
Abstract
In this paper, the composition and properties of two different activated calcinated clays were investigated under selected curing conditions. The tendency of metabentonite and metakaoline to form geopolymers and zeolites was investigated by FTIR spectroscopy, TGA and DSC techniques, PXRD, HT-PXRD, SEM, and BET-N2 adsorption analyses. The selection of the optimal sample compositions was done on a basis of preliminary results obtained from bentonite and kaolin clays activated at calcination temperatures (700, 750, 800 °C) with a holding time of 3 h in a combination with different NaOH concentrations (5, 10, 20 mass%). In a more detailed examination, the samples calcined at a temperature of 800 °C with different NaOH concentrations were studied. For metakaolin-based samples, dominant geopolymer formation was achieved using 5 mass% NaOH. The zeolite amount increased concomitantly with the concentration of the alkali activator, whereas zeolite A and zeolite P were determined as the predominant phases. For metabentonite-based samples, 10 mass% NaOH concentration was found to be optimal by the means of geopolymer formation. At higher alkaline activator concentrations, the presence of zeolitic phases faujasite and gobbinsite was confirmed. However, the determined degree of crystallinity was significantly lower than in the corresponding activated metakaolin samples. With a gradual increase in NaOH concentration, the specific surface area of the metabentonite-based samples increased reaching a value of 218.95 m2 g−1 promising for adsorption applications. The opposite effect was observed for the metakaolin-based samples, and this result is caused by the increased formation of zeolite phases and corresponding changes in their channel dimensions.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Alkali-activated binders or geopolymers are materials formed from a mixture of an aluminosilicate material with media solutions such as NaOH, sodium or potassium silicate, and phosphoric acid.[1, 2]. While metakaolin (MK) is generally very often used as supplementary cementitious materials [3,4,5,6] as well as for geopolymers or zeolites preparation [7,8,9,10], information about metabentonite (MB) is rather limited in the available literature [11, 12]. However, as kaolin deposits are restricted and the sources of high-purity kaolinite are utilized also by other industries, other clay minerals begin to attract attention as resources for alkali activated materials.
The main differences between metakaolin and metabentonite result from their chemical composition and atomic structure following their dominant parent minerals. MK is prepared by thermal treatment of source soil, kaolin, mainly consisting of kaolinite clay. Kaolinite belongs to the layered 1:1 clay minerals, in which each layer is formed by one tetrahedral (SiO4) and one octahedral (AlO6) sheet. As a consequence of hydrogen bonding between oxygen atoms and hydroxyl ions of the adjacent layers and a low extent of isomorphous substitution its specific surface and cation exchange capacity are rather low, therefore it does not possess any permanent layer charge, interlayer cations, or swelling behavior [13].
On the contrary, MB is obtained by heating bentonite, the main mineral of which is montmorillonite (accompanied by illite or kaolin clays depending on the sources). This 2:1 clay mineral is composed of layers containing one octahedral sheet sandwiched between two tetrahedral sheets [14]. Isomorphic substitutions with cations with comparable ionic radii can occur in octahedral as well as tetrahedral sites and determine the net layer charge on the unit cell. Very often, Si4+ ions in the tetrahedral sheet may be replaced by Al3+ ions and Al3+ in the octahedral sheet by Mg2+. Also, other larger cations, such as Ti4+, Fe3+, Fe2+, Zn2+, and Mn2+, tend to occupy the octahedral sites [15]. To balance the negative layer charges resulting from these substitutions, montmorillonite hosts interlayer cations (e.g. Ca2+, Na+, Mg2+, K+, and Sr2+) [16, 17]. Their hydration and subsequent osmotic behavior of their diffuse double layers result in swelling behavior. Various arrangements and compositions of sheets cause different physical and chemical properties [18].
As a consequence, MK displays higher aluminum content, while MB has higher silicon content and involves the mentioned ions.
Dehydration of both main minerals, kaolinite, and montmorillonite leads to the reduction of the Al coordination in the octahedral sheet from sixfold to the distribution of 5-, 4- and sometimes threefold coordination [14, 19, 20, 21], among which the Al sites in fivefold coordination are the most reactive [16, 22, 23].
As Al–OH groups of the kaolinite octahedral sheets are directly exposed to the interlayer, the structural disorder can be attained more easily upon heating than in the case of montmorillonite [24, 25]. The loss of the hydroxyl groups forms vacant sites, to which aluminum ions migrate. Layers delaminate and buckle. Dehydroxylation and amorphization of kaolinite thus occur simultaneously, while montmorillonite dehydroxylation (and the associated reduction in coordination number of Al atoms) occurs without full amorphization [14], retaining the layer structure [26]. The temperature of montmorillonite dehydroxylation depends on the valency and ionic radius of the interlayer cations. Divalent exchangeable cations, e.g. Ca2+, bind more water and more tightly than the monovalent exchangeable cations, such as Na+ [27, 28]. Dehydroxylation temperature thus increases with ionic radius [27].
Consequently, the higher reactivity of MK than that of MB can be attributed to the more disordered structure consisting of higher amorphous aluminate content, highly reactive AlV coordination form of aluminum, and its larger specific surface area [14].
Besides, in both cases, 1:1 and 2:1 clay minerals, the dissolution rate is dependent on the edge surface area available rather than the total surface area [29]. However, while dissolving of kaolinite is approximately congruent [30], based on the study of Garg and Skibsted [31], calcination temperature influences dissolution of calcined montmorillonite. When it was calcined at 800 °C, its dissolution was incongruent, and it turned to congruent at 900 °C. The authors explained it by higher amount of amorphous aluminosilicate resulting in weaker Si–O–Si/Al bonds and subsequent faster dissolution of Si than Al in the case of calcinated clay at 800 °C.
Calcination temperature and duration significantly affect the process of alkali activation and relating phase formation [32, 33]. Among other important factors belong concentration and type of alkaline activator, amount of water, Si/Al ratio, and curing conditions as well. Higher temperatures and/or longer periods result in the transformation of the metastable geopolymers into more stable zeolitic structures [34]. Higher concentrations of activation solutions decrease crystallinity, resulting in the preferential formation of geopolymers [34, 35]. In general, lower Si/Al ratio (< 1.5) induces zeolite formation, on the contrary higher (Si/Al > 1.5) geopolymer formation [36]. During geopolymerization, water is inevitable for dissolution of solids and following hydrolysis of dissolved ions. The increasing amount of water thus accelerates these processes. High amount of water will have dilution effect and will also kinetically hinder polycondensation, in which water is a product.
Although the potential of MB results from its Si/Al ratio, which preclude the need for additional soluble silica in an activator, its lower reactivity due to reduced amorphousness is expected when compared with MK. The promising ways how to attain its dehydroxylation are mechanical [37, 38] and acid treatment [16, 39, 40]. The second, chemical treatment, can, however, lead to the undesired aluminum leaching.
Because of variable composition of the source soils, especially those of bentonite, process parameters for geopolymer preparation need to be adjusted properly. The purpose of our work was to study the impact of calcination temperature and concentration of alkaline activator on the geopolymerization process of metabentonite and metakaolin-based geopolymers. The combination of PXRD, HT-PXRD, TGA/DSC, BET, SEM, and FTIR spectroscopy was used to evaluate the impact of process parameters thoroughly. To the best of our knowledge, such a comparative study has not yet been performed.
Materials and methods
Samples’ composition and raw materials
A composition of studied samples is depicted in Table 1. Kaolin OT 76 (Sedlecký kaolin, a. s., Božičany, Czech republic) and Bentonite P 030 (ENVIGEO, a.s., Banská Bystrica, Slovakia) were used as raw materials. Their chemical composition is shown in Table 2 [41, 42]. Sodium hydroxide with 99% purity (Slavus, s.r.o., Bratislava, Slovakia) was used as an alkali activator.
Calcination of raw materials
Kaolin and bentonite powder samples were calcined at three different temperatures (700, 750, and 800 °C) in a muffle furnace (heating rate 12 °C min−1, holding time 3 h). After spontaneous cooling, the samples were stored in sealed plastic bags under argon. FTIR confirmed total dehydroxylation of calcined samples (Fig. 1).

