
Abstract
Two series of new sulfur-containing thermoplastic poly(carbonate-urethane)s (PCURs) were prepared by a one-step melt polyaddition from diphenylethane derivative diol, i.e., 4,4′-(ethane-1,2-diyl)bis(benzenethiohexanol) as a nonconventional chain extender, 1,1′-methanediylbis(4-isocyanatobenzene) (MDI) or 1,6-diisocyanatohexane (HDI) and 20–60 mol% poly(hexane-1,6-diyl carbonate) diol of \(\overline{M}_{\text n} = 860\) g mol−1 as a soft segment. The FTIR and XRD as well as DSC, TG, and TG coupled with FTIR were employed to characterize the PCURs’ structure and thermal properties. Their physicochemical, adhesive, and tensile properties as well as Shore A/D hardness were also studied. The amorphous MDI-based PCURs were transparent materials (transmittance at 800 nm: 83–88%), while the HDI-based ones, showing partially crystalline structures (the degree of crystallinity: of 20–28%), were opaque. The polymers derived from HDI exhibited lower glass-transition temperatures than those obtained from MDI (from – 29 to – 12 °C vs. from 14 to 46 °C) as well as a higher degree of microphase separation. In turn, the PCURs based on MDI revealed higher tensile strength (up to 50.8 MPa vs. up to 33.4 MPa) and better thermal stability (e.g., the temperature of 1% mass loss: 294–305 °C vs. 262–278 °C). These sulfur-containing PCURs exhibited better adhesive properties to copper compared with the conventional PCURs synthesized from butane-1,4-diol as a chain extender.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Thermoplastic polyurethane elastomers (TPUs) are included in the important family of polymeric materials. Combination of such properties as high tensile strength and hardness with large elongation at break as well as excellent tear, abrasion, and chemical resistance results in a wide array of their applications ranging from footwear and ski boots to gaskets and seals. TPUs show mechanical properties which are typical of rubbers but can be processed by the methods applied for thermoplastics, i.e., injection, extrusion, calendaring. Moreover, they can be reshaped and recycled without affecting their physical properties negatively. This is possible owing to the absence of chemical cross-links, which usually occur in conventional rubbers [1,2,3,4]. The functional properties of TPUs are, in part, due to their segmented structure. They are multiblock copolymers formed by thermodynamically incompatible hard and soft segments. The hard segments are built from the alternating diisocyanate short-chain diol (chain extender) sequences, while the soft segments are constituted by a polymer diol. The former acts as reinforcement sites and provides physical cross-links by molecular association through hydrogen bonding, performing a similar role to chemical cross-links in rubbers. Thus, they exert strong influence on the modulus, hardness, tear strength, and thermal stability. In turn, the latter control the elasticity, softness, elongation at break, and low-temperature resistance [5,6,7].
The selection of appropriate reacting components is crucial for obtaining TPUs of the desired properties that satisfy customers’ requirements. Applications of polyester diols as soft segments enable preparation of TPUs of relatively good physical properties; however, they are susceptible to hydrolytic cleavage of the ester linkages. On the other hand, TPUs based on polyether diols exhibit a greater hydrolysis resistance and are preferred in the uses where hydrolytic stability is desired. Unluckily, the poly(ether-urethane)s are prone to oxidation [6, 8]. Both these disadvantages can be overcome when replacing polyester or polyether segments by oxidatively and hydrolytically stable polycarbonate ones. The poly(carbonate-urethane)s (PCURs) owing to their superior mechanical and biological properties are favored as biopolymers for long-lasting implantation [9,10,11,12]. Aromatic diisocyanates, e.g., 1,1′-methanediylbis(4-isocyanatobenzene) (MDI) and 2,4-diisocyanato-1-methylbenzene, as a part of the hard segments, produce TPUs of high tensile strength and modulus as well as thermal resistance [6, 13, 14]. However, unfavorable yellowing upon exposure to heat or UV–VIS light confines their applications for outdoor usages. Aliphatic diisocyanates, such as 1,6-diisocyanatohexane (HDI) and 1,1′-methanediylbis(4-isocyanatocyclohexane), make it possible to obtain light and coloring stable materials with a higher degree of microphase separation and better resistance to hydrolytic degradation over aromatic ones, though mechanical properties may be deteriorated [6, 14, 15]. For the synthesis of TPUs as chain extenders, aliphatic diols, of which butane-1,4-diol (BD) is most frequently used, are generally chosen [16], whereas the aromatic chain extenders are preferable in the case of reactions that must be highly controlled. Due to electron delocalization, they are characterized by less reactivity than aliphatic ones [6, 7]. The aromatic and aliphatic–aromatic diols are also advantageous when the aim is to prepare materials of higher modulus of elasticity and hardness, enhanced thermal stability or liquid crystalline properties [7, 17,18,19,20,21]. The investigation interests of co-workers and myself include aliphatic–aromatic diols with sulfur atoms, being the derivatives, among others, of diphenylmethane [22,23,24], diphenyl ether [25, 26], and diphenyl sulfide [25,26,27,28,29]. They enable preparation of thermoplastic polyurethanes of good thermal stability and mechanical properties and simultaneously of higher adhesive strength to metals and refractive index compared with the aliphatic ones [23, 26,27,28].
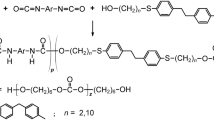
This paper deals with the synthesis and characterization of new thermoplastic PCURs based on the diphenylethane derivative diol. The main purpose of the research was to determine the effect of aliphatic and aromatic diisocyanate on the structure and physicochemical, thermal, mechanical, and adhesive properties of new PCURs differing in soft-segment contents. The PCURs were synthesized from HDI or MDI, 4,4′-(ethane-1,2-diyl)bis(benzenethiohexanol) and 20–60 mol% poly(hexane-1,6-diyl carbonate) diol (PHCD) of \(\overline{M}_{\text n} = 860\) g mol−1, and examined by Fourier transform infrared (FTIR) and ultraviolet–visible (UV–VIS) spectroscopies, X-ray diffraction analysis (XRD), thermogravimetry (TG), TG-FTIR, and differential scanning calorimetry (DSC). The measurements of Shore hardness, tensile properties, and lap shear strength to copper were performed. Of significant importance was also finding out which of the prepared polymers are characterized by elastomeric properties.
Experimental
Materials
The diol H (m.p. = 116–117 °C) was obtained by the condensation reaction of 4,4′-(ethane-1,2-diyl)bis(benzenethiol) with 6-chlorohexan-1-ol. The general procedure was as follows: 4,4′-(ethane-1,2-diyl)bis(benzenethiol) (14.78 g, 0.06 mol) was dissolved in 250 cm3 of 10% aqueous of sodium hydroxide, and the resulting solution was heated to 60 °C. 6-chlorohexan-1-ol (17.97 g, 0.125 mol) in 10 cm3 of ethanol was then added dropwise under vigorous stirring. The reaction mixture was heated and stirred in a water boiling bath for 2 h. Then it was cooled, and the condensation product separated as a white precipitate was collected by filtration, washed thoroughly with water, and dried in air. The crude compound (22.5 g, 84% yield) was recrystallized twice from benzene and dried in vacuo to a constant mass [30]. Sodium hydroxide, ethanol, and benzene (POCh, Poland) and 6-chlorohexan-1-ol (Fluka, Switzerland) were analytical reagent grade and used as received. Also the reagents such as MDI (98%) from Sigma-Aldrich (Germany) and HDI (99%) and dibutyltin dilaurate (DBTDL) from Merck–Schuchardt (Germany) were used without further purification. In turn, prior to the use, PHCD of \(\overline{M}_{\text n} = 860\) g mol−1 from Aldrich (USA) was heated at 90 °C in vacuo for 10 h (to about 0.005% water content).
Synthesis of PCURs
The PCURs, containing 20, 40, 50 or 60 mol% of the soft segment, were synthesized according to Scheme 1 by a one-step melt polyaddition of MDI or HDI, diol H, and PHCD. The syntheses were conducted at the NCO/OH molar ratio of 1 (from MDI) or 1.05 (from HDI) in the dry nitrogen atmosphere. In case less reactive aliphatic diisocyanate was used, the syntheses were carried out in the presence of DBTDL as a catalyst.

