
Abstract
Porous microspheres obtained from 2,3-epoxypropyl methacrylate (GMA) and 1,4-di(methacryloyloxymethyl) naphthalene (DMN) were modified by the reaction of active oxirane group with 1-(2-hydroxyethyl)-2-pyrrolidone (HP). Spherical shape of the copolymers was confirmed by scanning electron microscopy whereas low-temperature nitrogen adsorption–desorption method was applied to determine their porous structure. It was found that the modification process did not change noticeably the internal structure of the materials. The specific surface area for both types of materials ranged from 41 to 106 m2g−1 depending on the molar ratio of the monomers. Changes, however, were observed in reference to thermal properties. They were studied using differential scanning calorimetry and thermogravimetry coupled with FTIR in inert and oxidative conditions. The reaction with HP reinforced thermal stability of the modified microspheres in helium atmosphere. Their initial decomposition temperatures (IDTs) increased to 273–281 °C. In air atmosphere, the values of IDTs were at the same level (248–269 °C). The modified beads decomposed in three (helium) or two (air) stages.
Similar content being viewed by others

Introduction
Highly porous polymeric materials are very challenging class of materials. They have gained considerable attention with a wide range of applications including bioengineering fields [1,2,3], environmental purification [4], solid phase supports for catalysis [5,6,7,8,9], architectures with promising potential in gas storage [10, 11] and separation [12, 13].
In the majority of the mentioned above applications, the polymer surface character is of paramount importance. Particularly attractive are adsorbents of a polar nature that have affinity for a wide range of organic compounds. The presence of polar groups provides possibility to create fairly strong interaction adsorbent-adsorbate in the separation system. The attempts to synthesize adsorbent of polar character are the subject of many studies [14,15,16,17,18,19,20,21,22]. However, the number of polar monomers that are able to copolymerize with appropriate cross-linkers that offer good mechanical and thermal stability is quite limited. This limitation can be overcome by modification of the polymer matrix that contain reactive moiety. One of the most reactive functional groups is epoxide ring. Its modification with pyrrolidone and amines was presented in our previous papers [23,24,25,26,27]. Another reagent suitable for this purpose is 1-(2-hydroxyethyl)-2-pyrrolidone (HP) which contains in its structure not only strongly polar pyrrolidone unit but also hydroxyl one. This fact was the main reason that we decided to perform modification reaction of porous microspheres of 2,3-epoxypropyl methacrylate (GMA) cross-linked with 1,4-di(methacryloyloxymethyl) naphthalene (DMN) by HP. In the next step of the investigation, thermal properties of the modified beads were conducted by the means of differential scanning calorimetry (DSC) and thermogravimetry (TG) coupled with Fourier transform infrared (FTIR) spectroscopy. Additionally, the porous copolymers were characterized using elemental analysis, FTIR, scanning electron microscopy (SEM), atomic force microscopy (AFM) and low-temperature nitrogen adsorption–desorption method.
Experimental
Chemicals
GMA was purchased from Sigma-Aldrich. To remove inhibitors, it was washed with 5% aqueous NaOH whereas α,α′-azoisobutyronitrile and bis(2-ethylhexyl) sulfosuccinate sodium salt (from Fluka, AG, Switzerland) and HP (from Sigma-Aldrich) were used as received. Methyl alcohol, toluene and tin(IV) chloride (reagent grade) were acquired from POCh (Poland). DMN was synthesized in our department in accordance with the method described previously [28,29,30].
Synthesis of the modified GMA-co-DMN microspheres
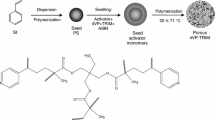
Copolymerization of GMA with DMN was performed according to the procedure reported earlier [23, 27]. Next, the polymer network containing the reactive epoxy rings was subjected to reaction with HP (Fig. 1). The reaction was conducted in the following way: 10 g of the obtained microspheres was immersed in 50 cm3 of HP and placed in a 100-cm3 round-bottomed three-necked reactor equipped with a thermometer, mechanical stirrer and condenser. Then, 0.05 g of tin(IV) chloride was added, and the content of the flask was heated at 120 °C for 8 h. The received in this process modified beads were washed with water and filtered. Next, they were purified in a Soxhlet extractor with hot toluene and dried under reduced pressure at 60 °C.