FTIR spectra of (a) kaolin, calcinated, and alkali activated metakaolin samples; b bentonite, calcinated and alkali activated metabentonite samples
Preparation of geopolymers
Geopolymer samples were prepared using metakaolin (MK) and metabentonite (MB) as binders and sodium hydroxide (NaOH) in three different mass concentrations (5, 10, and 20%) as alkali activator.
NaOH was added to MK and MB while stirring. Homogenization was performed by high-speed mixer (Standmixer 10,027,051, Klarstein, CHAL-TEC Vertriebs–Handels GmbH, 38 000 rpm for 5 min. Fresh mixtures were poured into 8 × 2 × 2 cm moulds and quickly placed in the oven over water bath. After 7 days at 80 °C, the samples were removed from the oven and cooled down to room temperature in a desiccator. The cooled samples were grounded to a fine powder and placed in Eppendorf tubes until the time of analysis.
Characterization of samples
Powder X-ray diffraction patterns (PXRD) of raw, calcined, and alkali-activated MB and MK samples were recorded under ambient conditions in Bragg–Brentano geometry using Ni-filtered CuKα radiation (λ = 0.1541 nm) with a Panalytical Empyrean diffractometer equipped with a PIXcel3D detector (Malvern Panalytical, Kassel, Germany). The PXRD patterns of powder samples were recorded in a range of 2–45° 2Θ with a 0.0263° 2Θ step size and scan speed 0.0223° 2Θ·s−1. All patterns were analyzed using Panalytical’s Highscore Plus software.
Phase transitions were monitored in situ using an HTK 16N high-temperature chamber (Anton Paar, Graz, Austria). Few drops of mechano-activated sample dispersed in ethanol were dropped onto the Pt strip. After ethanol evaporation and thin film formation, the sample was heated in air at a rate of 50 °C·min−1 up to selected temperature. The high-temperature (HT) PXRD pattern scans were recorded in a range of 5–35° 2Θ with a 0.0263° 2Θ step size and scan speed 0.135° 2Θ·s−1. Degree of crystallinity was calculated as the ratio of the integrated area of crystalline diffraction peaks and the sum of all the integrated area including the amorphous phase. The latter was determined separately by measuring a background (holder without sample) and then subtracted from the integrated area omitting the crystalline phase.
The specific surface areas (SSA) of the samples were determined by nitrogen adsorption using a Quantachrome Autosorb iQ apparatus. Before each measurement, the samples were 12 h degassed at 105 °C. The specific surface area calculation was based on the Brunauer--Emmett--Teller (BET) formalism [43].
The IR spectra were measured on a Nicolet 6700 Fourier Transform Infrared (FTIR) spectrometer from Thermo Scientific™. For the MIR region (4000–400 cm−1), the spectrometer was equipped with a DTGS detector, IR source and KBr beam splitter. One milligram of a sample was mixed with 200 mg KBr in an agate mortar and pressed into a pallet. The spectra were normalized to the stretching Si–O band occurring approximately at 1040 cm−1. The spectra were collected using Thermo Scientific OMNIC™ software package. The second derivative function, with the Savitsky-Golay derivative filter, was applied to find the exact position of the vibrational bands appearing as inflections/shoulders in the IR spectra. In order to eliminate the water present in the KBr pellets as much as possible, samples were dried overnight at 105 °C before measurement.
The thermal stability of individual geopolymers was investigated by TGA/DSC analysis (TGA/DSC-1, STARe software 9.30, Mettler Toledo). The 50 ± 0.1 mg of powder sample was used for analysis. The samples were placed in platinum crucibles and gradually heated to 1000 °C at the heating rate of 10 °C min−1 in the atmosphere of synthetic air (purity 5.0).
Scanning electron microscope (SEM) Vega 3, Tescan, Czech Republic equipped with SE and BSE detectors was used for observing samples morphology at a voltage of 30 kV. The samples were coated with gold.
Results and discussion
Structural characterization of raw and modified samples by FTIR spectroscopy
Figure 1a shows the MIR spectra of kaolin, metakaolin, and particular alkali-activated samples. The spectrum of kaolin shows four absorption bands in the OH stretching region. The first three vibrations at 3690, 3668, and 3652 cm−1 are stretching vibrations of outer surface hydroxyl groups. These three OH groups are located on the octahedral surface of the layers and form weak hydrogen bonds with the oxygen of the Si–O-Si bonds on the lower surface of the next layer [44, 45]. The absorption band nearby 3621 cm−1 belongs to inner hydroxyl groups, lying between the tetrahedral and octahedral sheets. Stretching vibrations in Si–O groups in kaolin occur between 1150 and 980 cm−1. Bands due to AlOH bending vibrations for Al in octahedral coordination can be observed at 911 and 700 cm−1. At 798 cm−1, the absorption band belonging to the bending Al-O in AlO6 octahedral coordination occurs and overlaps with Si–O vibration from quartz. Another band of lower intensity can be noted at 754 cm−1 and it is assigned to Si–O–AlIV bending vibration. On the contrary, more intense absorption band at 539 cm−1 is caused by the bending vibration of Si–O–AlVI and the one at 468 cm−1 by Si–O–Si bending vibrations. The last absorption band observed in the IR spectrum of kaolin at 432 cm−1 is assigned to δ(Si–O).
After calcination (Fig. 1a), dehydroxylation can be clearly observed in the IR spectra. In addition, instead of well-defined and sharp absorptions in the region of νSi–O vibrations, a broad band was formed between 1250 and 940 cm−1. Structural disordering resulted also in the formation of a wide band between 516 and 400 cm−1 and the disappearance of absorption bands connected with octahedral coordination of Al3+ in kaolin due to its calcination. The maximum around 802 cm−1 can be attributed to the Al–O bending vibration in the tetrahedral coordination [46].
After alkali activation, the formation of hydrated aluminosilicates confirms the occurrence of the stretching and bending absorption bands of OH groups in the region between 3700 and 3100 cm−1 and around 1655 cm−1, respectively [47]. The intensity of particular bands increases especially with increasing NaOH concentration. The effect of calcination temperature seems to be less evident. As the concentration of alkaline activator (5, 10, and 20 mass%) and calcination temperature (700, 750, 800 °C) increases, the band attributed to νSi–O and νAl–O gradually shifts to lower wavenumber. Based on the literature [47, 48, 49, 50], the shift is caused by the presence of non-bridging oxygen in the network, the incorporation of Si and Al into geopolymeric gel, and the state of hydration of the reaction products. Since Al–O bond is weaker than Si–O bond, the shift is more significant when the content of Al in tetrahedral positions in the network is higher. The higher the NaOH concentration, the narrower this most intense band is, which indicates a growing share of crystalline phases [51].
Unlike activated metabentonite samples, the presence of carbonates appeared only in the case of the highest calcination temperature and the highest NaOH concentration, which could again suggest zeolitization process. Król et al. [52] claimed that the high content of the zeolite phase is related to the lack of carbonate bands since Na+ ions are preferentially bound in the zeolite structure and cannot depolymerize amorphous skeleton or break the Si–O–Si bridges. In the MIR spectrum of the particular sample, in addition to the maximum at 1450 cm−1, two weak absorption bands at 1560 and 1400 cm−1 point out the presence of bicarbonate.
Besides, the intensity of the absorption band at 802 cm−1 decreases with increasing concentration of NaOH and it is no longer observed, when the highest amount of NaOH was applied. In the range of the so-called pseudolattice vibrations between 800 and 500 cm−1, originating from ring structures characteristic for zeolites, a significant increase in absorption band at 560 cm−1 can be observed. Several studies [51, 52] assigned this band to the vibration from zeolite A. Its presence and preferential formation in these systems were confirmed also by PXRD analyses (Section “XRD phase characterization”). As was mentioned, the occurrence of this band was recorded also in the spectra of calcined samples, the PXRD patterns of which have, except for muscovite and quartz, amorphous character (Fig. 6). It can be interpreted as an indicator of the condensation of the tetrahedra to 4-membered rings [52].
