
Abstract
The paper presents a method of the preparation and functionalization of polymer microspheres consisting of glycidyl methacrylate (GMA) and crosslinking agents: 1,4-dimethacryloyloxybenzene (1,4DMB) and trimethylolpropane trimethacrylate (TRIM). Poly(GMA-co-1,4DMB) and poly(GMA-co-TRIM) microspheres were obtained by seed swelling polymerization. To introduce thiol groups into the microspheres structure, the reaction with thiocarboxylic acids was performed. The chemical structure of parent and modified microspheres was confirmed by FTIR and Raman spectroscopy. Elemental composition of microspheres after functionalization was determined by elemental analysis. The analysis showed the percentage of sulfur in the range of 2.78–4.51%, which corresponds to a concentration of thiol group in the range of 0.87–1.41 mmol g−1. Additionally, the porous structure of the copolymers was investigated using the low-temperature nitrogen adsorption–desorption method. The starting microspheres are characterized by a specific surface in the range of 150–160 m2 g−1, whereas functionalized copolymers indicate slightly lower surface area, of about 130 m2 g−1. The thermal stability of the materials was determined by the method of differential scanning calorimetry and thermogravimetric analysis. The course of the thermal degradation under oxidative conditions of modified microspheres is different from the starting copolymers. The functionalized microspheres showed much higher thermal stability (approximately 270 °C) compared to the starting microspheres (230–250 °C).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Porous polymer microspheres, due to their variety of properties, are widely used, among others, as sorbents in the packing of chromatographic columns [1,2,3,4], resins for sorption of heavy metals [5,6,7], substrates for the preparation of silver and gold nanoparticles used in catalytic reactions, or in the synthesis of quantum dots and nanocomposites [6]. Polymer microspheres can be functionalized by direct synthesis of monomers containing functional groups [1, 8,9,10] or by chemical modification carried out with previously obtained polymeric particles [11, 12]. The first method is simpler and faster than the second one. It enquires only the use of appropriate functional monomer that possess functional groups, e.g., 2-hydroxyethyl methacrylate introduces the OH groups into the polymer structure [13, 14], (meth)acrylic acid (MMA)–COOH groups[15, 16], glycidyl methacrylate (GMA) oxirane groups [17]. Moreover, crosslinking agent is necessary, the most commonly used are aromatic divinylbenzene or aliphatic methacrylates, e.g., trimethylolpropane trimethacrylate (TRIM) or ethylene glycol dimethacrylate (EGDMA) [18,19,20]. The second method of modification is more complicated and can be performed only when in the structure of microspheres reactive functional groups are present, like for example oxirane ring that can react with many strong and weak nucleophiles [21]. Microspheres bearing oxirane groups were reacted for example with amines [11, 22,23,24], pyrrolidone [25], pyrazole derivatives [26] or reagents allowing to introduce carboxyl groups [12, 27]. Despite that this approach is demanding, some chemistries can be introduced into microspheres structure only in this route. Such example is thiol group (–SH). In the literature, methods of obtaining polymer microspheres containing thiol groups in their chemical structure are known. Most often, these are multistage methods, consisting in the reaction of the halomethyl group attached to aromatic structures, mainly derived from styrene, with the appropriate sulfur compound, leading to the formation of an intermediate adduct, which is hydrolyzed in the next stage to obtain the desired thiol [6, 7, 28,29,30]. Garamszegi et al. [31] presented a method of incorporation of –SH groups onto the surface of polystyrene microspheres obtained by atom transfer radical polymerization (ATRP). On the other hand, Li et al. [32] and Wenchao et al. [33] described the method of introducing -SH groups by opening the oxirane group with cystamine dihydrochloride. The presented methods for incorporating -SH groups on the surface of microspheres are multistage, time-consuming, and require the use of a solvent and/or catalyst. Therefore, attempts were made to develop a new way of obtaining crosslinked, porous copolymer microspheres based on GMA functionalized with thiol groups derived from thiocarboxylic acids in a one-step process, in a solvent and catalyst free environment, in a relatively short time and in a way that ensures the preservation of the spherical structure of the polymer and the crosslinked internal porous structure. The method of using thioglycolic acid (TGA) for oxirane ring was described Karadeniz et al. [34]. However, in this case, TGA was used to open oxirane rings present in epoxidized soybean oil in the environment of a strong oxidant, i.e., chloric acid (VII). Moreover, this reaction was performed within 18 h at 120 °C. Such conditions for porous microspheres should be regarded as drastic, could lead to destroy porous structure and even the whole microsphere. Additionally, the chloric acid (VII) is explosive in contact with organic substances.
In this paper, we present the new method of synthesis of polymer microspheres with embedded thiol groups. Starting copolymers of GMA and methacrylate crosslinkers (1,4DMB and TRIM) were obtained by seed swelling polymerization. The epoxide number of the obtained microspheres was determined, and then functionalization with thiocarboxylic acids was performed. The structure of the microspheres was examined using spectroscopic methods (Fourier transform infrared spectroscopy—attenuated total reflectance (FTIR-ATR) and Raman), elemental analysis, and porosimetric analysis. Finally, thermal stability as well as phase and structural transformations occurring in the studied microspheres were examined by means of thermogravimetric analysis coupled with FTIR (TG-FTIR) and differential scanning calorimetry (DSC).