Scheme of modification reaction of DMN-co-GMA copolymers with HP
Method of copolymers characterization
In order to perform the elemental analysis (CHN) of the synthesized copolymers, the Perkin–Elmer 2400 analyzer (USA) was used.
The main parameters of the porous structure were determined on the basis of the low-temperature nitrogen adsorption–desorption method. The isotherms at −196 °C were established volumetrically using ASAP 2405 N apparatus (Micromeritics Corp., Norcross, USA). Before the experiment, the adsorbents were degassed (10−2 mm Hg) at 140 °C. The linear Brunauer–Emmett–Teller (BET) plots of adsorption data were used to evaluate the specific surface area (SBET). The total pore volume (V) was estimated from a single-point adsorption at a relative pressure p/po = 0.985. The calculations of pore-size distributions (PSD) followed the Barrett, Joyner and Halenda (BJH) procedure. The pore diameters (DBJH) were estimated from the PSD maxima.
The FTIR spectra were recorded with a Bruker Tensor 27 FTIR spectrometer (Germany) using the attenuated total reflectance (ATR) method. To collect the spectra, absorbance mode was used. The following parameters: a spectral resolution of 4 cm−1, 16 scans per each spectrum and 4000–600 cm−1 spectral range were applied.
SEM studies were carried out on a Tesla BS-301 microscope (Czechoslovakia) operating at 15.0 keV whereas AFM ones on AFM Nanoscope III (Digital Instruments, USA) working in contact mode.
TG and DSC measurements were conducted with a Netzsch STA 449 F1 Jupiter apparatus (Germany) in the same way as described previously [27]. Also, the analysis of the volatiles formed during the thermal degradation was done in an analogous manner as presented in this paper.
Furthermore, the DSC measurements were also carried out with a Netzsch 204 calorimeter (Germany). They were done in Al crucibles with a perforated lid. As a reference, an empty one was used. A dry argon gas with a flow rate of 20 cm3 min–1 was purged through the sample cell. The dynamic scans were performed at the heating rate of 10 °C min−1 from room temperature to 500 ºC. The mass of the copolymer sample was about 10 mg.
Results and discussion
The suspension–emulsion polymerization was successfully applied for the preparation of DMN-co-GMA copolymers. This method has been widely used in our laboratory, and it has been established that is especially suitable for synthesis of porous microspheres with diameter in the micrometric range (Figs. 2 and 3). In the course of the synthesis, DMN was utilized as a cross-linker. It provided thermal and mechanical properties of the resulted polymeric matrix. Sequentially, GMA delivered reactive oxirane rings. In order to increase the number of epoxy group in the polymer network, the molar ratio of the monomers was changed from 1:1 to 5:1.

SEM micrograph of DMN-co-GMA4 + HP copolymer

AFM image of DMN-co-GMA4 + HP copolymer
As the main goal of the research was the modification of the surface of DMN-co-GMA beads, a subsequent reaction with 1-(2-hydroxyethyl)-2-pyrrolidone was carried out. The oxirane rings existed in the copolymer were opened, and materials with pendant hydroxyl and pyrrolidone functionalities were synthesized. The codes of newly created copolymers were based on the molar ratio of DMN to GMA and are given in the list of symbols. The results of the elemental analysis conducted for these materials are presented in Table 1. As can be seen, the amount of nitrogen in the structure of the copolymers grows along with the increasing GMA in the polymerization mixture. Additionally, ATR-FTIR spectroscopy was applied to confirm the proper course of the modification reaction. Figure 4 displays the spectra of modified (DMN-co-GMA5 + HP) and parent (DMN-co-GMA5) copolymers. It was observed that absorbance peaks of epoxide ring (992, 906 and 841 cm−1) disappeared. At the same time, new peaks characteristic of pyrrolidone unit at 1656 cm−1 and of hydroxyl group at 3408 cm−1 (ν O–H) and 1058 cm−1 (ν C–OH) appeared.