Synthesis of PCURs
To synthesize the HDI-based PCURs, the diol H and PHCD (0.01 mol together) and HDI (0.0105 mol) were weighed out into a three-necked round-bottom flask fitted with a mechanical stirrer, a gas inlet tube and a calcium chloride drying tube, and placed in an oil bath of a temperature 130 °C. Next to the clear and mixed melt DBTDL (0.03 g) was added. In the case of the MDI-based PCURs, diisocyanate (0.01 mol) was put into a flask only after melting and mixing of dihydroxy compounds at 130 °C. The reactions were continued until the viscosity increase made stirring impossible. Then the formed colorless product was conditioned at 140 °C for 2 h.
Measurements
Reduced viscosities (ηreds, dL g−1) of 0.5 mass% PCUR solutions in a phenol (Ph)/1,1,2,2-tetrachloroethane (TChE) (Ph/TChE) mixture with a mass ratio of 1:3 were determined in an Ubbelohde viscometer (Poland) at 25 °C.
The FTIR spectra were recorded with a Bruker Tensor 27 FTIR spectrometer (Germany) using the attenuated total reflectance (ATR) technique. All spectra were obtained within the range of 600 and 4000 cm−1 in the absorbance mode. The spectral resolution was 4 cm−1 with 32 scans taken for each spectrum. The infrared carbonyl stretching region of the polymers was resolved into the Gaussian curves using OPUS 7.2 software.
TG was done on a Netzsch STA 449 F1 Jupiter thermal analyzer (Germany) in helium atmosphere (flow = 20 cm3 min−1) from room temperature up to 1000 °C at the heating rate of 10 °C min−1. The TG analysis was conducted in Al2O3 crucibles. The mass of the sample was ~ 10 mg. The composition of a gas evolved during the decomposition process was detected and analyzed by a Bruker Tensor 27 FTIR spectrometer (Germany) coupled online to a Netzsch STA instrument by the Teflon transfer line with a 2 mm diameter heated to 200 °C. The FTIR spectra were recorded in the spectral range of 600–4000 cm−1 with a 4 cm−1 resolution and 16 repetitions.
DSC was performed by means of a Netzsch 204 calorimeter (Germany). The measurements were done in aluminum pans with pierced lid (mass of 40 ± 1 mg). As a reference an empty aluminum crucible was used. Dry nitrogen gas with a flow rate of 30 mL min−1 was purged through the sample cell. Cooling was accomplished with liquid nitrogen. About 10 mg samples (± 0.2 mg) were cooled to – 100 °C, heated to 200 °C, cooled again to – 100 °C and reheated to 200 °C. The heating/cooling rate of 10 °C min−1 was applied. The reported transitions were taken from the first and second heating cycles. The inflection point on the curves of the heat capacity changes has been chosen to represent the glass transition temperatures (Tgs) for the polymer samples. Melting temperatures (Tms) were read at the endothermic peak maxima.
The XRD patterns of the PCUR samples were collected using a DRON-3 X-ray apparatus (Russia) with a copper tube and nickel filter. The data collection was recorded in the range of 8–50° with a step of 0.04°. The obtained patterns were analyzed applying the WAXSFIT computer program [31]. The program resolves a diffraction curve on diffraction peaks and amorphous halo which allows to estimate the degree of crystallinity.
Press molding was carried out with a Carver hydraulic press (USA) at 120–140 °C (for the PCURs based on HDI) and 150–165 °C (for the PCURs based on MDI) under 10–30 MPa pressure.
The transparency of the compression-molded 1-mm-thick plates was measured using a UV-1800 (Shimadzu, Japan) UV–VIS spectrophotometer in the 300–900 nm wavelength range with a sampling interval of 0.5 nm.
The hardness of the PCURs was measured by the Shore A/D method on a Zwick 7206/H04 analog hardness testing apparatus (Germany) at 23 °C according to ISO 828 standard; readings were taken after 15 s.
The mechanical properties of the PCURs were determined by tensile testing on the specimens cut from the pressed sheets (1 mm thick and 6 mm wide for the section measured) at the head speed of 100 mm min−1 at 23 °C using a Zwick/Roell Z010 (Germany) apparatus. The Polish Standard PN-81/C-89034 (EN ISO Standard 527-1:1996 and 527-2:1996) was applied.
The single-lap shear strength of the PCURs to the copper plate sized 100 mm × 25 mm × 1.5 mm was measured using a Zwick/Roell Z010 (Germany) machine following the Polish Standard PN-EN 1465:2009. The adhesive joint, 12.5 mm × 25 mm × 0.2 mm, was done by pressing the PCUR between the ends of two copper sheets at 120–160 °C (following PN-EN-13887:2005) and then leaving them under a pressure of 30 MPa to cool to room temperature. Next, the plates were fixed by the tensile-testing machine clips and underwent tensile testing, the head speed of 2 mm min−1 at 23 °C.
Results and discussion
Table 1 presents the designations, ηred and transmittance data for the PCURs.
The PCURs synthesized from HDI turned out to be opaque materials, while those from MDI exhibited good transparency. Transmittances at 800 and 500 nm determined for the latter polymers were contained in the range of 83.0–87.8% and 69.8–74.1%, respectively (see Table 1 and Fig. 1). The PCURs of both types exhibited high resistance against the common organic solvents at room temperature. All of them did not dissolve in dimethyl sulfoxide, tetrahydrofuran, chloroform, N,N-dimethylacetamide, N,N-dimethylformamide, and N-methyl-2-pyrrolidone. The MDI-derived ones were also insoluble in TChE and partially soluble only in a Ph/TChE mixture whereas the HDI-based ones completely dissolved in a Ph/TChE mixture and partially in TChE. The ηred values received in this mixture point to high molar masses of all the PCURs.