Materials and methods
Chemicals
Styrene (99%), glycidyl methacrylate (GMA; 97%), trimethylolpropane trimethacrylate (TRIM), thioglycolic acid (TGA; 98%), 3-mercaptopropionic acid (3-MPA; ≥ 99%), 2,2′-azobis(2-methylpropionitrile) (AIBN; 98%), polyvinylpyrrolidone with average molecular mass of 40,000 g mol−1 (PVP40) and sodium lauryl sulfate (SDS; 99%) were obtained from Sigma-Aldrich. Toluene, tetrahydrofuran (THF), acetone, methanol, and ethanol were from POCh (Gliwice, Poland). 1,4-dimethacryloyloxybenzene (1,4DMB, IUPAC name benzene-1,4-diylbis(2-methylprop-2-enoate) was synthesized in the laboratory of the Department of Polymer Chemistry, MCS University in Lublin according to the earlier described procedure [2].
Synthesis of porous polymer microspheres
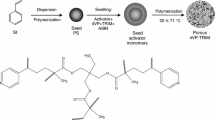
Porous polymer microspheres were obtained with the use of functional monomers of GMA and crosslinking monomers: 1,4DMB and TRIM by seed swelling polymerization (Fig. 1). The molar ratio of the functional monomer to the crosslinker was 3:1. In the first step, a shape precursor, polystyrene (PS), was obtained as a result of styrene dispersion polymerization carried out in an ethanolic PVP solution in the presence of the AIBN initiator. The swelling of the PS beads was carried out in the presence of an activator (toluene), a mixture of monomers, and the initiator. The resulting emulsion was placed in a three-neck round bottom flask and kept at 35 °C for 24 h with continuous agitation (100 rpm). The polymerization was then carried out for 24 h at 71 °C. Theobtained microspheres were isolated by filtration, washed with 1000 cm3 of hot water and finally washed with 50 cm3 of acetone and methanol, respectively. To remove residual linear PS chains, extraction was performed with hot THF for 6 h under continuous stirring (250 rpm). After extraction, the microspheres were filtered off, washed with 20 cm3 of THF and dried at room conditions.

Scheme of the synthesis of primary microspheres
Functionalization of polymer microspheres
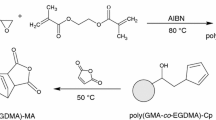
The functionalization of poly(GMA-co-1,4DMB) and poly(GMA-co-TRIM) microspheres was carried out according to the scheme presented in Fig. 2. The previously obtained polymer microspheres and TGA (or 3-MPA) were introduced into a glass test tube in a volume ratio of 1:1.5, respectively. The mixture prepared in this way was sealed with a Teflon plug and placed in a microwave reactor for 4 min at a temperature of 100 °C under constant stirring. After completion of the reaction, the contents of the tube were centrifuged, the supernatant containing the unreacted acid was decanted, followed by extraction of the microspheres with acetone and methanol to wash away any remaining acid.

Scheme of modification of primary microspheres
Analysis methods
FTIR analysis was performed on a Tensor 27 FTIR spectrophotometer (Bruker, Germany) using the ATR technique. The spectra consisted of 16 scans with a resolution of 4 cm−1 in the wavenumber range 4000–600 cm−1. Raman analysis was performed with an in Via Reflex Raman microscope (Renishaw, UK). Elemental analysis was performed with a EuroEA Elemental Analyzer (CHNS) (HEKAtech, Germany). The parameters characterizing the porosity of the copolymer were determined by nitrogen adsorption using an ASAP 2420 volumetric analyzer (Micromeritics, USA). Prior to the measurement, the samples were degassed overnight under vacuum at 60 °C. The SBET specific surface area was estimated by the standard Brunauer–Emmett–Teller (BET) method.
The content of oxirane groups in the copolymer structure was determined by back titration. To a known amount of copolymer, a dioxane HCl solution was introduced, and the sample was left for 24 h. Then it was titrated with 0.2 M methanolic NaOH solution in the presence of cresol red. The epoxide number was determined from the difference between the volume of the blank and that found for the titrated copolymer sample.
The morphology and the internal structure of microspheres were examined using scanning electron microscope (SEM) (FEI Quanta 3D FEG) working at 5 kV. Before analysis, polymeric particles were coated with a thin layer of gold.
Calorimetric measurements were made with a DSC 204 calorimeter (Netzsch, Germany) operating in dynamic mode. Dynamic scans were performed in two steps with a heating rate of 10 K min−1 from room temperature to 500 °C under air atmosphere with flow rate of 30 mL min−1. The first scan was performed to remove adsorbed moisture from room temperature to 150 °C and the second scan from room temperature to 500 °C. Samples were analyzed in a pierced aluminum crucibles.