ATR-FTIR spectra of parent and modified with HP copolymers
The newly modified materials have been designated for adsorption processes. This fact implies the necessity of internal structure investigation. The basic parameters of porous structure of the investigated copolymers are listed in Table 2. The reaction with HP does not lead to significant changes of the internal structure of new copolymers in comparison with the parent ones [23]. The values of specific surface area decrease from 105 to 41 m2g−1 depending on the molar ratio of the cross-linker to the functional monomer. This fact can be straightforwardly explained on the basis of the phase separation physical process which intensively depends on the concentration of the cross-linker. In the case of high amount of DMN, the phase separation is shifted to the point of greater conversion of monomer to polymer. In a consequence, a network of interconnected separate microglobules is formed. It possesses high surface area and broad pore-size distribution with a maximum in the region of micro- to mesopore. Diminished cross-linker concentration leads to early phase separation at lower conversion. This process stimulates not only aggregation of microglobules but also filling of small pores. Therefore, polymer network with lower surface area is formed.
Interestingly, the process of modification of the DMN-co-GMA copolymers with HP contributes to the changes of the thermal properties of the newly obtained materials. In helium, the process of thermal decomposition of the modified copolymers progress in three stages comparing to one or two for the parent ones. As an example, Fig. 5 presents TG and DTG curves obtained for DMN-co-GMA1 and DMN-co-GMA1 + HP copolymers. Additionally, it was observed that the IDTs increased from 258–269 °C for parent copolymers [23] to 273–281 °C for modified ones (Table 3). Simultaneously, the T20% and T50% decreased. In turn, the measurement data obtained in synthetic air show two steps for both the parent and functionalized copolymers (Fig. 6). In this atmosphere, the modified copolymers exhibited similar IDTs: 248–269 °C (Table 4) vs. 249–269 °C [23]. Most of them, however, revealed lower values of T20% and T50%.

TG and DTG curves of parent (DMN-co-GMA1) and modified with HP (DMN-co-GMA1 + HP) copolymers obtained in helium

TG and DTG curves of parent (DMN-co-GMA1) and modified with HP (DMN-co-GMA1 + HP) copolymers obtained in synthetic air
The alterations in thermal degradation of DMN-GMA copolymers have been visible not only between initial and functionalized ones but also in the entire series of modified copolymers where they vary in the molar ratio of GMA to DMN. The copolymer with the largest amount of functional monomer (DMN-co-GMA5 + HP) showed the smaller thermal resistance both in helium and in synthetic air. Simultaneously, the most cross-linked copolymer (DMN-co-GMA1 + HP) had the best thermal resistance in air. Figures 7 and 8 present TG and DTG curves for the selected copolymers. Looking at DTG curves determined in synthetic air atmosphere, an individual peak with the max. at ~ 500 °C is evidently visible. In the case of investigation completed in helium, DTG curves do not show such a peak. Moreover, the residue of the sample in helium is three times higher than in air.