UV–VIS spectra of the PCURs based on MDI
ATR-FTIR
The chemical structures of the polymers were examined by ATR-FTIR spectroscopy. All the spectra exhibit significant absorptions of the urethane and carbonate groups as well as the benzene ring and methylene group as is shown below and in Fig. 2. On the other hand, the absorption characteristics of the isocyanate group at ~ 2270 cm−1 are not displayed, which is a sign of its full conversion. In order to investigate the hard–hard and hard–soft segments interactions by H-bonding, the deconvolution of the carbonyl stretching band was done. The studies were carried out not only for all the PCURs, but also for the regular polyurethanes (RPURs) synthesized from the diol H and MDI (M) or HDI (H) in melt, being the models of hard segments in the PCURs, and PHCD as the representative of a soft segment. Only on the basis of the results, which are given in Tables 2 and 3 and Figs. 3–5, it is possible to ascribe peaks belonging to the non- and H-bonded carbonyls originating from the carbonate and urethane groups.

ATR–FTIR spectra of selected PCURs

Deconvolution of the carbonyl stretching region of the PCURs based on HDI (dashed line: recorded spectra, solid line: resolved peaks)

Deconvolution of the carbonyl stretching region of the PCURs based on MDI (dashed line: recorded spectra, solid line: resolved peaks)

Deconvolution of the carbonyl stretching region of the RPURs and PHCD (dashed line: recorded spectra, solid line: resolved peaks)
ATR-FTIR for the PCURs from MDI (cm−1): 1529–1528 (N–H bending) and 3337–3325 (N–H stretching) of the urethane group; 1736–1699 (C=O stretching of the urethane and carbonate groups); 1249–1247 (C–O stretching of the carbonate group); 2934–2928 and 2859–2856 (asymmetric and symmetric C–H stretching of CH2); 1596 and 1494–1493 (C–C stretching of benzene ring); 815–814 (C–H bending of p-disubstituted benzene ring).
ATR-FTIR for the PCURs from HDI (cm−1): 1533–1532 (N–H bending) and 3330–3328 (N–H stretching) of the urethane group; 1741–1683 (C=O stretching of the urethane and carbonate groups); 1253–1248 (C–O stretching of the carbonate group); 2932–2928 and 2859–2857 (asymmetric and symmetric C–H stretching of CH2); 1494–1493 (C–C stretching of benzene ring); 815–814 (C–H bending of p-disubstituted benzene ring).
From the deconvolution of carbonyl stretching region for the HDI series PCURs (see Table 2 and Fig. 3), it results that it is composed of six bands. Taking into account the deconvolution of semicrystalline RPUR H [30] and PHCD, presented in Fig. 5, the band at the highest wavenumber, i.e., 1743–1740 cm−1, should correspond to the non-H-bonded carbonate carbonyl groups and the next at 1721–1717 cm−1 should be attributed to the H-bonded carbonate and non-H-bonded urethane ones. In turn, the remaining bands are connected with the H-bonded urethane carbonyl groups in the amorphous (at 1712–1705 cm−1) or crystalline (at 1693–1645 cm−1) hard-segment domains [9]. As can be seen, an increase in the soft-segment content was accompanied by an increase in the fraction of the non-H-bonded carbonate carbonyl groups (from 20.7 to 50.4%), indicating a drop in the extent of urethane–carbonate interactions. The consequence is a growth of the degree of microphase separation. Furthermore, it can be noticed that the fraction of the H-bonded urethane carbonyl groups in the amorphous hard-segment domains (in the range of 3.3–11.8% for the samples H-4–H-6 and not detected for the sample H-2) was significantly lower in comparison with that in the crystalline ones (35.3–59.6%), and this points out that all polymers of this series were characterized by a high degree of crystallinity. These inter-urethane interactions were also dependent on the PCUR composition. Thus, with the increase in the content of PHCD soft segment, the share of the latter fraction decreased indicating a decrease in hard-segment ordering. These results are in accordance with the DSC and XRD data presented further in the paper.
The deconvolution of carbonyl stretching region performed for the MDI series PCURs (see Table 3 and Fig. 4) revealed that this region is built from three or four bands: I at 1742–1738 cm−1, II 1724–1721 cm−1, III 1705–1696 cm−1 and IV 1680–1662 cm−1 (except for the sample M-6). Band I can be related to the non-H-bonded carbonate carbonyl groups, band II to the H-bonded carbonate carbonyl groups and the non-H-bonded urethane carbonyl groups, while bands III and IV to the H-bonded urethane ones. Considering the DSC curves shown in Fig. 6, which present very low endothermic peaks and only for polymers M-2, M-4 and M-5, band III should be associated with the presence of completely amorphous hard-segment domains, and band IV of low-ordered one. However, in the other papers concerning MDI-derived polyurethanes, the band at 1705–1695 cm−1 was attributed to the H-bonded urethane carbonyls in the ordered crystalline phase [32, 33]. Similarly to the PCURs of HDI series, with the increase in the soft-segment content the share of band I increased, pointing to the rise of the degree of microphase separation. The biggest difference was observed between the M-2 polymer (9.7%) and M-4 (39.0%). This suggests that these polymers differ most in the degree of microphase separation as confirmed by the DSC analysis.