The 10 mass% NaOH seems to be the optimal concentration for alkali activation of metakaolin samples leading to the dominant geopolymer formation since increased concentration of alkaline activator also led to the appearance of the band at about 678 cm−1 and the weak one at 748 cm−1. They descend from the structure of crystalline zeolite P (Fig. 1a) because of double-ring external linkage and symmetric stretch vibration of the internal tetrahedron, respectively [53]. Also, the degree of crystallinity (DOC) was observed very high in the case of MK800 activated with 20 mass% NaOH (Fig. 8).
The MIR spectrum of bentonite (Fig. 1b) shows a characteristic absorption band at 3630 cm−1 belonging to stretching vibrations of structural OH groups coordinated to Al [54]. The absorption band at 3424 cm−1 due to stretching vibrations of OH in water molecules can come from both bentonite and also from hygroscopic KBr.
Slightly visible absorption bands appearing at 2925 and 2854 cm−1 are assigned νasCH2 and νsCH2 stretching, respectively. The reason for the presence of weak organic groups in inorganic bentonite is probably the handling of the sample before the measurement itself. This phenomenon has also been observed in kaolin samples.
The bending vibrations of water molecules occur at 1637 cm−1. The absorption bands appearing around 1500–1380 cm−1 relate to the asymmetric stretching of the O–C–O bonds of CO32− due to carbonation on the surface [55, 56].
The most intense and complex band in the infrared (IR) spectrum of bentonite at 1041 cm−1 is attributed to the stretching vibration of Si–O groups of the tetrahedral sheets. Absorption bands at 918, 890, and 848 cm−1 belong to the bending vibration of δAl–Al–OH, δAl–Fe–OH, and δAl–Mg–OH groups, respectively. The characteristic absorption band of silica (α-quartz) is observed at 797 cm−1 and the band at 625 cm−1 is assigned to the bending vibration of Al–O groups [57]. The band at 693 cm−1 is probably due to the vibrations of six-membered rings, which can be also present in the quartz [58]. The last two absorption bands in the IR spectrum of bentonite at 523 and 467 cm−1 belong to Al–O–Si and Si–O–Si bending vibrations, respectively.
Calcination at 700 °C led to the dehydration and partial dehydroxylation of bentonite manifesting by a significant decrease of absorption bands in the regions of stretching and bending vibrations of OH in water and the disappearance of the band at 3630 cm−1. Instead, as a result of hydroxyl groups rearrangement, the new one manifested at 3675 cm−1. Higher calcination temperature caused total dehydroxylation of bentonite and no sign of a metal-bonded hydroxyl group is visible. The dominant band due to Si–O stretching vibration broadened and its maximum shifted to higher wavenumbers, which indicates structure disordering. Already after calcination at 700 °C, a shoulder appeared around 1136 cm−1 and after treatment of bentonite at 800 °C, the additional one at 1084 cm−1 was developed. Thermal treatment resulted also in the decomposition of carbonates. Absorption bands coming from Al–Al–OH, Al–Fe–OH, and Al–Mg–OH bending vibrations disappeared. In the same region, an absorption band at 797 cm−1 increases with temperature, which together with the occurrence of the band at 1084 cm−1 demonstrates the increasing ratio of amorphous silica. Besides, the formation of other new absorption bands can be observed after thermal treatment of bentonite, namely at 864, 733, 644, 569, 484, low intensity 438, and 423 cm−1. The band at 484 cm−1 comes from the shift of the original one from the position at 467 cm−1 because of the disturbances in the silicate structure. An increase in calcination temperature caused a further shift of this band back to 463 cm−1, which again confirmed the formation of amorphous silica. Both absorption bands, at 484 and 569 cm−1, were after calcination at 700 °C observed also by Holtzer et al. [59], but their origin was marked as unknown. Here, the occurrence of the second one is attributed to the shift of the absorption band located in the bentonite sample at 524 cm−1 because of the Al3+ coordination reduction leading to the shortening of Al–O distances [60]. The intensity of this band together with that at 733 cm−1 decreased with increasing calcination temperature. Further newly appeared absorption bands persist after the increase in temperature, which indicates that OH− is not involved in their formation. They can be assigned also to the reduction of aluminum coordination number and formation of different silicate and aluminosilicate ring structures [58, 61], while that at 423 cm−1 to bending vibrations of O–Al–O [62].
As in the case of alkali-activated metakaolin samples, absorption bands due to vibrations of structural OH as well as OH in water increase with increasing NaOH concentration and calcination temperature. However, in the case of 20% NaOH, particular intensities decrease with increasing temperature. Besides, in the case of samples calcined at 800 °C, the higher the NaOH concentration, the lower are OH absorption bands. Already 5% NaOH led to the appearance of stretching vibrations in the region of metal bonded OH. An increase in the calcination temperature, however, caused that the well-defined bands changed to shoulders. This is also the case with higher NaOH concentrations. It suggests that only short distance order exists in the forming prevailing geopolymer structures. The maximum in the region of OH stretching vibrations shifts to higher wavenumbers with increasing calcination temperature.
The presence of carbonated phases is confirmed in the case of all alkali-activated metabentonite samples. Based on the MIR spectra, their amount increases with NaOH concentration. Especially in the case of 20% NaOH, particular stretching absorption bands decrease with increasing calcination temperature.
The position of the main νSi–O band moves to lower wavenumbers mainly as a consequence of higher NaOH amount, less significantly also with increasing calcination temperature demonstrating the formation of geopolymer structures. An increase in NaOH concentration for samples calcined at 700 °C resulted in the spread of this dominant absorption band, while activation of samples calcined at 800 °C led to its narrowing with increasing concentration of alkaline activator. Similarly, the higher the calcination temperature, the wider the band is in the MIR spectra of samples activated by two lower NaOH concentrations. On the contrary, the band narrows in the case of 20% NaOH with increasing calcination temperature. These findings agree with the observations in the area of OH and C=O vibrations occurrence and similarly to the results for activated metakaolin samples confirm the increasing amount of crystalline zeolites in the samples calcined at the highest temperature and by using higher alkaline activator concentrations. The degree of crystallinity determined based on the results of PXRD is around 20%, thus significantly lower than in the case of corresponding activated metakaolin samples (Section “XRD phase characterization”).
The absorption band that appeared after calcination at 1136 cm−1 persisted also after the process of alkali activation. It only changed from the well defined to the shoulder, which relates to the broadening of the main band. Also, the bands between 800 and 500 cm−1, which occurred following the process of amorphization, can be still observed after activation with 5% NaOH. An increase in temperature and concentration of alkaline activator caused their diminishing. The band at 797 cm−1 assigned to amorphous silica and quartz disappeared after alkali activation with 10 and 20% NaOH. Instead, the absorption band around 780 cm−1 appeared and moved to lower wavenumber with increasing NaOH concentration as well as calcination temperature. Besides, other new absorption bands can be observed around 570 and 690 cm−1 already in the MIR spectra of samples activated by 5% NaOH. As in the case of metakaolin-based samples, changes in this region of MIR spectra reflect the formation of different ring structures. In the samples activated by 10 and 20% NaOH, the presence of zeolitic faujasite-Na and gobbinsite was proven by PXRD (Fig. 7), while the lowest concentration of alkaline activator did not result in the formation of any zeolite phase. Based on the studies [63, 64], the mentioned band around 780 cm−1, the newly forming one at 688 cm−1, and 998 cm−1 represent symmetrical stretching vibrations corresponding to the external and internal TO4 structure (T = Si, Al), respectively, all from the structure of prevalent faujasite. Also, the hump at 1384 cm−1 can be attributed to the double-ring external link in the faujasite structure.
In the region of lower wavenumber, the absorption band occurring at about 423 cm−1 gradually disappeared, which, together with the shift of the main νSi–O band, reflects the incorporation of Al3+ into the structure of geopolymer and zeolites. Previously observed bands around 436 and 484 cm−1 merged. When 5 and 10% NaOH were used for activation of metabentonite samples, the position of the new main maximum moved to lower wavenumber as the calcination temperature increased. In the case of 20% NaOH, the opposite trend can be observed. This again points to the increasing amount of crystalline zeolites with more rigid structures than geopolymers, which are more resistant to the change of bending angle.