TG and DTG curves of selected copolymers modified with HP obtained in helium

TG and DTG curves of selected copolymers modified with HP obtained in synthetic air
In light of the chemical structure of DMN-co-GMA + HP copolymers, it can be assumed that formation and evolution of aldehydes along with carboxylic and vinyl compounds may take place during thermal treatment of the materials. The former compounds can be created as a result of α-hydrogen bond scission while the latter ones of β-hydrogen bond scission. It should also be taken into consideration that the highly cross-linked copolymers can undergo the random chain scission.
First decomposition step may be connected with the degradation process of residual oxirane rings and included hydroxypyrrolidone units. T1max has the lowest value (340 °C) for the copolymer with the greatest amount of functional monomer (DMN-co-GMA5 + HP). It indicates that the degradation process of this copolymer proceeds at the earliest among all the copolymers under the study. In the case of the most cross-linked and consequently the least enriched in oxirane groups, copolymer (DMN-co-GMA1 + HP) T1max is the highest (353 °C). At second step with T2max in the range of 379–384 °C, mostly chain scission of ester linkages may take place and the degradation of the cross-linked parts of the microspheres begins. At third step (407–420 °C), further thermal degradation of cross-linked parts of the copolymers proceeds. The highest value of T3max is observed for the most cross-linked copolymer. It is worth to mention that the three stages of the decomposition are partially superimposed.
The suggested course of the thermal decomposition process is reflected in the analysis of the emitted gaseous products. It was performed for the chosen copolymer, i.e., DMN-co-GMA1-HP, and the received 3D plots of FTIR spectra of gases evolved during its decomposition, both in helium and synthetic air, are given in Fig. 9. For the detailed discussion of the obtained results, the maxima were extracted and shown in Figs. 10 and 11. As can be seen, the maxima of gases emissions are in accordance with the maxima determined from DTG curves.

3D plots of FTIR spectra of volatile products evolved during thermal decomposition of DMN-co-GMA1 + HP copolymer in helium and synthetic air atmospheres

FTIR spectra of volatile products evolved from DMN-co–GMA1 + HP copolymer at the maxima of decomposition in helium

FTIR spectra of volatile products evolved from DMN-co–GMA1 + HP copolymer at the maxima of decomposition in synthetic air
Based on the FTIR spectra obtained in inert conditions ( Figs. 9 and 10), one may state that in first decomposition step (maximum at 353 °C), the emission of unsaturated aliphatic compounds manifested by bands at 3105–2870 cm−1 (ν C–H of –CH3, = CH2 and –CH = CH2 groups) and at 943 cm−1 (δoop C–H of –CH = CH2 group) took place. These compounds can imply the β-elimination decomposition of ester groups. Moreover, bands that are characteristic of organic carbonyl compounds (ν C = O at 1764 and 1711 cm−1), especially esters ( ν C–O of ester group at 1125 cm−1) and carboxylic acids (ν O–H of carboxylic group at 3582 cm−1) can be found. They confirm β-elimination degradation reaction. Also, CO2 (adsorption peaks at 2360–2311 and 669 cm−1), CO (adsorption peaks at 2181 and 2114 cm−1) and H2O (adsorption peaks at ~ 4000–3500 and ~ 1800–1300 cm−1) can be detected in first decomposition step. In second degradation step (T2max at 379 °C), larger amounts of CO2, CO and organic carbonyl products were found. In turn, in third step ( T3max at 420 °C), decreasing of the carbonyl compounds extent comparing with the previous stage was noticed. On account of the above data, one may state, that the highest amount of volatile products was emitted in the second step. This fact is in accordance with TG and DTG curves. Namely, the highest mass loss (~ 39%) was observed in this step.
The FTIR spectra of DMN-co-GMA1 + HP recorded during first decomposition stage (T1max at 313 °C) in synthetic air (Figs. 9 and 11), similarly to that received in inert conditions, show bands pointing to the presence of H2O, CO2, CO and organic carbonyl compounds, including unsaturated ones. In second stage (T2max at 521 °C), mainly H2O, CO2 and CO were evolved.
Thermal properties of the DMN-co-GMA copolymers were also monitored by the DSC method. Figure 12 displays DSC curves for three selected copolymers ascertained in helium. As one can see, both exothermic as well as endothermic thermal events took place. The first endothermic peak (the maximum at about 100 °C) is correlated with the moisture vaporization from the copolymers. With the increase in the quantity of HP functionalities in the copolymer, the volume of adsorbed water considerably increases. Second endothermic peak whose maximum increases with the degree of cross-linking (from 323 to 340 ºC) can be referred to the degradation reaction of hydroxyl and ester groups as well as pyrrolidone units. It should also be taken into consideration that the decomposition of unreacted epoxide rings can occur. What is noteworthy, notable changes with respect to the enthalpy of this process were noticed. Its value increases together with the increasing amount of functional groups present in the polymeric matrix.