DSC curves of the MDI-based PCURs
From the above analysis, it follows that the HDI-based PCURs are more prone to hydrogen bonding within the hard segments than the MDI-based counterparts. Linear aliphatic nature of HDI favors the urethane groups in the same plane resulting in the creation of highly ordered hard-segment domains. In turn, the aromatic nature of MDI introduces steric hindrances and restricts the formation of highly ordered structures.
DSC
The numerical data [definite Tg, Tm and heat of melting (ΔHm) values] obtained for all the PCURs after one-month conditioning at room temperature are summarized in Table 4, whereas the DSC curves are presented in Figs. 6 and 7. To facilitate the interpretation of the results Table 4 and Figs. 6 and 7 contain the earlier presented data determined for both the RPURs [30] and PHCD [34].

DSC curves of the HDI-based PCURs as well as of PHCD
The curves received for the PCURs based on MDI (Fig. 6) in the first heating scans exhibit a small endothermic peak (except for the M-6) with the maximum ranging from 106 to 124 °C, connected with the melting of hard-segment domains, which disappears in the second heating scans. This points to a low tendency to form ordered structures by these polymers. Moreover, the curves display a glass transition in the range of 14–46 °C (I heating scan) and 12–37 °C (II heating scan). In the case of polymer M-2, just as it was observed for the RPUR M [30], a sharp relaxation peak superimposed on this transition can be seen in the first heating cycle. In turn, the second heating cycle of this polymer shows the glass transition in the normal shape. Such endothermic peak results from the relaxation of the enthalpy occurring during the storage of the amorphous polymers at a temperature not much lower than that of the glass-transition region. Unfortunately, the Tg values limit the applicability of the MDI-based PCURs as elastomers at room temperature. Similar or somewhat higher Tg values were discovered for the analogous polymers produced from the other aliphatic–aromatic chain extenders [22, 26].
The curves obtained for the PCURs based on HDI (Fig. 7) in the first heating scans show two or three endothermic peaks with the maxima in the range of 47–129 °C. Considering the data received for PHCD and RPUR H, it can be stated that these peaks are associated with the melting of hard-segment domains. However, the lower-temperature peaks at 47–53 °C could be ascribed to the melting of the short-range-ordered structures, whereas those at 112–129 °C correspond to the melting of the more ordered structures, including long-range-ordered ones. The maxima of the higher-temperature endothermic peaks shift toward lower values, and the ∆Hm values decrease with the increasing soft-segment content, which indicates a decrease in the ordering degree. Nevertheless, these relatively sharp and high peaks are a sign that all the HDI-derived PCURs possessed a high degree of ordering within the hard-segment domains. The presence of these peaks also in the second heating scans (with the maxima at 109–133 °C) confirms the high tendency of these PCURs to form ordered structures. In order to detect the presence of the crystalline phase in these polymers, XRD studies are necessary. Apart from the above-mentioned peaks, steps connected with a glass transition appear on the DSC curves. In the first heating cycle, Tgs were contained in the range of – 29 to – 12 °C, while in the second heating one, they generally moved to somewhat higher values (– 26 to – 9 °C) as a result of the mutual miscibility of the soft and hard segments during the first heating. The lower Tgs and closer to that of pure oligomerol used as a soft segment (– 69 °C) indicate that the PCURs synthesized from HDI showed a higher degree of microphase separation in comparison with those obtained from MDI. The higher degree of microphase separation is caused by weaker interactions of the N–H urethane groups of the hard segments with the carbonate C=O groups of soft-segment chains and stronger interactions between the hard segments, resulting in the higher degree of ordering of the hard-segment domains.
XRD
Considering the DSC data, the XRD analysis was made only for the PCURs obtained from HDI. The received patterns are presented in Fig. 8, while the results of the analysis of these patterns by the WAXFIT program are given in Table 5 and on the sample plot in Fig. 9. From these data, it follows that all the examined polymers exhibited semicrystalline structures. Namely, on all XRD patterns crystalline peaks were found. As these peaks were assumed those whose full width at half-maximum (FWHM) was contained in the range of 0.4–1.8°. The degree of crystallinity calculated for the polymers, as the ratio of the sum of crystalline to the total sum of crystalline and amorphous peaks areas, decreased as the soft-segment content grew from 27.6 to 19.9%. This is consistent with the results obtained using the DSC method.