Thermal analysis was performed with a STA 449 F1 Jupiter (Netzsch, Germany) thermal analyzer at a synthetic air atmosphere with flow rate of 25 mL min−1. Heating rate was of 10 K min−1, the temperature range 35–850 °C, the sample mass was about 10 mg. Samples were analyzed in open Al2O3 crucibles. An empty Al2O3 crucible was used as a reference. Gaseous decomposition products of the polymer samples were analyzed with a Tensor 27 Bruker FTIR spectrometer (Germany) coupled directly to an STA instrument. The spectra were collected every 10 K (1 scan) in the wavenumber range 4000–600 cm−1.
Results
Chemical structure characterization
Polymer microspheres poly(GMA-co-1,4DMB) and poly(GMA-co-TRIM) were obtained by seed swelling polymerization. This method allows to obtain crosslinked microspheres with narrow size distribution and permanent developed porous structure. The aim of the research was to prepare highly porous microspheres using GMA and various crosslinking agents: 1,4DMB and TRIM. In the course of the oxirane ring opening reaction present in the structure of these copolymers, the –SH groups were incorporated onto the surface of the microspheres employing thiocarboxylic acids. Since the presence of oxirane groups in the primary microspheres is extremely important to this modification, firstly the reverse titration with HCl/dioxane was done to assess the epoxide number. The results are presented in Table 1 together with the theoretically calculated values for the epoxide number. The experimentally determined epoxide numbers are lower than the theoretical ones, which may result from the course of the reaction of oxiranes with HCl only on the surface of the microspheres, or some of the oxirane groups could be opened by hydrolysis during the polymerization, as reported by Park et al. [25]. In addition, this assumption is confirmed by the FTIR-ATR spectra (Figs. 3, 4), where a wide band of -OH group vibrations are observed for starting copolymers. But it should be taken into account that this band can be also from water molecules absorbed in copolymers pores, which is characteristic of porous polymers [12, 26,27,28].

FTIR-ATR spectra (a) and Raman spectra (b) of poly(GMA-co-TRIM) and poly(GMA-co-TRIM)-SH

FTIR-ATR spectra (a) and Raman spectra (b) of poly(GMA-co-1,4DMB) and poly(GMA-co-1,4DMB)-SH
The use of FTIR-ATR and Raman techniques allowed determining the chemical structure of the copolymers before and after incorporation of the -SH groups onto the surface. Figure 3 shows the FTIR-ATR and Raman spectra of the poly(GMA-co-TRIM) copolymers before and after functionalization with thiol groups derived from TGA and 3-MPA acids. In the FTIR spectrum of starting microspheres based on GMA and TRIM, the characteristic absorption band derived from oxirane ring vibration is present at 908 cm−1. Moreover, the broad absorption band visible at 3460 cm−1 derives from -OH groups vibrations. This confirms that some amount of oxirane rings was opened during the polymerization process and is convergent with determined epoxide number. The FTIR and Raman spectra of microspheres after functionalization reveal the new characteristic absorption bands at 2578 cm−1 derived from the –SH stretching vibration. In the FTIR spectra this band is of very low intensity, but it is very clearly visible in the Raman spectra. Also the band at 3460 cm−1 (the hydroxyl groups vibrations) are of higher intensities than in the spectrum of starting copolymer due to formation of -OH groups during the functionalization (Fig. 2). At the same time, the disappearance of the band at 908 cm−1 is observed. The presence on the spectra of all copolymers bands at 2950–3000 cm−1 (methyl and methylene groups vibration), 1772 cm−1 (carbonyl group vibration), 1145 cm−1 (–C–O–C– group vibration) confirms that the crosslinked structure of copolymeric network was not destroyed during the modification reaction.
In the FTIR spectra (Fig. 4) for poly(GMA-co-1,4DMB) microspheres after functionalization, also like in the case of poly(GMA-co-TRIM), a wide band is observed at the wavenumber of 3460 cm−1, which is derived from hydroxyl groups formed during oxirane ring opening. Additionally, both the FTIR (low-intensity) and Raman (high intensity) spectra show bands characteristic for the vibrations of the -SH group at 2578 cm−1. The band at 908 cm−1 disappeared after the reaction with thiocarboxylic acids, whereas bands at 3000–2880 cm−1 (–CH3; –CH2–), 1732 cm−1 (C=O), 1597 cm−1 (aromatic ring), 1173, 1121 cm−1 (–C–O–C–) remained unchanged.
The results of the elemental analysis (Table 2) also confirm that the functionalization of starting microspheres with the use of TGA and 3-MPA acids run successfully according with the scheme presented in Fig. 2. The elemental analysis showed that the functionalized microspheres contain 2.78–4.51% of sulfur, which corresponds to the concentration of -SH groups in the range of 0.87–1.41 mmol g−1. Modification of microspheres with TGA acid was performed with better efficiency than with 3-MPA acid.