DSC curves of the selected copolymers modified with HP determined in helium
The exothermic peak following the first step of endothermic decomposition can refer to a thermal cross-linking reaction of double bounds, which are formed in the first step of thermal degradation by the β-bond scission reactions. The enthalpy of the cross-linking reaction is the greatest for the copolymers with the biggest amount of the ester bonds which are the most susceptible to the scission (DMN-co-GMA1 + HP). In the case of DMN-co-GMA5 + HP (the copolymer with the smallest amount of the ester groups), this process is hardly visible.
The last region (above 400 °C) demonstrates the thermal degradation of the cross-linked parts of the copolymers.
In regard to experiments conducted in air, only the exothermic peaks are visible on the DSC curves (Fig. 13). They are connected with the degradation of the investigated polymers that occur via oxidation processes. The peaks with the maxima at 320–348 °C, analogically as in the case of helium atmosphere, can be associated with the degradation of hydroxyl and ester groups as well as pyrrolidone units. The higher amount of these functional groups the higher enthalpy of the degradation reaction was observed. Its value increased from about 90 μV mg−1 for DMN-co-GMA1 + HP to 390 μV mg−1 for DMN-co-GMA5 + HP copolymer. In the next step, oxidative degradation of cross-linked parts takes place. As a result, the second exothermic peaks can be observed. For the less cross-linked copolymer (DMN-co-GMA5 + HP), the maximum is at 496 °C. Along with the increasing amount of cross-linker, the maximum shifts to higher temperatures. For the most cross-linked copolymer (DMN-co-GMA1 + HP) equals 520 °C. The same dependence can be perceived in the case of the enthalpy values of this process (from about 280 to 690 μV mg−1).

