isothermal calorimetry. Cem Concrete Res. 2021;141: 106344.
Persson C. GUM – Guide to the Expression of Uncertainty in Measurement. In: Q-KEN, Riga, Latvia, 2011.
Anon. Standard reference material 720. National Bureau of Standards, Washington DC, 1982.
Acknowledgements
We would like to acknowledge the financial support provided by The Swedish Research Council (MH) and Calmetrix Inc. (LW) for this research study.
Funding
Open access funding provided by Lund University.
Author information
Authors and Affiliations
Contributions
Both authors contributed to the idea of the study and the design of how to perform it. Sample preparations, measurements and analysis were performed by L.W. and M.H. Both authors contributed to the writing of the article and the supplementary material by writing different sections of the draft. Both authors read and approved the final manuscript as well as the supplementary material.
Corresponding author
Ethics declarations
Conflict of interests
MH has no known competing financial interests or personal relationships that have influenced the work reported in this paper. LW is an active co-owner of Calmetrix who produced the instrument used in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hothar, M., Wadsö, L. Accurate heat capacity determination of solids and liquids using a heat conduction calorimeter. J Therm Anal Calorim 149, 2179–2188 (2024). https://doi.org/10.1007/s10973-023-12824-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-023-12824-8