Porous structure
The use of the low-temperature nitrogen adsorption–desorption method allowed to study the porous structure of poly(GMA-co-1,4DMB) and poly(GMA-co-TRIM) microspheres before and after functionalization with thiocarboxylic acids. The specific surface area (SBET), pore volume (Vp), and pore diameter at the top of the PSD were determined, and the measurement results are presented in Table 1. The specific surface area of poly(GMA-co-TRIM) microspheres is slightly higher (165 m2 g−1) compared to poly(GMA-co-1,4DMB) microspheres, the surface area of which is 156 m2 g−1. This difference is connected with the crosslinker nature: TRIM possess three methacrylate groups whereas 1,4DMB—two and hence TRIM can build more crosslinked copolymeric network. It is known that the more crosslinked network is, the more developed porous structure possess the polymeric microspheres [32, 35]. After the functionalization process, for both copolymers the drop in the value of specific surface area is observed to about 130 m2 g−1. Some reorganization of porous structure take place during the reaction. On the other hand, the pore volumes of the microspheres both before and after functionalization remain almost unchanged. Based on the PSD diagrams (Fig. 5), it can be concluded that the pores are in the range of micro- and mesopores. Two peaks are visible on the PSDs. The first one is observed at about 4 nm and is also characteristic of other methacrylic copolymers [18, 36]. As reported by Flodin et al. [37], the presence of this peak is related to the regular structure of copolymers in the micro scale. Regardless of the crosslinker used, the PSDmax of the mesopore range is around 32 nm. The pore distribution remained unchanged after the modification process.

Pore size distributions of the investigated polymeric materials
In addition, the morphology and porous structure was also examined by means of SEM analysis. As it is seen from images presented in Fig. 6 copolymer particles retained microspheric shape after the chemical modification. The surface of microspheres reveals porous structure, however larger voids are present on the surface of poly(GMA-co-TRIM) than poly(GMA-co-1,4DMB). The porous nature of the surface is also visible for modified microspheres. Thus, it can be concluded that the functionalization performed with the use thiocarboxylic acids does not destroy the created during polymerization microspheres porous structure.

SEM micrographs of microspheres: poly(GMA-co-1,4DMB) (a); poly(GMA-co-1,4DMB)-SH (b); poly(GMA-co-TRIM) (c); poly(GMA-co-TRIM)-SH (d)
Thermal behavior studies
The thermal properties of the copolymers were determined by differential scanning calorimetry and thermogravimetric analysis. The TG, DTG, and DSC curves for poly(GMA-co-1,4DMB) and poly(GMA-co-1,4DMB)-SH copolymers are presented in Fig. 7. Table 3 shows the temperature of 5% and 50% mass loss (T5%, T50%), temperature of the maximum mass loss (Tmax), and the corresponding mass loss (Δm). The poly(GMA-co-1,4DMB) polymer shows a three-step degradation. The first maximum on DTG curve is observed at 272 °C and corresponds to 36.5% mass loss (Δm1), the second one at 348 °C and the last one at 456 °C. From the DSC curve it is visible that every degradation step is exothermic. However, in the case of introducing –SH groups into the structure of the microspheres, a two-stage degradation process is observed, with the first maximum at 355 °C. It is accompanied by an 84% mass loss (Δm2). The second step indicates the maximum at 479 °C and coincides with the last degradation step of starting copolymer. DSC curve reveals two exothermic peaks corresponding to the maxima on the DTG curve. The T5% value of the SH-copolymer is 30 °C higher than that of the starting copolymer. The T50% is also higher for the modified copolymer. Thus, it is clearly visible that the introduction of -SH groups into the copolymer network increases its thermal stability compared to the starting material. Taking into account the chemical structure of both copolymers, it can be assumed that the oxirane rings limit the thermal stability. Similar increase of thermal stability of crosslinked copolymers derived from GMA after modification within the oxirane ring were reported by in literature [12, 23, 25, 38].