Thermal analysis (TG/DTG/DSC)
An inorganic structure and resulting internal stability make the geopolymers promising materials for high-temperature applications, e. g. fire-retardant coatings or structural concrete in fire-prone areas [65]. Alkali-activated samples prepared using metakaolin and metabentonite thermally treated at optimal calcination temperature, 800 °C, were therefore investigated using thermal analysis. Particular TG and DTG curves are depicted in Figs. 2, 3.

TG/DTG analysis of metakaolin-based geopolymers/zeolites

TG/DTG analysis of metabentonite-based geopolymers/zeolites
Thermal analysis proved a very high degree of both metakaolin and metabentonite dehydroxylation after calcination at 800 °C. Only small mass losses can be observed in the area of interparticle, adsorbed, and interlayer water dehydration (up to ~ 400 °C), while DTG peaks in the temperature interval of kaolinite and bentonite dehydroxylation occurring between 350–580 °C and 400–800 °C, respectively, are totally missing.
On the contrary, the higher the alkali activator amount, the higher the total mass loss of the formed geopolymers/zeolites is detected. The same trend was observed also by other researchers [66,67,68,69] and relates to the finer pore structure able to maintain more free and adsorbed water. Whereas activation of metakaolin by 20 mass% NaOH (GP20_MK800) resulted in a 19% total mass loss, 27% total mass loss was determined for metabentonite-based geopolymer prepared with the same amount of activator (GP20_MB800).
Obviously, higher mass loss due to the release of water especially from the structure of alkali-activated samples resulted in the shift of maxima, corresponding to the DTG peaks, to higher temperatures. The maxima of the most intense DTG peak is located around 155 °C in the case of GP20_MK800. Besides, a shoulder and two small peaks appeared at 103 °C, 323 °C, and 500 °C, respectively (Fig. 3). For the sample GP20_MB800, the maximum of the main DTG peak lies at 68 °C, followed by the one at 122 °C. In addition, several very small mass losses can be observed in the temperature interval between 280 and 630 °C (Fig. 4). In the case of both, metakaolin- and metabentonite-based geopolymers/zeolites samples, the mass losses observed at lower temperatures can be attributed to the dehydration of “free” water and water from the interlayer space. Water release from cavities and cations hydration spheres is expected to takes place up to approximately 400 °C [70, 71]. Khan et al. [72] assigned mass loss between 300 and 450 °C to the water present in Si–O–H bond.

DSC curves of metakaolin-based geopolymers/zeolites
These findings indicate that the binding between the water and the network is stronger in the metakaolin-based geopolymers/zeolites. Besides, these samples demonstrate higher amount of chemically bonded than physically adsorbed water.
The discussed mass losses, related to evaporation, dehydration, and dehydroxylation processes, correspond with the endothermic effects observed in the DSC curves (Figs. 4, 5). In addition to them, the exothermic effects appeared in the metakaolin-based samples above 700 °C that are caused by crystallization. PXRD analysis of GP20_MK800 (Fig. S1) after heating to 1000 °C confirmed the presence of nepheline and carnegieite with traces of quartz, also observed by Kuenzel et al. [73]. The second exothermic maximum at about 970 °C (Fig. 4), observed in the calcined metakaolin sample and geopolymer prepared by its activation with 5 mass% NaOH, can be assigned to the transformation of the metakaolin to the spinel phase followed by the crystallization of mullite [65]. Its presence thus confirms incomplete transformation of metakaolin to geopolymer and/or zeolite structures in the case of the lowest alkaline activator amount. Consequently, it gradually disappeared with increasing alkaline activator amount. On the contrary, the DSC curve of the sample prepared with the highest NaOH concentration exhibits the most intense maximum around 845 °C as well as the new one at 890 °C, which can be explained by the formation of sodium containing aluminosilicate.

DSC curves of metabentonite-based geopolymers/zeolites
Contrary to the metakaolin-based geopolymers/zeolites, the DSC curves of the metabentonite-based alkali-activated samples (Fig. 5) are more complex. Several exothermic effects appeared already above 200 °C. In the case of GP5_MB800 and GP10_MB800 samples, the most intense exothermic effect displayed a maximum of around 300 °C. Metabentonite activated by 20 mass% NaOH has the maximum of the highest exothermic effect at 690 °C, followed by the one at 830 °C. PXRD analysis of this sample (Fig. S1) revealed the presence of nepheline with a small amount of quartz.
BET analysis
The results of gas adsorption analyses are summarized in Table 3. In general, geopolymer formation strongly depends on the specific surface area of starting calcined clay. The higher the surface area, the higher the amount of formed geopolymer can be expected. Too high temperature or too long heating of clays, however, result in the sintering of grains, which will hinder their dissolution by adding an alkaline solution [74]. In our case, the specific surface area of metakaolin (SSAMK800 = 15.18 m2 g−1) is almost three times higher than that of metabentonite (SSAMB800 = 5.08 m2 g−1).
Despite this, the decrease of specific surface area with increasing concentration of alkaline activator was determined in the case of metakaolin-based samples. It relates to the formation of ordered zeolite structures at the selected curing temperature. Not only the crystallization degree (Fig. 8), but also the character of the formed zeolite influenced obtained values. Zeolite A [75], comprising of 8-membered rings with the dimensions 4.1 × 4.1 Å, is opened for the adsorption of nitrogen, the input of nitrogen molecules is strongly restricted by the dimensions of zeolite P channels (3.9 × 3.9 Å), also formed by 8 rings [76]. Hou et al. [66] observed an increasing reaction rate and extent of geopolymerization processes with an increasing Na/Al ratio for the metakaolin-based Na-geopolymer. However, in accordance with our observations, they pointed out that too excessive reaction rate will cause rapid setting and hardening, which in turn will hinder geopolymerization.
On the contrary, the specific surface area of metabentonite-based samples significantly increases with increasing NaOH amount following the process of geopolymerization. The frameworks of zeolite phases detected by PXRD are formed by channels, which do not present an obstacle to the gas used for adsorption analyses, as opposed to zeolites formed in activated metakaolin [76, 77]. Sample GP20_MB800 showed the largest specific surface area 218.95 m2 g−1, which should be a promising value for adsorption processes. Adsorption tests and detailed evaluation of pore characteristics of the prepared geopolymer-zeolite samples will be the focus of further study.
XRD phase characterization
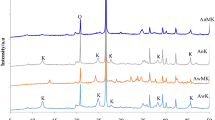
Kaolinite is the dominant phase in kaolin, while minor crystalline phases that are present are quartz and muscovite (Fig. 6). As a result of kaolinite amorphization induced by dehydroxylation at 800 °C for 3 h, the degree of crystallinity (DOC) before and after calcination changes from 32.3 to 4.4% (Fig. 8). After calcination, the kaolinite structure is completely disrupted, and only muscovite and quartz show increased durability under applied conditions. Obtained metakaolin was consequently treated with NaOH solutions with different OH− activities [78]. After treatment with NaOH, quartz and muscovite crystalline phases are still present. Increased OH− activity results in the formation of new crystalline phases, zeolite A and zeolite P (Fig. 6), which is also manifested by an increase of DOC (Fig. 8).

PXRD patterns of kaolin (a), MK800 (b), GP5_MK800 (c), GP10_MK800 (d), and GP20_MK800 (e). Crystalline phases are marked as follows: ●—muscovite (ref. code 00-007-0032), ○—kaolinite (ref. code 01-078-1996), ♦—quartz-low (ref. code 01-083-2469), ⃰—zeolite A (ref. code 00-039-0222), and ◊—zeolite P (ref. code 00-016-0354)
Bentonite is composed mainly of montmorillonite, less dominant crystalline phases are illite and quartz (Fig. 7). Amorphization of montmorillonite induced by dehydroxylation at 800 °C for 3 h decreases DOC from 35.7 down to 5.8% (Fig. 8). After calcination, the montmorillonite structure is completely disrupted, and only illite and quartz remain present under applied conditions [12]. After treatment of metabentonite with 5 mass% NaOH, quartz and illite crystalline phases are still present and the formation of carnegieite is favoured. At 10 and 20 mass% NaOH illite is diminished, while faujasite and gobbinsite are the new dominant zeolitic crystalline phases (Fig. 7).