DSC curves of the selected copolymers modified with HP determined in synthetic air
Conclusions
Based on the conducted studies, one can state that the process of functionalization of DMN-co-GMA copolymers with HP does not change their basic parameters of porous structure. On the other hand, it has got considerable impact on thermal properties of the newly obtained functionalized materials. These properties were evaluated by TG/DSC/FTIR methods in oxidative (synthetic air) and inert (helium) conditions. The conducted studies indicate that in an inert atmosphere the IDTs increase from 258–269 °C for parent copolymers to 273–281 °C for modified ones. In oxidative conditions, they are kept practically on the same level. The examination of the DTG curves showed that the thermal decomposition of the functionalized copolymers carried out in helium proceeded in three stages whereas in synthetic air in two ones. Generally, the thermal decomposition of the functionalized microspheres exhibited multistage decomposition patterns different from parent DMN-co-GMA copolymers.
Furthermore, comprehensive analysis of the volatile decomposition products demonstrates that both α-hydrogen and β-hydrogen bond scission mechanism are possible in the degradation process of the functionalized DMN-co-GMA + HP copolymers. They may also decompose according to random chain scission mechanism.
Abbreviations
- D BJH :
-
Pore diameter (Å)
- DMN:
-
1,4-di(methacryloyloxy methyl) naphthalene
- FDT:
-
Final decomposition temperature (°C)
- GMA:
-
2,3-epoxypropyl methacrylate (glycidyl methacrylate)
- HP:
-
1-(2-hydroxyethyl)-2-pyrrolidone
- IDT:
-
Initial decomposition temperature (°C)
- S BET :
-
Specific surface area (m2 g−1)
- T 1max :
-
Temperature of the first maximum rate of mass loss (°C)
- T 2max :
-
Temperature of the second maximum rate of mass loss (°C)
- T 3max :
-
Temperature of the third maximum rate of mass loss (°C)
- T 20% :
-
Temperature of 20% mass loss (°C)
- T 50% :
-
Temperature of 50% mass loss (°C)
- DMN-co-GMA1+HP:
-
The copolymer modified with HP, molar ratio of DMN to GMA equals 1:1
- DMN-co-GMA2+HP:
-
The copolymer modified with HP, molar ratio of DMN to GMA equals 1:2
- DMN-co-GMA3+HP:
-
The copolymer modified with HP, molar ratio of DMN to GMA equals 1:3
- DMN-co-GMA4+HP:
-
The copolymer modified with HP, molar ratio of DMN to GMA equals 1:4
- DMN-co-GMA5+HP:
-
The copolymer modified with HP, molar ratio of DMN to GMA equals 1:5
- V:
-
Pore volume (cm3 g−1)
References
Hu Y, Gu X, Yang Y, Huang J, Hu M, Chen W, Tong Z, Wang C. Facile fabrication of poly(L-lactic acid)-grafted hydroxyapatite/poly(lactic-co-glycolic acid) scaffolds by pickering high internal phase emulsion templates. ACS Appl Mater Interfaces. 2014;6:17166–75.
Busby W, Cameron NR, Jahoda C. Emulsion-derived foams (polyHIPEs) containing Poly(epsilon-caprolactone) as matrixes for tissue engineering. Biomacromol. 2001;2:154–64.
Engelmayr GC, Cheng MY, Bettinger CJ, Borenstein JT, Langer R, Freed LE. Accordion-like honeycombs for tissue engineering of cardiac anisotropy. Nat Mater. 2008;7:1003–100.
Yang Y, Tong Z, Ngai T, Wang C. Nitrogen-rich and fire resistant carbon aerogels for the removal of oil contaminants from water. ACS Appl Mater Interfaces. 2014;6:6351–60.
Desforges A, Backov R, Deleuze H, Mondain-Monval O. Generation of palladium nanoparticles within macrocellular polymeric supports: application to heterogeneous catalysis of the Suzuki−Miyaura coupling reaction. Adv Funct Mater. 2005;15:1689–95.
Zhang S, Chen J, Lykakis IN, Perchyonok VT. Streamlining organic free radical synthesis through modern molecular technology: from polymer supported synthesis to microreactors and beyond. Curr Org Synth. 2010;7:177–88.
Sundell Mats J, Pajunen Esko O, Hormi Osmo EO, Nasman JH. Synthesis and use as a catalyst support of porous polystyrene with bis(phosphonic acid)-functionalized surfaces. Chem Mater. 1993;5:372–6.
Ruckenstein E, Hong L. Binding catalytic sites to the surface of porous polymers and some catalytic applications. Chem Mater. 1992;4:122–7.