TG (a), DTG (b), and DSC (c) curves of poly(GMA-co-1,4DMB) and poly(GMA-co-1,4DMB)-SH copolymers obtained under air atmosphere
It is known from the literature that the thermal degradation of poly(GMA) begins with depolymerization reaction, followed by isomerization of the oxirane group [12, 39]. The degradation of the poly(GMA-co-1,4DMB) copolymer is identical in the initial phase as in the case of the homopolymer, but also oxidation process take place, which is confirmed by the analysis of the degradation gaseous products of this copolymer. 3D FTIR spectra of emitted gasses are presented in Fig. 8, whereas Fig. 9 shows the extracted at the maxima of decomposition single spectra for poly(GMA-co-1,4DMB) and poly(GMA-co-1,4DMB)-SH. In the case of spectrum collected at 271 °C corresponding to the maximum in the DTG curve for poly(GMA-co-1,4DMB), significant absorption band is observed in the region of C=O group vibration. It is visible that this band is complex and three maxima can be distinguish: at 1712, 1736 and 1744 cm−1. They relate to the vibrations of the carbonyl group in aldehydes and esters. Another characteristic of the C–O–C group vibration band is observed at 1159 cm−1. In addition, at 1638, 945 and 939 cm−1, bands of =CH2 vibrations are present, and at 850 cm−1 band of the oxirane group vibrations is visible. The presence of mentioned absorption bands confirms that in the first degradation step of parent copolymer the emission of GMA monomer takes place by depolymerization mechanism. In order to confirm this suspicion, the GMA spectrum from the NIST base is presented in Fig. 9 [40]. Moreover, in the spectrum region of 2800–2680 cm−1, the characteristic of acrolein [41] bands are visible what point that isomerization of oxirane compounds also was possible. Besides, at 2360 cm−1, bands derived from carbon dioxide, which is a product of ester bond degradation, and oxidation are observed. Spectrum collected at 343 °C reveals broad absorption bands in the region of about 1800–1700 cm−1 (C=O vibration), and at 1200–1110 cm−1 (C–O–C vibration) indicating emission of carbonyl and alcohol compounds derived from degradation of crosslinked part of copolymer. Nevertheless, in this step mainly oxidation takes place what is evidenced by the emission of carbon dioxide (2357, 2311, and 670 cm−1), carbon monoxide (2104 and 2180 cm−1) and water (3700–3600 and 1600–1400 cm−1). Finally, at the highest temperature, mainly emission of carbon dioxide, carbon monoxide and water is observed.

3D FTIR diagrams of gases evolved from the poly(GMA-co-1,4DMB) (a) and poly(GMA-co-1,4DMB)-SH (b) materials under air atmosphere

FTIR spectra of gases evolved from poly(GMA-co-1,4DMB) (a) and poly(GMA-co-1,4DMB)-SH (b) at the maxima of emission
On the other hand, for poly (GMA-co-1,4DMB)-SH in the spectrum collected at 342 °C and 355 °C, there are no absorption bands derived from GMA or acrolein. The course of these spectra is similar to that of starting copolymer collected at 343 °C. Attention should be drawn to the spectrum collected at 342 °C, before the Tmax2. In this spectrum the new band appeared at 2072 and 2047 cm−1 and is associated with the vibrations of the C=O group in carbonyl sulfide [42, 43]. The evolution of carbonyl sulfide may be the result of the decomposition of a thiol unit itself or temporarily formed thioester linkages as a product of nucleophilic attack of the end -SH group onto carbonyl ester group. What is interesting, in the spectrum collected at 355 °C the emission of carbon sulfide is not observed.
The TG, DTG, and DSC curves for poly(GMA-co-TRIM) and poly(GMA-co-TRIM)-SH copolymers are shown in Fig. 10 whereas in Table 3 the temperature of 5% and 50% mass loss (T5%, T50%), temperature of the maximum mass loss (Tmax), and the corresponding mass loss (Δm) are given. The DTG curve shows that the poly(GMA-co-TRIM) polymer undergoes a three-stage degradation process. The first maximum is observed at 233 °C and corresponds to a mass loss of 21.3% (Δm1), the second one at 345 °C, and the third one at 458 °C. Compared to the poly(GMA-co-1,4DMB) microspheres, TRIM crosslinked copolymer is less thermally stable, its T5% is of about 22 °C lower. For poly(GMA-co-TRIM)-SH microspheres, the two-stage degradation process is observed for which the first maximum is observed at 368 °C and is accompanied by 87.8% mass loss (Δm2). The polymer after functionalization possess a much higher thermal stability compared to the starting one. The value of T5% for the starting copolymer is 227 °C, and for the one polymer is 273 °C. In the DSC curve for the poly(GMA-co-TRIM) copolymer, three exothermic peaks with maxima at 235 °C, 355 °C and 460 °C are observed, whereas in the DSC curve of poly(GMA-co-TRIM)-SH copolymer only two exothermic peaks are present, with maxima corresponding to the maxima of DTG curves. This signifies that studied TRIM-crosslinked copolymers undergo exothermic degradation under air atmosphere. FTIR analysis of gaseous products of poly(GMA-co-TRIM) decomposition (Figs. 11, 12) reveals that its degradation in the initial step is of an identical path like in the case of poly(GMA) homopolymer. The absorption bands at: 1736 cm−1 coming from the vibrations of the C=O group, at 1159 cm−1 derived from the C–O–C vibration in the esters and at 850 cm−1 derived from oxirane ring vibration suggest the emission of GMA monomer. Moreover, the absorption bands at 2357, 2311, and 670 cm−1 derived from carbon dioxide and bands of carbon monoxide (2104 and 2180 cm−1) are more significant than in the spectrum collected at the first step degradation of poly(GMA-co-1,4DMB) copolymer. This suggest that the TRIM copolymer is more susceptible on the oxidation reactions than 1,4DMB copolymer. In the course of poly(GMA-co-TRIM)-SH thermal degradation, the spectrum collected at 371 °C reveals notable absorption bands derived from carbon dioxide, carbon monoxide and water (3700–3600 and 1600–1400 cm−1) indicating that the oxidation reaction is the main process. However, bands of small intensities at 1772 cm−1 (C=O group), 1163 cm−1 (C–O–C group) are also visible and inform that some esters species are emitted. Moreover, like in the case of poly(GMA-co-1,4DMB)-SH, the band at 2072 cm−1 associated with C=O vibrations from carbon sulfide is observed on the spectrum collected at 344 °C, before the maximum of emission. Its low intensity can be due to the lower sulfur content in the copolymer compared with poly(GMA-co-1,4DMB)-SH.