XRD patterns of the PCURs based on HDI

XRD curves (points) of PCUR H-2 resolved into crystalline and amorphous peaks (solid lines)
Taking into account the obtained results, it can be said that the studied PCURs were characterized by more ordered structures than the earlier synthesized PCURs from the diol containing diphenylmethane unit, i.e., 6,6′-[methylenebis(1,4-phenylenemethylenethio)]dihexan-1-ol, which exhibited the crystallinity degree in the range of 0.8–5.1% [35]. In view of the possibility of rotation around the central –C–C– bond, the diphenylethane units allow more compact packing and crystallization between the hard segments in comparison with the diphenylmethane ones [6].
TG
The TG method was used to determine the thermal stability of both the PCURs and RPURs under the inert conditions. Based on the course of TG curves, the temperatures of 1, 5, 10 and 50% mass loss (T1, T5, T10, T50) were designated, whereas the temperature of the maximum rate of mass loss (Tmax) for particular decomposition stages was designed on the basis of differential TG (DTG) curves. Table 6 gives all the determined values, while Figs. 10–13 display the obtained curves. Moreover, for the RPURs and PCURs with 50 mol% PHCD content (M-5 and H-5) an analysis of volatile decomposition products was conducted by the TG coupled with FTIR spectroscopy (TG-FTIR). Figure 14 presents the 3D FTIR plots for PCURs M-5 and H-5, whereas Figs. 15–17 show the FTIR spectra obtained at the maximum rate of the thermal decomposition of all examined polymers. In order to make the interpretation of the results easier, the TG-FTIR studies were also carried out for diol H. The data obtained are presented in Fig. 18.

TG and DTG curves of RPURs

TG and DTG curves of PCURs with 50 mol% soft-segment content

DTG curves of PCURs based on MDI

DTG curves of PCURs based on HDI

3D plots of FTIR spectra of volatile products obtained during the thermal decomposition of PCURs M-5 and H-5

FTIR spectra of volatile products obtained at the maximum rate of the thermal decomposition of RPURs H and M (for the first and second stages)

FTIR spectra of volatile products obtained at the maximum rate of the thermal decomposition of PCUR M-5 (for the first and second stages)

FTIR spectra of volatile products obtained at the maximum rate of the thermal decomposition of PCUR H-5 (for the first, second and third stages)