PXRD patterns of bentonite (a), MB800 (b), GP5_MB800 (c), GP10_MB800 (d), and GP20_MB800 (e). Crystalline phases are marked as follows: ●—montmorillonite (ref. code 96-900-2780), ○—illite (ref. code 96-900-9666), ♦—quartz-low (ref. code 96-901-0147), ⃰—carnegieite (ref. code 00-052-1342), ◊—faujasite (ref. code 00-038-0240), and ×—gobbinsite (ref. code 01-082-1867)

Degree of crystallinity of raw and calcined materials, and selected samples
To bring more light into the better understanding of processes which took place during DSC analyses samples GP20_MK800 and GP20_MB800 were investigated by in situ high-temperature PXRD (HT-PXRD) (Figs. 9, 10). In the case of GP20_MK800 sample (Fig. 9) at room temperature, the particular diffraction peaks can be attributed to muscovite, zeolite A, zeolite P, and quartz. With a gradual increase in temperature, the peaks attributed to zeolite P disappeared. In accordance with findings of Borodina et al. [79] and Huo et al. [80], the peaks of phillipsite became more apparent. This corresponds well with exothermic maximum of GP20_MK800 at around 250 °C on DSC curve. On the other hand, endothermic peak located around 350 °C can be assigned to the phillipsite decomposition. Beyond 650 °C, the diffraction peaks attributed to zeolite P and muscovite are less distinguishable up to their total decomposition. Contrary to this, the nepheline phase begins to crystallize, which is also confirmed by DSC peak located around 850 °C [80, 81].

a and b PXRD patterns of GP20_MK800 at selected temperatures. Crystalline phases are marked as follows: ●—muscovite (ref. code 00-007-0032), ○—kaolinite (ref. code 01-078-1996), ♦—quartz-low (ref. code 01-083-2469), ⃰—zeolite A (ref. code 00-039-0222), ◊—zeolite P (ref. code 00-016-0354), †—phillipsite (ref. code 00-024-1046), and ∞—nepheline (ref. code 01-076-2467)

a and b PXRD patterns of GP20_MB800 at selected temperatures. Crystalline phases are marked as follows: ●—montmorillonite (ref. code 96-900-2780), ○—illite (ref. code 96-900-9666), ♦—quartz-low (ref. code 96-901-0147), ⃰—carnegieite (ref. code 00-052-1342), ◊—faujasite (ref. code 00-038-0240), ×—gobbinsite (ref. code 01-082-1867), ∞—nepheline (ref. code 01-076-2467), and ‡—mullite (ref. code 01-083-1881)
For GP20_MB800 sample (Fig. 10) at room temperature, the particular diffraction peaks can be attributed to faujasite and gobbinsite, whereas the latter is a natural analogue of synthetic zeolite P. With gradual temperature increase, the diffraction peaks attributed to gobbinsite disappeared, similarly to GP20_MK800 sample. The DSC exothermic peak located at 300 °C can be associated with the re-arrangement of less stable zeolite X phase to a more stable zeolite Y phase. Both of these phases belong to family of faujasite-type structure zeolites, differing in their Si/Al atomic ratio. For zeolite X the typical range of Si/Al ratio is from 1 to 1.5, while as the Y-type zeolite has higher ratio [82]. According to the literature data [83, 84], the found temperature range between 700 and 800 °C corresponds well with the gradual decomposition of faujasite. The crystallisation of nephelline at 800 °C, which can be seen in the HT-PXRD diffractogram, corresponds well with the third DSC exothermic peak located around 830 °C. The pronounced shoulder of the last DSC peak located around 970 °C can be assigned to mullite formation [83].
SEM analysis
SEM particle images of MK800 and its modification are presented in Fig. 11. SEM image of MK 800 (Fig. 11a) shows that the MK particles contain scaly plate-like and multilayered with some degree of roughness, also noted on the surface of the MK particles [85]. The flaky, layered particles began to aggretate into larger clusters after treatment of metakaolin with 5 mass% NaOH (Fig. 11b). The sponge-like gel formed in GP10_MK800 (Fig. 11c) indicates the growth of the geopolymer structure. This indicates that a geopolymerization reaction has taken place and in the case of sample GP20_MK800 (Fig. 11d), the zeolitization process is the most observed by SEM micrograph. This was manifested in the microstructure by the formation of cuboidal formations with larger and clear cavities. The results from SEM correlate well with the results obtained from PXRD and FTIR. SEM micrographs of MB800 and and its individual modifications are presented in Fig. 12. At first glance, the morphology of MB800 (Fig. 12a) is already clearly distinguished from MK 800 by its larger and more ordered particles. Microcracks are visible on the surface of these particles, which could be due to the initial calcination of the sample. After modification with 5 mass% of alkaline activator (Fig. 12b), cobweb-like gels begin to form on the surface of the particles, which could be a manifestation of incipient geopolymerization. Distinct crystals on the surface of sample GP10_MB800 (Fig. 12c) as well as exceptionally occurring cuboidal formations are a sign of incipient crystallization of the zeolite. However, compared to GP20_MK800, sample GP20_MB800 (Fig. 12d) does not show as clearly defined regular cuboidal formations, which would indicate a lower proportion of zeolite and hence crystalline phase, which correlates well with DOC (Fig. 8).

SEM images of MK800 (a), GP5_MK800 (b), GP10_MK800 (c), and GP20_MK800 (d)

SEM images of MB800 (a), GP5_MB800 (b), GP10_MB800 (c), and GP20_MB800 (d)
Conclusions
Metakaolin and metabentonite were prepared by thermal treatment of the source clays, kaolin and bentonite, and their potential for geopolymer/zeolite formation was evaluated by FTIR, TGA/DSC, PXRD, HT PXRD, SEM, and BET. The following preparation conditions were used (calcination temperature: 700, 750, 800 °C; concentration of NaOH: 5, 10, 20 mass%, curing conditions: 7 d at 80 °C). Dehydroxylation of kaolin was attained already after 3 h of calcination at 700 °C; however, higher temperatures were needed for amorphization of bentonite. The higher the NaOH concentration, the more promoted formation of hydrated aluminosilicates and the more enhanced Al3+ incorporation can be observed regardless of the initial material. The optimal preparation conditions, however, depend on the intended target material: geopolymer or zeolite. The 10% NaOH seems to be the optimal concentration for alkali activation of metakaolin samples leading to the dominant geopolymer formation. Further increase in alkaline activator concentration resulted in a significant rise of zeolite ratio (predominant zeolite A and P). The final material structure of samples activated by 10 and 20% NaOH did not differ significantly depending on the calcination temperature. Following the bentonite character, the activity of metabentonite is much more strongly conditioned by temperature. Geopolymer formation was promoted mainly using 800 °C and 10% NaOH. A higher alkaline activator concentration is required in the case of lower calcination temperature. The use of 800 °C and 20% NaOH seems to be the optimal concentration for alkali activation of metabentonite. Although resulted in enhanced zeolite formation, however, the determined degree of crystallinity (21%) is significantly lower than in corresponding activated metakaolin samples (32%). The specific surface areas (SSA) of the samples increased in the series MB800 < GP5_MB800 < GP10_MB800 < GP20_MB800. Conversely, the opposite effect was observed for samples of metakaolin-based geopolymers. SSA of activated samples, prepared by calcination at 800 °C, strongly depended on the process of geopolymerzation, the degree of crystallinity, and the character of formed zeolitic phases. The value of 218.95 m2 g−1, determined for metabentonite activated by 20 mass% NaOH seems to be very promising for adsorption processes. The specific surface areas of activated metakaolin samples were negatively influenced by increased zeolite formation and the dimensions of their channels restricting nitrogen entry.