TG (a), DTG (b), and DSC (c) curves of poly(GMA-co-TRIM) and poly(GMA-co-TRIM)-SH copolymers obtained under air atmosphere

3D FTIR diagrams of gases evolved from the poly(GMA-co-TRIM) (a) and poly(GMA-co-TRIM)-SH (b) materials under air atmosphere

FTIR spectra of gases evolved from poly(GMA-co-TRIM) (a) and poly(GMA-co-TRIM)-SH (b) at the maxima of emission
Conclusions
The use of seed swelling polymerization made it possible to obtain porous methacrylic copolymers based on GMA and crosslinking agents: 1,4DMB and TRIM. The oxirane ring present in the structure of the starting microspheres can be reacted with the use of thiocarboxylic acids. The proposed method of introducing thiol groups into the structure of polymeric porous microspheres is simple, fast and does not require the use of additional solvents or catalysts. It allows to maintain the spherical structure of the starting copolymers and preserve their developed porous structure. The FTIR, Raman and CHNS results confirm the introduction of the thiol groups into the structure of the microspheres. Elemental analysis shows that the functionalization of the microspheres with TGA acid was more efficient than with 3-MPA acid. The thermal stability of the obtained microspheres under air atmosphere was tested with the use of DSC and TG methods. The starting materials are characterized by a three-stage exothermic degradation process. Their thermal stability is in the range of 230–250 °C under an oxidative conditions. The degradation of the starting microspheres begins with the depolymerization of the glycidyl fragments in the copolymer network, and is accompanied with oxidation reactions. The introduction of -SH groups into the copolymers networks improves their thermal stability to about 280 °C, and the degradation of the copolymers takes place in two exothermic stages. Among the gases released during the decomposition, mainly CO2 was observed. Additionally, carbonyl sulfide and gaseous organic compounds containing carbonyl groups, and aliphatic fragments were detected.
References
Ferreira A, Bigan M, Blondeau D. Optimization of a polymeric HPLC phase: poly(glycidyl methacrylate-co-ethylene dimethacrylate): influence of the polymerization conditions on the pore structure of macroporous beads. React Funct Polym. 2003;56:123–36. https://doi.org/10.1016/S1381-5148(03)00049-X.
Grochowicz M, Gawdzik B. Preparation and characterization of porous crosslinked microspheres of new aromatic methacrylates. J Porous Mater. 2013;20:339–49. https://doi.org/10.1007/s10934-012-9603-0.
Unsal E, Çamli ST, Irmak T, Tuncel M, Tuncel A. Monodisperse poly (styrene-co-divinylbenzene) particles (3.2 μm) with relatively small pore size as HPLC packing material. Chromatographia. 2004;60:553–60. https://doi.org/10.1365/s10337-004-0416-4.
Çelebi B. A simple synthetic route for the preparation of a reversed-phase stationary phase based on monosized-porous hydrogel beads and its chromatographic use for separation of small molecules. Acta Chromatogr. 2017;29:143–59. https://doi.org/10.1556/1326.2017.29.2.00.
Salih B, Denizli A, Kavakli C, Say R, Pişkin E. Adsorption of heavy metal ions onto dithizone-anchored poly (EGDMA-HEMA) microbeads. Talanta. 1998;46:1205–13. https://doi.org/10.1016/S0039-9140(97)00362-7.
Tank R, Pathak U, Singh A, Gupta A, Gupta DC. A convenient one step preparation of crosslinked polystyrene mercaptomethyl resin. React Funct Polym. 2009;69:224–8. https://doi.org/10.1016/j.reactfunctpolym.2008.12.022.
Podkościelna B, Kolodyńska D. A new type of cation-exchange polymeric microspheres with pendant methylenethiol groups. Polym Adv Technol. 2013;24:866–72. https://doi.org/10.1002/pat.3155.
Podkościelna B, Bartnicki A, Gawdzik B. New crosslinked hydrogels derivatives of 2-hydroxyethyl methacrylate: Synthesis, modifications and properties. Express Polym Lett. 2012;6:759–71. https://doi.org/10.3144/expresspolymlett.2012.81.
Prasath RA, Gokmen MT, Espeel P, Du Prez FE. Thiol-ene and thiol-yne chemistry in microfluidics: a straightforward method towards macroporous and nonporous functional polymer beads. Polym Chem. 2010;1:685–92. https://doi.org/10.1039/c0py00041h.