FTIR spectrum of volatile products obtained at the maximum rate of the thermal decomposition of the diol H (393 °C)
The DTG curves obtained for both RPURs (Fig. 10) show two overlapping peaks indicating a two-stage decomposition process. In the case of polymer based on MDI these peaks, with the maxima at 378 and 419 °C, have almost the same intensity, although they relate to ~ 65 and 32% mass losses, respectively. In turn, on the curves of the HDI-based polymer the peaks of significantly different intensity are observed with the maxima at 343 and 421 °C. They correspond to ~ 26 and 72% mass losses. The peaks with the maxima in the range of 343–378 °C can relate to the decomposition of urethane and sulfide bonds [22], while those with the maxima in the range of 419–421 °C are probably associated with the decomposition of aliphatic chains and aromatic units [34, 36]. These assumptions require the analysis of volatile decomposition products. From the analysis of TG data (Table 6), it follows that RPUR M was characterized by higher T1 (307 vs. 290 °C), T5 (334 vs. 316 °C), T10 (348 vs. 330 °C) but lower T50 (387 vs. 409 °C) than RPUR H.
The FTIR spectrum of RPUR H (Fig. 15) from the first decomposition step (Tmax at 343 °C) shows significant absorption of carbon dioxide (bands at 2359–2310 cm−1 associated with the asymmetric stretching vibrations and at 669 cm−1 with the degenerate bending vibrations) and very small of water (at ~ 4000–3500 and ~ 1800–1300 cm−1, connected with the stretching and bending vibrations, correspondingly). In turn, the spectrum recorded during the second decomposition step (Tmax at 421 °C) does not exhibit the bands typical of carbon dioxide, and shows those characteristic of carbonyl sulfide and aliphatic unsaturated compounds. The creation of the former product is evidenced by the bands at 2072 and 2047 cm−1, characteristic of asymmetric and symmetric C=O stretching vibrations, respectively. The formation of the latter products is manifested by the bands at 930 and 965 cm−1 as well as at 3086 cm−1 (the C–H out-of-plane deformation and stretching vibrations, respectively, of –CH=CH2 group), at 2935 and 2877 cm−1 (the asymmetric and symmetric C–H stretching vibrations of –CH3 and =CH2 groups, correspondingly), at 1642 cm−1 (the C=C stretching vibrations of –CH=CH2 group) and at 1452 and 1390 cm−1 (the C–H bending vibrations of –CH3 and =CH2 groups). Thus it can be assumed that in the first step only urethane linkages decomposed, whereas in the second one, the sulfide linkages and aliphatic fragments of polymer chains were susceptible to decomposition. In addition, the absence of bands coming from the aliphatic unsaturated alcohols (probably 5-hexen-1-ol [37], being the product of diol H decomposition (see Fig. 18) and amines points to the fact that the urethane linkage degradation was not attached to its dissociation as well as did not proceed according to the mechanism, as a result of which volatile primary amines, alkenes and carbon dioxide were generated. This suggests, however, that nonvolatile secondary amines and carbon dioxide were formed according to the following way [38, 39]:
The spectrum of RPUR M (Fig. 15) from the first decomposition step (Tmax at 378 °C) besides the bands typical of carbon dioxide (also of high intensity), displays the bands of low intensity which indicate formation of carbonyl sulfide (at 2072 and 2047 cm−1), unsaturated aliphatic compounds (at 3086, 2937, 2872, 996 and 915 cm−1) and alcohols (at 1045 cm−1, connected with the C–OH stretching vibrations). Here, the band at ~ 3700–3550 cm−1 can originate from both alcohols (the O–H stretching vibrations) and carbon dioxide. The spectrum collected during the second decomposition step exhibits the same bands as that from the first step. Nevertheless, the bands coming from carbon dioxide and carbonyl sulfide have lower intensities whereas those of unsaturated alcohols are of higher intensities. Moreover, the spectrum exhibits additionally the bands confirming the evolution of unsaturated compounds (at 1641, 1452 and 1379 cm−1). In the spectrum there are also bands visible at 2181 and 2110 cm−1 which are a sign of the presence of carbon monoxide. Taking into account these results it can be stated that urethane and sulfide linkages decomposed in both stages, however, the main decomposition of urethane linkages occurred in the first one. Subsequently, the presence of the bands characteristic of aliphatic unsaturated alcohols indicates that the decomposition of urethane bonds was connected with their dissociation to MDI and diol H followed by the carbodiimization of isocyanate and accompanied by the emission of carbon dioxide [38, 39]:
All the PCURs being the derivatives of MDI, just as RPUR M, decomposed in two stages. The DTG curves show two overlapping peaks with the maxima at 359–366 °C and 402–414 °C. The increase in the PHCD content caused an increase in the peak intensity at a lower temperature, and a decrease at a higher temperature (as is illustrated in Fig. 12). Thus, mass loss from ~ 73 (for M-2) to ~ 93% (for M-6) was observed in the first decomposition stage, whereas in the second decomposition stage the range was from ~ 16 (for M-2) to ~ 5% (for M-6). The comparison of the DTG data obtained for the PCURs, RPUR M and PHCD reveals that the first step of the decomposition of hard segments was simultaneous with that of soft segments. In turn, the PCURs synthesized from HDI decomposed in three stages. The DTG curves exhibit three peaks, also partially overlapped, with the maxima at 330–339, 363–383 and 403–406 °C (see Fig. 13). The peak at 363–383 °C, not visible on the curve obtained for RPUR H, should therefore be ascribed to the decomposition of PHCD soft segments. Due to the fact that the peak at 330–339 °C has higher intensity than that at 403–406 °C, contrary to the situation of RPUR H decomposition, it can be assumed that the decomposition of the hard segments built into the structure of PCURs took place following a different mechanism than that of the isolated hard segment. The differences in peak intensities increased with the growth of the soft-segment content. The mass loss of the first step was in the range of ~ 41–64% whereas of the third one: ~ 8–26%.
As it is evident from the data contained in Table 6, the PCURs based on MDI showed higher T1, T5, T10, T50 than those based on HDI, as already found for RPURs (with the exception of T50). In both series, these temperature indicators decreased as the PHCD content increased, due to the worse thermal stability of the polycarbonate soft segments than that of the hard segments. Nevertheless, the thermal stability of the newly obtained PCURs is similar (in the case of HDI series) [27] or better (in the case of MDI series) [26, 29] than that of PCURs prepared from the other sulfur-containing aliphatic–aromatic chain extenders.
The FTIR spectrum from the first stage of PCUR M-5 decomposition (Fig. 16, Tmax at 360 °C) is very similar to the spectrum received in the first step of RPUR M decomposition, i.e., there are absorption bands typical of carbon dioxide, carbonyl sulfide and aliphatic unsaturated alcohols. Additionally, only the bands at 1745 and 2185 cm−1 appear, indicating the formation of carbonyl compounds (C=O stretching vibrations) and carbon monoxide, respectively. Thus, it can be supposed that carbon monoxide and carbonyl compounds are the products of the PHCD soft-segment decomposition. Moreover, a greater intensity of the bands characteristic of carbon dioxide indicates that carbon dioxide originates from the decomposition of soft segments, which is confirmed by the spectrum obtained for the PHCD [28]. In the second decomposition stage of this polymer (Fig. 16, Tmax at 414 °C) bands characteristic of the above-mentioned compounds can be also seen but they are of higher intensity. There is also a band suggesting the formation of aldehydes (at 2720 cm−1) which was not seen in the decomposition products of RPUR M. Summing up, aldehydes originate from the decomposition of only polycarbonate chains, whereas carbon dioxide, carbon monoxide, and aliphatic unsaturated alcohols can come from the decomposition of both polycarbonate chains and hard segments.
The FTIR spectra from the three stages of PCUR H-5 decomposition (Fig. 17) show the bands characteristic of carbon dioxide, carbon monoxide, aromatic compounds (at 1508–1501 cm−1, the C–C stretching vibrations), carbonyl compounds, including aldehydes (at ~ 1800–1700 cm−1, the C=O stretching vibrations, at 2822 and 2720 cm−1, C–H stretching vibrations), ethers (at 1139–1134 cm−1, the C–O stretching vibrations), alcohols (at 1040 cm−1, the C–OH stretching vibrations), aliphatic products (at 2937 and 2864 cm−1, the asymmetric and symmetric C–H stretching vibrations of –CH3 and =CH2 groups, at 1456 and 1379 cm−1, the C–H bending vibrations of –CH3 and =CH2 groups). In the first two stages, the peaks typical of carbon dioxide have almost the same intensity and significantly higher than in the third one. Taking into account the spectra obtained for the RPUR H and PHCD it can be supposed that the first step of PCUR H-5 decomposition included mainly the decomposition of urethane bonds and the second was related mainly to the decomposition of polycarbonate chains. The analysis of the results does not give a unequivocal reply to the question whether hard segments in this polymer decomposed in a different way from that of the RPUR H. The decomposition stages overlap and the decomposition of polycarbonate soft segments proceeds partly together with the decomposition of hard segments. Moreover, unsaturated aliphatic alcohols are the products of both the diol H and PHCD decomposition. The spectra do not show bands pointing to the presence of volatile amines.
Mechanical properties
The Shore A/D hardness and tensile tests were performed for all the PCURs, except for H-2, which was a brittle material. The numerical data are shown in Table 7.
Based on these results, it is possible to conclude that the kind of diisocyanate used had important influence on the modulus of elasticity, tensile strength, and elongation at break of the PCURs. The PCURs derived from MDI showed a significantly higher tensile strength (41.2–50.8 vs. 23.5–33.4 MPa), but much lower elongation at break (130–360 vs. 340–680%) than those derived from HDI. The latter polymers showing semicrystalline structures exhibited higher values of the modulus of elasticity. Moreover, it can be noticed that the increased PHCD soft-segment content resulted in decreased tensile strength (only in the HDI series), whereas elongation at break increased. In the MDI series, the polymer containing 50 mol% PHCD turned out to possess the best tensile strength. The stress–strain curve obtained for this polymer at 23 °C had the appearance typical of elastomers as it is presented in Fig. 19. PCUR M-6 also displayed a typical elastomeric stress–strain curve, whereas the remaining polymers of this series, with Tgs equaling to or higher than room temperature, showed yield stress, characteristic of plastics (see Fig. 19).