References
Kaze CR, Nana A, Lecomte-Nana GL, Deutou JGN, Kamseu E, Melo UC, Andreola F, Leonelli C. Thermal behaviour and microstructural evolution of metakaolin and meta-halloysite-based geopolymer binders: a comparative study. J Therm Anal Calorim. 2022;147:2055–71. https://doi.org/10.1007/s10973-021-10555-2.
Kaze CR, Naghizadeh A, Tchadjie L, Djobo JNY, Nemaleu JGD, Kamseu E, Melo UC, Tayeh BA. Lateritic soils based geopolymer materials: a review. Constr Build Mater. 2022;344:128157. https://doi.org/10.1016/j.conbuildmat.2022.128157.
Siddique R, Cachim P. Waste and supplementary cementitious materials in concrete: characterisation, properties, and applications. Duxford: Woodhead Publishing; 2018. https://doi.org/10.1016/C2016-0-04037-8
Badogiannis E, Kakali G, Tsivilis S. Metakaolin as supplementary cementitious material. J Therm Anal Calorim. 2005;81:457–62. https://doi.org/10.1007/s10973-005-0806-3.
Namnoum YC, Hilloulin B, Grondin F, Loukili A. Determination of the origin of the strength regain after self-healing of binary and ternary cementitious materials including slag and metakaolin. J Build Eng. 2021;41:102739. https://doi.org/10.1016/j.jobe.2021.102739.
Zhan P, He Z, Ma Z, Liang C, Zhang X, Abreham AA, Shi J. Utilization of nano-metakaolin in concrete: a review. J Build Eng. 2020;30:101259. https://doi.org/10.1016/j.jobe.2020.101259.
Bowen F, Jiesheng L, Jing W, Chen Y, Tongtong Z, Tan X, Sun Z. Investigation on the impact of different activator to solid ratio on properties and micro-structure of metakaolin geopolymer. Case Stud Constr Mater. 2022;16:e01127. https://doi.org/10.1016/j.cscm.2022.e01127.
Luo Y, Meng J, Wang D, Jiao L, Xue G. Experimental study on mechanical properties and microstructure of metakaolin based geopolymer stabilized silty clay. Constr Build Mater. 2022;316:125662. https://doi.org/10.1016/j.conbuildmat.2021.125662.
Sudagar A, Andrejkovičová S, Patinha C, Velosa A, McAdam A, Ferreira da Silva E, Rocha F. A novel study on the influence of cork waste residue on metakaolin-zeolite based geopolymers. Appl Clay Sci. 2018;152:196–210. https://doi.org/10.1016/j.clay.2017.11.013.
Król M, Mozgawa W. Zeolite layer on metakaolin-based support. Microporous Mesoporous Mater. 2019;282:109–13. https://doi.org/10.1016/j.micromeso.2019.03.028.
Aygörmez Y. Assessment of performance of metabentonite and metazeolite-based geopolymers with fly ash sand replacement. Constr Build Mater. 2021;302:124423. https://doi.org/10.1016/j.conbuildmat.2021.124423.
Živica V, Palou MT. Physico-chemical characterization of thermally treated bentonite. Compos Part B Eng. 2015;68:436–45. https://doi.org/10.1016/j.compositesb.2014.07.019.
Kumari N, Mohan C. Basics of clay minerals and their characteristic properties. In: Morari Do Nascimento G, editor. Clay and clay minerals. London: IntechOpen; 2021. pp. 15–43. https://doi.org/10.5772/intechopen.97672
Khalifa AZ, Cizer Ö, Pontikes Y, Heath A, Patureau P, Bernal SA, Marsh ATM. Advances in alkali-activation of clay minerals. Cem Concr Res. 2020;132:106050. https://doi.org/10.1016/j.cemconres.2020.106050.
Schulze DG. Clay minerals. In: Hillel D, Hatfield JL, editors. Encyclopedia of soils in the environment. 1st ed. Oxford: Elsevier; 2005. p. 246–54.
Marsh A, Heath A, Patureau P, Evernden M, Walker P. Alkali activation behaviour of un-calcined montmorillonite and illite clay minerals. Appl Clay Sci. 2018;166:250–61. https://doi.org/10.1016/j.clay.2018.09.011.
Wei J, Gencturk B. Hydration of ternary Portland cement blends containing metakaolin and sodium bentonite. Cem Concr Res. 2019;123:105772. https://doi.org/10.1016/j.cemconres.2019.05.017.
Brigatti MF, Galán E, Theng BKG. Structure and mineralogy of clay minerals. In: Bergaya F, Lagaly G, editors. Handbook of clay science. 2nd ed. Amsterdam: Elsevier; 2013. pp. 21–81. https://doi.org/10.1016/B978-0-08-098258-8.00002-X
Massiot D, Dion P, Alcover JF, Bergaya F. 27Al and 29Si MAS NMR study of kaolinite thermal decomposition by controlled rate thermal analysis. J Am Ceram Soc. 1995;78(11):2940–4. https://doi.org/10.1111/j.1151-2916.1995.tb09067.x.
White CE, Provis JL, Proffen T, Riley DP, van Deventera JSJ. Combining density functional theory (DFT) and pair distribution function (PDF) analysis to solve the structure of metastable materials: the case of metakaolin. Phys Chem Chem Phys. 2010;12(13):3239–45. https://doi.org/10.1039/B922993K.
Sperinck S, Raiteri P, Marks N, Wright K. Dehydroxylation of kaolinite to metakaolin—a molecular dynamics study. J Mater Chem. 2011;21(7):2118–25. https://doi.org/10.1039/C0JM01748E.
Provis JL. Alkali-activation of calcined clays–past, present and future. In: Martinera F, Favier A, Scrivener K, editors. Calcined clays for sustainable concrete: proceedings of the 2nd international conference on calcined clays for sustainable concrete. RILEM bookseries, vol. 16. Dordrecht: Springer; 2018. pp. 372–6. https://doi.org/10.1007/978-94-024-1207-9_60
Duxson P, Provis JL, Lukey GC, van Deventer JSJ. Structural ordering in geopolymers derived from metakaolin. In: Davidovits J, editor. Geopolymer: green chemistry and sustainable development solutions. Proceedings of the World Congress Geopolymer. Saint-Quentin: Geopolymer Institute; 2005. p. 21–5.
Fernandez R, Martirena F, Scrivener KL. The origin of the pozzolanic activity of calcined clay minerals: a comparison between kaolinite, illite and montmorillonite. Cem Concr Res. 2011;41(1):113–22. https://doi.org/10.1016/j.cemconres.2010.09.013.
Snellings R, Mertens G, Elsen J. Supplementary cementitious materials. Rev Mineral Geochem. 2012;74(1):211–78. https://doi.org/10.2138/rmg.2012.74.6.
Heller-Kallai L. Thermally modified clay minerals. In: Bergaya F, Lagaly G, editors. Handbook of clay science. 2nd ed. Amsterdam: Elsevier; 2013. p. 411–33. https://doi.org/10.1016/B978-0-08-098258-8.00014-6.
Emmerich K, Madsen FT, Kahr G. Dehydroxylation behavior of heat-treated and steam-treated homoionic cis-vacant montmorillonites. Clay Clay Miner. 1999;47(5):591–604. https://doi.org/10.1346/CCMN.1999.0470506.
Derkowski A, Kuligiewicz A. Rehydroxylation in smectites and other clay minerals observed in-situ with a modified thermogravimetric system. Appl Clay Sci. 2017;136:219–29. https://doi.org/10.1016/j.clay.2016.11.030.
Yokoyama S, Kuroda M, Sato T. Atomic force microscopy study of montmorillonite dissolution under highly alkaline conditions. Clay Clay Miner. 2005;53(2):147–54. https://doi.org/10.1346/ccmn.2005.0530204.
Aldabsheh I, Khoury H, Wastiels J, Rahier H. Dissolution behavior of Jordanian clay-rich materials in alkaline solutions for alkali activation purpose. Part I. Appl Clay Sci. 2015;115:238–47. https://doi.org/10.1016/j.clay.2015.08.004.