Grochowicz M, Bartnicki A, Gawdzik B. Preparation and characterization of porous polymeric microspheres obtained from multifunctional methacrylate monomers. J Polym Sci Part A Polym Chem. 2008;46:6165–74. https://doi.org/10.1002/pola.22927.
Podkościelna B. Synthesis, modification, and porous properties of new glycidyl methacrylate copolymers. J Appl Polym Sci. 2011;120:3020–6. https://doi.org/10.1002/app.33420.
Grochowicz M, Pączkowski P, Gawdzik B. Investigation of the thermal properties of glycidyl methacrylate–ethylene glycol dimethacrylate copolymeric microspheres modified by Diels-Alder reaction. J Therm Anal Calorim. 2018;133:499–508. https://doi.org/10.1007/s10973-017-6785-3.
Bílková Z, Slováková M, Lyka A, Horák D, Lenfeld J, Turková J, et al. Oriented immobilization of galactose oxidase to bead and magnetic bead cellulose and poly(HEMA-co-EDMA) and magnetic poly(HEMA-co-EDMA) microspheres. J Chromatogr B Anal Technol Biomed Life Sci. 2002;770:25–34. https://doi.org/10.1016/S0378-4347(01)00439-X.
Sivakumar M, Rao KP. Synthesis, characterization, and in vitro release of ibuprofen from poly(MMA-HEMA) copolymeric core-shell hydrogel microspheres for biomedical applications. J Appl Polym Sci. 2002;83:3045–54. https://doi.org/10.1002/app.10310.
Xie G, Zhang Q, Luo Z, Wu M, Li T. Preparation and characterization of monodisperse magnetic poly(styrene butyl acrylate methacrylic acid) microspheres in the presence of a polar solvent. J Appl Polym Sci. 2003;87:1733–8. https://doi.org/10.1002/app.11483.
Gao Q, Luo D, Ding J, Feng Y-Q. Rapid magnetic solid-phase extraction based on magnetite/silica/poly(methacrylic acid-co-ethylene glycol dimethacrylate) composite microspheres for the determination of sulfonamide in milk samples. J Chromatogr A. 2010;1217:5602–9. https://doi.org/10.1016/j.chroma.2010.06.067.
Xiao J, Lu Q, Cong H, Shen Y, Yu B. Microporous poly(glycidyl methacrylate-co-ethylene glycol dimethyl acrylate) microspheres: synthesis, functionalization and applications. Polym Chem. 2021;12:6050–70. https://doi.org/10.1039/d1py00834j.
Grochowicz M, Gawdzik B. Permanently porous copolymeric microspheres based on aromatic methacrylates. React Funct Polym. 2011;71:625–33. https://doi.org/10.1016/j.reactfunctpolym.2011.03.001.
Šmigol V, Švec F. Synthesis and properties of uniform beads based on macroporous copolymer glycidyl methacrylate–ethylene dimethacrylate: a way to improve separation media for HPLC. J Appl Polym Sci. 1992;46:1439–48. https://doi.org/10.1002/app.1992.070460814.
Petro M, Svec F, Fréchet JMJ. Monodisperse hydrolyzed poly(glycidyl methacrylate-co-ethylene dimethacrylate) beads as a stationary phase for normal-phase HPLC. Anal Chem. 1997;69:3131–9. https://doi.org/10.1021/ac970365a.
Hansen T, Vermeeren P, Haim A, van Dorp MJH, Codée JDC, Bickelhaupt FM, et al. Regioselectivity of epoxide ring-openings via S<inf>N</inf>2 reactions under basic and acidic conditions. European J Org Chem. 2020;2020:3822–8. https://doi.org/10.1002/ejoc.202000590.
Zhang WL, Piao SH, Choi HJ. Facile and fast synthesis of polyaniline-coated poly(glycidyl methacrylate) core-shell microspheres and their electro-responsive characteristics. J Colloid Interface Sci. 2013;402:100–6. https://doi.org/10.1016/j.jcis.2013.04.011.
Maciejewska M. Thermal properties of TRIM–GMA copolymers with pendant amine groups. J Therm Anal Calorim. 2016;126:1777–85. https://doi.org/10.1007/s10973-016-5617-1.
Sobiesiak M, Podkościelna B, Podkościelny P. New functionalised polymeric microspheres for multicomponent solid phase extraction of phenolic compounds. Adsorption. 2016;22:653–62. https://doi.org/10.1007/s10450-015-9749-6.
Maciejewska M, Rogulska M. Porous DMN-co-GMA copolymers modified with 1-(2-hydroxyethyl)-2-pyrrolidone. J Therm Anal Calorim. 2021;144:699–711. https://doi.org/10.1007/s10973-020-09496-z.
Podkościelna B, Klimek K, Karczmarzyk Z, Wysocki W, Brodacka M, Serafin K, et al. Polymer microspheres modified with pyrazole derivatives as potential agents in anticancer therapy—preliminary studies. Bioorg Chem. 2022. https://doi.org/10.1016/j.bioorg.2022.105765.