Stress–strain curves of the PCURs based on MDI
The type of diisocyanate also had an effect on Shore hardness. Lower hardness, in both A and D scales, was shown by the polymers of MDI series possessing a small degree of ordering. In this series, the differences in hardness in the A scale were noticeably larger (where for measurements the indenter with a truncated cone was used and lower load was applied), which may be due to the fact that along with the increase in the PHCD content there was a transition from plastomers to elastomers. However, all polymers of the HDI series were typical elastomers with a similar degree of crystallinity. In turn, the differences in hardness in the D scale (where a sharp-ended indenter and higher load were used) were almost the same as in the HDI series.
These newly synthesized polymers revealed much higher tensile strength than the analogous materials obtained from diol H, PTMO with a similar \(\overline{M}_{\text n}\) and MDI (21.2–32.5 MPa [40]) or HDI (11.7–17.0 MPa [41]). This can be a result of higher polarity of the carbonate groups than of the ether ones which contributes to stronger hydrogen bonding between the hard segments and the polycarbonate soft segments in comparison with those between the hard segments and the polyether ones [6, 42]. The studied PCURs also exhibited a higher tensile strength compared to the corresponding PCURs based on diol with the diphenylmethane unit, i.e., 6,6′-[methylenebis(1,4-phenylenemethylenethio)]dihexan-1-ol [22, 35]. In the case of polymers derived from HDI, this can be caused by better ordering of hard-segment domains.
Adhesive properties
The influence of sulfur-containing chain extender on the lap shear strength to copper (adhesion) of the obtained PCURs was investigated for the samples with 50 mol% PHCD content, i.e., H-5 and M-5 and their conventional counterparts synthesized under the same conditions from BD as a chain extender. The obtained results are presented in Fig. 20. They reveal that polymers H-5 and M-5 containing more polar groups showed about twice higher adhesive strength than those from BD (10.0 ± 0.32 and 16.0 ± 0.37 MPa vs. 4.6 ± 0.25 and 7.0 ± 0.30 MPa). In addition, it is evident that the polymers derived from MDI exhibited a higher value of this parameter.