Garg N, Skibsted J. Heated montmorillonite: structure, reactivity, and dissolution. In: Scrivener K, Favier A, editors. Calcined clays for sustainable concrete: proceedings of the 1st international conference on calcined clays for sustainable concrete. RILEM bookseries, vol. 10. Dordrecht: Springer; 2015. pp. 117–24. https://doi.org/10.1007/978-94-017-9939-3_15
Hu N, Bernsmeier D, Grathoff GH, Warr LN. The influence of alkali activator type, curing temperature and gibbsite on the geopolymerization of an interstratified illite-smectite rich clay from Friedland. Appl Clay Sci. 2017;135:386–93. https://doi.org/10.1016/j.clay.2016.10.021.
Diffo KBB, Elimbi A, Cyr M, Manga JD, Kouamo HT. Effect of the rate of calcination of kaolin on the properties of metakaolin-based geopolymers. J Asian Ceram Soc. 2015;3(1):130–8. https://doi.org/10.1016/j.jascer.2014.12.003.
Johnson EBG, Arshad SE. Hydrothermally synthesized zeolites based on kaolinite: a review. Appl Clay Sci. 2014;97–98:215–21. https://doi.org/10.1016/j.clay.2014.06.005.
Alzeer M, MacKenzie KJD. Synthesis and mechanical properties of new fibre-reinforced composites of inorganic polymers with natural wool fibres. J Mater Sci. 2012;47:6958–65. https://doi.org/10.1007/s10853-012-6644-3.
Duxson P, Mallicoat SW, Lukey GC, Kriven WM, van Deventer JSJ. The effect of alkali and Si/Al ratio on the development of mechanical properties of metakaolin-based geopolymers. Colloids Surf A Physicochem Eng Asp. 2007;292(1):8–20. https://doi.org/10.1016/j.colsurfa.2006.05.044.
Borges R, Dutra LM, Barison A, Wypych F. MAS NMR and EPR study of structural changes in talc and montmorillonite induced by grinding. Clay Miner. 2016;51:69–80. https://doi.org/10.1180/claymin.2016.051.1.06.
Fernández M, Alba MD, Sánchez TRM. Effects of thermal and mechanical treatments on montmorillonite homoionized with mono- and polyvalent cations: insight into the surface and structural changes. Colloids Surf A Physicochem Eng Asp. 2013;423:1–10. https://doi.org/10.1016/j.colsurfa.2013.01.040.
Richardson CK, Markuszewski R, Durham KS, Bluhm DD. Effect of caustic and microwave treatment on clay minerals associated with coal. In: Vorres KS, editor. Mineral matter and ash in coal. Washington, DC: American Chemical Society; 1986. pp. 513–23. https://doi.org/10.1021/bk-1986-0301.ch038
Belviso C, Cavalcante F, Niceforo G, Lettino A. Sodalite, faujasite and A-type zeolite from 2:1dioctahedral and 2:1:1 trioctahedral clay minerals. A singular review of synthesis methods through laboratory trials at a low incubation temperature. Powder Technol. 2017;320:483–97. https://doi.org/10.1016/j.powtec.2017.07.039.
Živica V, Palou M, Kuzielová E, Žemlička M. Super high strength metabentonite based geopolymer. Procedia Eng. 2016;151:133–40. https://doi.org/10.1016/j.proeng.2016.07.354.
Trník A, Štubňa I, Moravčíková J. Sound velocity of kaolin in the temperature range from 20 °C to 1100 °C. Int J Thermophys. 2009;30:1323–8. https://doi.org/10.1007/s10765-009-0634-y.
Jaroniec M, Kruk M, Sayari A. Adsorption methods for characterization of surface and structural properties of mesoporous molecular sieves. Stud Surf Sci Catal. 1998;117:325–32. https://doi.org/10.1016/S0167-2991(98)81008-2.
Farmer VC. Transverse and longitudinal crystal modes associated with OH stretching vibrations in single crystals of kaolinite and dickite. Spectrochim Acta Part A Mol Biomol Spectrosc. 2000;56(5):927–30. https://doi.org/10.1016/S1386-1425(99)00182-1.
Madejová J. FTIR techniques in clay mineral studies. Vib Spectrosc. 2003;31(1):1–10. https://doi.org/10.1016/S0924-2031(02)00065-6.
Autef A, Joussein E, Gasgnier G, Pronier S, Sobrados I, Sanz J, Rossignol S. Role of metakaolin dehydroxylation in geopolymer synthesis. Powder Technol. 2013;250:33–9. https://doi.org/10.1016/j.powtec.2013.09.022.
Burciaga-Díaz O, Durón-Sifuentes M, Díaz-Guillén JA, Escalante-García JI. Effect of waste glass incorporation on the properties of geopolymers formulated with low purity metakaolin. Cem Concr Compos. 2020;107:103492. https://doi.org/10.1016/j.cemconcomp.2019.103492.
Rożek P, Król M, Mozgawa W. Spectroscopic studies of fly ash-based geopolymers. Spectrochim Acta Part A Mol Biomol Spectrosc. 2018;198:283–9. https://doi.org/10.1016/j.saa.2018.03.034.
Fernández-Jiménez A, Palomo A. Mid-infrared spectroscopic studies of alkali-activated fly ash structure. Microporous Mesoporous Mater. 2005;86(1–3):207–14. https://doi.org/10.1016/j.micromeso.2005.05.057.
Criado M, Palomo A, Fernández-Jiménez A. Alkali activation of fly ashes. Part 1: effect of curing conditions on the carbonation of the reaction products. Fuel. 2005;84(16):2048–54. https://doi.org/10.1016/j.fuel.2005.03.030.
Król M, Mozgawa W, Morawska J, Pichór W. Spectroscopic investigation of hydrothermally synthesized zeolites from expanded perlite. Microporous Mesoporous Mater. 2014;196:216–22. https://doi.org/10.1016/j.micromeso.2014.05.017.
Król M, Minkiewicz J, Mozgawa W. IR spectroscopy studies of zeolites in geopolymeric materials derived from kaolinite. J Mol Struct. 2016;1126:200–6. https://doi.org/10.1016/j.molstruc.2016.02.027.
Pal P, Das JK, Das N, Bandyopadhyay S. Synthesis of NaP zeolite at room temperature and short crystallization time by sonochemical method. Ultrason Sonochem. 2013;20(1):314–21. https://doi.org/10.1016/j.ultsonch.2012.07.012.
Crăciun C. The study of some normal and abnormal montmorillonites by thermal analysis and infrared spectroscopy. Thermochim Acta. 1987;117:25–36. https://doi.org/10.1016/0040-6031(87)88097-8.
López FJ, Sugita S, Tagaya M, Kobayashi T. Geopolymers using rice husk silica and metakaolin derivatives; preparation and their characteristics. J Mater Sci Chem Eng. 2014;2(5):35–43. https://doi.org/10.4236/msce.2014.25006.
Wan Q, Rao F, Song S, Zhang Y. Immobilization forms of ZnO in the solidification/stabilization (S/S) of a zinc mine tailing through geopolymerization. J Mater Res Technol. 2019;8(6):5728–35. https://doi.org/10.1016/j.jmrt.2019.09.040.
Che C, Glotch TD, Bish DL, Michalski JR, Xu W. Spectroscopic study of the dehydration and/or dehydroxylation of phyllosilicate and zeolite minerals. J Geophys Res-Planet. 2011;116:E05007. https://doi.org/10.1029/2010JE003740.
Sitarz M, Handke M, Mozgawa W. Identification of silicooxygen rings in SiO2 based on IR spectra. Spectrochim Acta Part A Mol Biomol Spectrosc. 2000;56(9):1819–23. https://doi.org/10.1016/S1386-1425(00)00241-9.
Holtzer M, Bobrowski A, Żymankowska-Kumon S. Temperature influence on structural changes of foundry bentonites. J Mol Struct. 2011;1004(1–3):102–8. https://doi.org/10.1016/j.molstruc.2011.07.040.