Zasońska BA, Šálek P, Procházková J, Müllerová S, Svoboda J, Petrovský E, et al. Peroxidase-like activity of magnetic poly(glycidyl methacrylate-co-ethylene dimethacrylate) particles. Sci Rep. 2019. https://doi.org/10.1038/s41598-018-38012-5.
Kobayashi S, Hachiya I, Suzuki S, Moriwaki M. Polymer-supported silyl enol ethers. Synthesis and reactions with imines for the preparation of an amino alcohol library. Tetrahedron Lett. 1996;37:2809–12. https://doi.org/10.1016/0040-4039(96)00435-2.
Masquelin T, Meunier N, Gerber F, Rossé G. Solution- and solid-phase synthesis of combinatorial libraries of trisubstituted 1,3,5-triazines. Heterocycles. 1998;48:2489–505. https://doi.org/10.3987/COM-98-8231.
Becht J-M, Wagner A, Mioskowski C. A straightforward preparation of a polystyrene thiol resin. Tetrahedron Lett. 2004;45:7031–3. https://doi.org/10.1016/j.tetlet.2004.07.147.
Garamszegi L, Donzel C, Carrot GE, Nguyen TQ, Hilborn J. Synthesis of thiol end-functional polystyrene via atom transfer radical polymerization. React Funct Polym. 2003;55:179–83. https://doi.org/10.1016/S1381-5148(02)00232-8.
Li X, Liu Y, Xu Z, Yan H. Preparation of magnetic microspheres with thiol-containing polymer brushes and immobilization of gold nanoparticles in the brush layer. Eur Polym J. 2011;47:1877–84. https://doi.org/10.1016/j.eurpolymj.2011.07.010.
Zhang W, Sun Y, Zhang L. In situ synthesis of monodisperse silver nanoparticles on sulfhydryl-functionalized poly(glycidyl methacrylate) microspheres for catalytic reduction of 4-nitrophenol. Ind Eng Chem Res. 2015;54:6480–8. https://doi.org/10.1021/acs.iecr.5b01010.
Karadeniz K, Aki H, Sen MY, Çalikoʇlu Y. Ring opening of epoxidized soybean oil with compounds containing two different functional groups. JAOCS J Am Oil Chem Soc. 2015;92:725–31. https://doi.org/10.1007/s11746-015-2638-z.
Grochowicz M, Szajnecki Ł, Rogulska M. Crosslinked 4-vinylpyridine monodisperse functional microspheres for sorption of ibuprofen and Ketoprofen. Polymers (Basel). 2022. https://doi.org/10.3390/polym14102080.
Kierys A, Grochowicz M, Kosik P. The release of ibuprofen sodium salt from permanently porous poly(hydroxyethyl methacrylate-co-trimethylolpropane trimethacrylate) resins. Microporous Mesoporous Mater. 2015;217:133–40. https://doi.org/10.1016/j.micromeso.2015.06.009.
Rosenberg J-E, Flodin P. Macroporous gels. 1. polymerization of trimethylolpropane trimethacrylate in toluene. Macromolecules. 1986;19:1543–6. https://doi.org/10.1021/ma00160a011.
Chiniwalla P, Bai Y, Elce E, Shick R, Christopher McDougall W, Bidstrup Allen SA, et al. Crosslinking and decomposition reactions of epoxide functionalized polynorbornene Part I FTIR and thermogravimetric analysis. J Appl Polym Sci. 2003;89:568–77. https://doi.org/10.1002/app.12234.
Piracha A, Zulfiqar S, McNeill IC. The thermal degradation of copolymers of glycidyl methacrylate and vinylacetate. Polym Degrad Stab. 1996;51:319–26. https://doi.org/10.1016/0141-3910(95)00202-2.
NIST. https://webbook.nist.gov/cgi/inchi?ID=C106912&Mask=80. Accessed 16 Oct 2022
NIST. https://webbook.nist.gov/cgi/cbook.cgi?ID=C107028&Type=IR-SPEC&Index=1. Accessed 16 Oct 2022
NIST. https://webbook.nist.gov/cgi/inchi?ID=C463581&Type=IR-SPEC&Index=0#IR-SPEC. Accessed 16 Oct 2022
Rogulska M, Kultys A, Lubczak J. New thermoplastic polyurethane elastomers based on aliphatic-aromatic chain extenders with different content of sulfur atoms. J Therm Anal Calorim. 2015;121:397–410. https://doi.org/10.1007/s10973-015-4445-z.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Magdalena Maciejewska and Marta Grochowicz. The first draft of the manuscript was written by Magdalena Maciejewska and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Maciejewska, M., Grochowicz, M. Synthesis and thermal characterization of porous polymeric microspheres functionalized with thiol groups. J Therm Anal Calorim 148, 4195–4210 (2023). https://doi.org/10.1007/s10973-023-11972-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-023-11972-1