Liu HH, Wan DC, Du J, Jin M. Dendritic amphiphile mediated one-pot preparation of 3D Pt nanoparticles-decorated PolyHIPE as a durable and well-recyclable catalyst. ACS Appl Mater Interfaces. 2015;7:20885–92.
Furukawa H, Yaghi OM. Storage of hydrogen, methane, and carbon dioxide in highly porous covalent organic frameworks for clean energy applications. J Am Chem Soc. 2009;131:8875–83.
Li B, Huang X, Liang L, Tan B. Synthesis of uniform microporous polymer nanoparticles and their applications for hydrogen storage. J Mater Chem. 2010;20:7444–500.
Shirshova N, Johansson P, Marczewski MJ, Kot E, Ensling D, Bismarck A, Steinke JHG. Polymerised high internal phase ionic liquid-in-oil emulsions as potential separators for lithium ion batteries. J Mater Chem A. 2013;1:9612–9.
Hughes JM, Budd PM, Tiede K, Lewis J. Polymerized high internal phase emulsion monoliths for the chromatographic separation of engineered nanoparticles. J Appl Polym Sci. 2015;132:41229.
Maciejewska M. Characterization of macroporous 1-vinyl-2-pyrrolidone copolymers obtained by suspension polymerization. J Appl Polym Sci. 2012;124:568–75.
Maciejewska M, Osypiuk-Tomasik J. Sorption on porous copolymers of 1-vinyl-2-pyrrolidone-divinylbenzene. J Therm Anal Calorim. 2013;114:749–55.
Kolarz BN, Jermakowicz-Bartkowiak D. Poly(4-vinylpyridine) resins towards perrhenate sorption and desorption. React Funct Polym. 2011;71:95–103.
Trochimczuk AW, Streat M, Kolarz N. Highly polar polymeric sorbents Characterization and sorptive properties towards phenol and its derivatives. React Funct Polym. 2001;46:259–71.
Grochowicz M. Investigation of the thermal behavior of 4-vinylpyridine–trimethylolpropane trimethacrylate copolymeric microspheres. J Therm Anal Calorim. 2014;118:1603–11.
Grochowicz M, Szajnecki Ł, Gawdzik B. 4VP-TRIM composite polymer particles and their application as adsorbents. Adsorpt Sci Technol. 2015;33:609–16.
Zeng X, Yu T, Wang P, Yuan R, Wen Q, Fan Y, Wang C, Shi R. Preparation and characterization of polar polymeric adsorbents with high surface area for the removal of phenol from water. J Hazard Mater. 2010;177:773–80.
Maciejewska M, Osypiuk-Tomasik J. Studies of sorption properties of porous copolymers of 1-vinyl-2-pyrrolidone. J Therm Anal Calorim. 2013;111:1595–601.
Grochowicz M, Pączkowski P, Gawdzik B. Investigation of the thermal properties of glycidyl methacrylate–ethylene glycol dimethacrylate copolymeric microspheres modified by Diels–Alder reaction. J Therm Anal Calorim. 2018;133:499–508.
Maciejewska M. Synthesis and thermal properties of parent and modified DMN-co-GMA copolymers. J Therm Anal Calorim. 2018;133:969–80.
Maciejewska M. Study on thermal properties TRIM-GMA copolymers with pendant amine groups. J Therm Anal Calorim. 2016;126:1777–855.
Maciejewska M, Bolbukh YM. Study on synthesis and characterization of porous microspheres with pendant amine groups. Adsorpt Sci Technol. 2015;33:617–23.
Maciejewska M. Characterization of thermal properties of porous microspheres bearing pyrrolidone units. J Therm Anal Calorim. 2015;119:1147–55.
Maciejewska M, Rogulska M. Insight into functionalized DMN-co-GMA copolymers. I. Modification with diethylenetriamine. J Therm Anal Calorim. 2019. https://doi.org/10.1007/s10973-019-08327-0.
Gawdzik B, Czerwińska-Bil U. Porous copolymer-based cation exchanger for the off-line preconcentration of aromatic amines from water. Chromatogr. 1991;32:167–70.
Matynia T, Gawdzik B. Influence of synthesis conditions of porous copolymers of 1,4-di(methacryloyloxymethyl) naphthalene with divinylbenzene on their structure. I. Influence of mol composition of monomers. Angew Makromol Chem. 1987;147:123–32.
Gawdzik B, Matynia T. Influence of synthesis conditions of porous copolymers of 1,4-di(methacryloyloxymethyl) naphthalene with divinylbenzene on their structure. II. Influence of diluent composition. Angew Makromol Chem. 1987;125:33–5.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Maciejewska, M., Rogulska, M. Porous DMN-co-GMA copolymers modified with 1-(2-hydroxyethyl)-2-pyrrolidone. J Therm Anal Calorim 144, 699–711 (2021). https://doi.org/10.1007/s10973-020-09496-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-020-09496-z