Sayin M, von Reichenbach HG. Infrared spectra of muscovites as affected by chemical composition, heating and particle size. Clay Miner. 1978;13(3):241–54. https://doi.org/10.1180/claymin.1978.013.3.01.
Sykes D, Kubicki JD. Four-membered rings in silica and aluminosilicate glasses. Am Mineral. 1996;81(3–4):265–72. https://doi.org/10.2138/am-1996-3-401.
Lubas M, Sitarz M, Fojud Z, Jurga S. Structure of multicomponent SiO2–Al2O3–Fe2O3–CaO–MgO glasses for the preparation of fibrous insulating materials. J Mol Struct. 2005;744:615–9. https://doi.org/10.1016/j.molstruc.2004.12.011.
Mekki A, Benmaati A, Mokhtar A, Hachemaoui M, Zaoui F, Habib Zahmani H, Sassi M, Hacini S, Boukoussa B. Michael addition of 1, 3-dicarbonyl derivatives in the presence of zeolite Y as an heterogeneous catalyst. J Inorg Organomet Polym Mater. 2020;30(7):2323–34. https://doi.org/10.1007/s10904-019-01424-5.
Yin H, Zhou T, Liu Y, Chai Y, Liu C. Synthesis of high-quality nanocrystalline zeolite Y using pseudoboehmite as aluminum source. J Porous Mater. 2012;19(3):277–81. https://doi.org/10.1007/s10934-011-9471-z.
Barbosa VFF, MacKenzie KJD. Synthesis and thermal behaviour of potassium sialate geopolymers. Mater Lett. 2003;57(9–10):1477–82. https://doi.org/10.1016/S0167-577X(02)01009-1.
Hou L, Li J, Lu ZY. Effect of Na/Al on formation, structures and properties of metakaolin based Na-geopolymer. Constr Build Mater. 2019;226:250–8. https://doi.org/10.1016/j.conbuildmat.2019.07.171.
Shoaei P, Ameri F, Karimzadeh M, Atabakhsh E, Zareei SA, Behforouz B. Difference between geopolymers and alkali-activated materials. In: Colangelo F, Cioffi R, Farina I, editors. Handbook of sustainable concrete and industrial waste management. Duxford: Woodhead Publishing; 2022. pp. 421–35. https://doi.org/10.1016/B978-0-12-821730-6.00018-8
Payá J, Agrela F, Rosales J, Morales MM, Borrachero MV. Application of alkali-activated industrial waste. In: de Brito J, Agrela F, editors. New trends in eco-efficient and recycled concrete. Duxford: Woodhead Publishing; 2018. pp. 357–424. https://doi.org/10.1016/B978-0-08-102480-5.00013-0
Živica V, Palou MT, Ifka T, Bágeľ Ľ. High strength metahalloysite based geopolymer. Compos Part B Eng. 2014;57:155–65. https://doi.org/10.1016/j.compositesb.2013.09.034.
Ursu AV, Jinescu G, Gros F, Nistor ID, Miron ND, Lisa G, Silion M, Djelveh G, Azzouz A. Thermal and chemical stability of Romanian bentonite. J Therm Anal Calorim. 2011;106:965–71. https://doi.org/10.1007/s10973-011-1414-z.
Bayram H, Önal M, Yılmaz H, Sarıkaya Y. Thermal analysis of a white calcium bentonite. J Therm Anal Calorim. 2010;101:873–9. https://doi.org/10.1007/s10973-009-0626-y.
Khan MI, Azizli K, Sufian S, Man Z. Sodium silicate-free geopolymers as coating materials: effects of Na/Al and water/solid ratios on adhesion strength. Ceram Int. 2015;41(2):2794–805. https://doi.org/10.1016/j.ceramint.2014.10.099.
Kuenzel C, Grover LM, Vandeperre L, Boccaccini AR, Cheeseman CR. Production of nepheline/quartz ceramics from geopolymer mortars. J Eur Ceram Soc. 2013;33(2):251–8. https://doi.org/10.1016/j.jeurceramsoc.2012.08.022.
Dietel J, Warr LN, Bertmer M, Steudel A, Grathoff GH, Emmerich K. The importance of specific surface area in the geopolymerization of heated illitic clay. Appl Clay Sci. 2017;139:99–107. https://doi.org/10.1016/j.clay.2017.01.001.
International Zeolite Association. In: Database of zeolite structures. 2017. https://europe.iza-structure.org/IZA-SC/framework.php?STC=LTA. Accessed 31 Mar 2023
International Zeolite Association. In: Database of zeolite structures. 2017. https://europe.iza-structure.org/IZA-SC/framework.php?STC=GIS. Accessed 31 Mar 2023
International Zeolite Association. In: Database of zeolite structures. 2017. https://europe.iza-structure.org/IZA-SC/framework.php?STC=FAU. Accessed 31 Mar 2023
El Alouani M, Alehyen S, El Achouri M, Taibi M. Preparation, characterization, and application of metakaolin-based geopolymer for removal of methylene blue from aqueous solution. J Chem. 2019;2019:4212901. https://doi.org/10.1155/2019/4212901.
Borodina U, Goryainov S, Krylova S, Vtyurin A, Krylov A. The behavior of zeolites wairakite and phillipsite at high PT parameters. Spectrochim Acta Part A-Mol Biomol Spectrosc. 2022;273:120979. https://doi.org/10.1016/j.saa.2022.120979.
Huo Z, Xu X, Lv Z, Song J, He M, Li Z, Wang Q, Yan L, Li Y. Thermal study of NaP zeolite with different morphologies. J Therm Anal Calorim. 2013;111:365–9. https://doi.org/10.1007/s10973-012-2301-y.
Kuzielová E, Žemlička M, Másilko J, Hudec P, Palou MT. Influence of hydrothermal treatment parameters on the phase composition of zeolites. J Therm Anal Calorim. 2020;142:37–50. https://doi.org/10.1007/s10973-020-09784-8.
Julbe A, Drobek M. Zeolite X: Type. In: Drioli E, Giorno L, editors. Encyclopedia of membranes. Berlin: Springer; 2014, pp. 2055–6, https://doi.org/10.1007/978-3-642-40872-4_607-1
Dos Santos de Castro PR, Maia AÁB, Angélica RS. Study of the thermal stability of faujasite zeolite synthesized from kaolin waste from the Amazon. Mater Res-Ibero-Am J Mater. 2020;22(5):20190321. https://doi.org/10.1590/1980-5373-MR-2019-0321.
Li CY, Rees LVC. The thermal stability of faujasites with different SiAl ratios. Zeolites. 1986;6(1):60–5. https://doi.org/10.1016/0144-2449(86)90013-8.
Pillay DL, Olalusi OB, Kiliswa MW, Awoyera PO, Kolawole JT, Babafemi AJ. Engineering performance of metakaolin based concrete. Clean Eng Technol. 2022;6:100383. https://doi.org/10.1016/j.clet.2021.100383.
Acknowledgements
The authors acknowledge the financial support of this research by the Slovak Grant Agency VEGA (Grants 2/0032/21 and 2/0017/21) and the Slovak Research and Development Agency (APVV-19-0490, APVV-20-0175 and APVV-16-0341). The authors would also like to thank Dr. Vladimír Pavlík for his help in providing valuable advice and assistance in creating the SEM images.
Funding
Open access funding provided by The Ministry of Education, Science, Research and Sport of the Slovak Republic in cooperation with Centre for Scientific and Technical Information of the Slovak Republic.
Author information
Authors and Affiliations
Contributions
MS contributed to conceptualization; writing—original draft preparation; and writing—review and editing; MS and EK contributed to supervision; MS, EK, MŽ, and MM contributed to methodology; MS, MŽ, MM, and AS contributed to formal analysis and investigation; and MTP and EK contributed to funding acquisition.
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Slaný, M., Kuzielová, E., Žemlička, M. et al. Metabentonite and metakaolin-based geopolymers/zeolites: relation between kind of clay, calcination temperature and concentration of alkaline activator. J Therm Anal Calorim 148, 10531–10547 (2023). https://doi.org/10.1007/s10973-023-12267-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-023-12267-1