Lap shear strength of the PCURs with 50 mol% soft-segment content and their analogs obtained from BD
Conclusions
New thermoplastic MDI- and HDI-based PCURs were synthesized via a simple one-step melt polyaddition from diphenylethane derivative diol as a nonconventional chain extender. Their physicochemical, thermal, mechanical, and adhesive properties were determined. The results show that the diisocyanate structure has a significant influence on the properties of the obtained materials. The polymers being the derivatives of a symmetrical and linear aliphatic diisocyanate possessed semi crystalline structures, connected with the hard-segment crystallization, and thus they were opaque materials. The degrees of crystallinity calculated for these polymers ranged from 19.9 to 27.6%. On the contrary, the PCURs obtained from bulky aromatic diisocyanate with an angled structure were amorphous and transparent (transmittance at 800 nm: 83–88%). All the HDI series PCURs and two of the MDI one turned out to be TPUs. The DSC results show that the former polymers revealed lower Tgs (from – 29 to – 12 °C vs. from 14 to 46 °C) and better microphase separation. On the other hand, according to the mechanical properties analysis, the latter polymers possessed very good tensile strengths (42.2–50.8 vs. 23.5–33.4 MPa). They also showed lower moduli of elasticity, hardnesses, and smaller elongations at break. As is clear from the TG study of the PCURs, they all were characterized by a relatively good thermal stability, though dependent on both the kind of diisocyanate and the soft-segment content. The MDI series polymers exhibited higher T1, T5, T10, and T50 than their analogs of the HDI series (294–305 vs. 262–278 °C, 317–325 vs. 293–307 °C, 330–337 vs. 306–320 °C, 362–375 vs. 346–365 °C, respectively). These temperature indicators decreased as the PHCD content increased. From this study it also follows that the PCURs decomposed in two (from MDI) or three (from HDI) partially overlapped stages. Their decomposition was associated with the evolution of the following volatile products: carbon dioxide, carbon monoxide, carbonyl sulfide, aliphatic aldehydes and unsaturated alcohols as well as in the case of PCUR H-5 also aromatic compounds and ethers. Considering the adhesive properties to metals, it can be stated that the use of diol H was beneficial for the values of the lap shear strength to copper of the PCURs. These sulfur-containing PCURs exhibited about twice higher adhesive strength than those synthesized from BD.
References
Sonnenschein MF. Polyurethanes: science, technology, markets, and trends. New Jersey: Wiley; 2014.
The polyurethanes book. Randall D, Lee S, editors. Everberg, Belgium: Huntsman Polyurethanes; New York: distributed by John Wiley & Sons; 2002.
Qi HJ, Boyce MC. Stress–strain behavior of thermoplastic polyurethanes. Mech Mat. 2005;37:817–39.
Špírková M, Kredatusová J, Hodan J. The impact of conditions mimicking physiological environment on the thermal stability of aliphatic polycarbonate-based polyurethane elastomers. J Therm Anal Calorim. 2017;128:1699–709.
Kull KL, Bass RW, Craft G, Julien T, Marangon E, Marrouat C, Harmon JP. Synthesis and characterization of an ultra-soft poly(carbonate urethane). Eur Polym J. 2015;71:510–22.
Prisacariu C. Polyurethane elastomers: from morphology to mechanical aspects. Wien: Springer; 2011.
Wirpsza Z. Polyurethanes: chemistry, technology and applications. New York: Ellis Horwood; 1993.
Hepburn C. Polyurethane elastomers. London: Elsevier Science Publishers Ltd; 1992.
Špírková M, Poreba R, Pavličević J, Kobera L, Baldrian J, Pekarek M. Aliphatic polycarbonate-based polyurethane elastomers and nanocomposites. I. The influence of hard-segment content and macrodiol-constitution on bottom-up self-assembly. J Appl Polym Sci. 2012;126:1016–30.
Pavličević J, Špírková M, Sinadinović-Fišer S, Budinski-Simendić J, Govedarica O, Janković M. The influence of organoclays on the morphology, phase separation and thermal properties of polycarbonate-based polyurethane hybrid materials. Maced J Chem Chem Eng. 2013;32:151–61.
Christenson EM, Anderson JM, Hiltner A. Antioxidant inhibition of poly(carbonate urethane) in vivo biodegradation. J Biomed Mater Res. 2006;76A:480–90.
Rokicki G, Kowalczyk T. Synthesis of oligocarbonate diols and their characterization by MALDI-TOF spectrometry. Polymer. 2000;41:9013–31.
Govorčin Bajsić E, Rek V, Agić A. Thermal degradation of polyurethane elastomers: determination of kinetic parameters. J Elastom Plast. 2003;35:311–23.
Gomez CM, Gutierrez D, Asensio M, Costa V, Nohales A. Transparent thermoplastic polyurethanes based on aliphatic diisocyanates and polycarbonate diol. J Elastom Plast. 2016;49:77–95.
Datta J, Kasprzyk P. Thermoplastic polyurethanes derived from petrochemical or renewable resources: a comprehensive review. Polym Eng Sci. 2018;58:E14–35.
Datta J, Kasprzyk P, Błażek K, Włoch M. Synthesis, structure and properties of poly(ester-urethane)s obtained using bio-based and petrochemical 1,3-propanediol and 1,4-butanediol. J Therm Anal Calorim. 2017;130:261–76.
Liaw DJ. The relative physical and thermal properties of polyurethane elastomers: effect of chain extenders of bisphenols, diisocyanate, and polyol structures. J Appl Polym Sci. 1997;66:1251–65.
Mehdipour-Ataei S, Mahmoodi A. New polyurethane elastomers with enhanced thermal stability. Polym Plast Technol. 2014;53:1553–60.
Liu X, Wang T, Li J, Cheng J, Zhang J. Synthesis and properties of segmented polyurethanes with hydroquinone ether derivatives as chain extender. J Polym Res. 2015;22:149.
Oprea S, Joga A, Zorlescu B, Oprea V. Effect of the hard segment structure on properties of resorcinol derivatives-based polyurethane elastomers. High Perform Polym. 2014;26:859–66.
Padmavathy T, Srinivasan KSV. Studies on thermotropic main chain liquid crystalline segmented polyurethanes 1. Synthesis and properties of polyurethanes from high aspect ratio mesogenic diol as chain extender. J Macromol Sci Polym Rev. 2003;C43:45–85.
Kultys A, Rogulska M, Pikus S. New thermoplastic segmented polyurethanes with hard segments derived from 4,4′-diphenylmethane diisocyanate and methylenebis(1,4-phenylenemethylenethio)dialcanols. J Appl Polym Sci. 2012;123:331–46.
Puszka A, Kultys A. The influence of soft segments on some properties of new transparent segmented polyurethanes. Polym Adv Technol. 2017;28:1937–44.
Puszka A, Kultys A. New thermoplastic polyurethane elastomers based on aliphatic diisocyanate: synthesis and characterization. J Therm Anal Calorim. 2017;128:407–16.
Rogulska M, Kultys A, Lubczak J. New thermoplastic polyurethane elastomers based on aliphatic-aromatic chain extenders with different content of sulfur atoms. J Therm Anal Calorim. 2015;121:397–410.
Rogulska M, Kultys A, Puszka A. New thermoplastic poly(carbonate-urethane)s based on chain extenders with sulfur atoms. Chem Pap. 2017;71:1195–204.
Rogulska M, Kultys A. Aliphatic polycarbonate-based thermoplastic polyurethane elastomers containing diphenyl sulfide units. J Therm Anal Calorim. 2016;126:225–43.
Rogulska M. Transparent sulfur-containing thermoplastic polyurethanes with polyether and polycarbonate soft segments. Polym Bull. 2018;75:1211–35.
Rogulska M, Kultys A, Pikus S. The effect of chain extender structure on the properties of new thermoplastic poly(carbonate-urethane)s derived from MDI. J Therm Anal Calorim. 2017;127:2325–39.
Rogulska M, Podkościelny W, Kultys A, Pikus S, Poździk E. Studies on thermoplastic polyurethanes based on new diphenylethane-derivative diols. I. Synthesis and characterization of nonsegmented polyurethanes from HDI and MDI. Eur Polym J. 2006;42:1786–97.
Rabiej M, Rabiej S. Analysis of X-ray diffraction pattern of polymers by means of WAXSFIT computer program (in Polish). Bielsko-Biała: ATM; 2006.
Ugarte L, Fernández-d’Arlas B, Valea A, González ML, Corcuera MA, Eceiza A. Morphology—properties relationship in high-renewable content polyurethanes. Polym Eng Sci. 2014;54:2282–91.
Eceiza A, Martin MD, de la Caba K, Kortaberria G, Gabilondo N, Corcuera MA, Mondragon I. Thermoplastic polyurethane elastomers based on polycarbonate diols with different soft segment molecular weight and chemical structure: mechanical and thermal properties. Polym Eng Sci. 2008;48:297–306.
Rogulska M, Kultys A, Olszewska E. New thermoplastic poly(thiourethane-urethane) elastomers based on hexane-1,6-diyl diisocyanate (HDI). J Therm Anal Calorim. 2013;114:903–16.
Kultys A, Rogulska M, Pikus S, Skrzypiec K. The synthesis and characterization of new thermoplastic poly(carbonate-urethane) elastomers derived from HDI and aliphatic-aromatic chain extenders. Eur Polym J. 2009;45:2629–43.
Xu Y, Petrovic Z, Das S, Wilkes GL. Morphology and properties of thermoplastic polyurethanes with dangling chains in ricinoleate-based soft segments. Polymer. 2008;49:4248–58.
The website http://webbook.nist.gov/chemistry/.
Chattopadhyay DK, Webster DC. Thermal stability and flame retardancy of polyurethanes. Prog Polym Sci. 2009;34:1068–133.
Simon J, Barla F, Kelemen-Haller A, Farkas F, Kraxner M. Thermal stability of polyurethanes. Chromatographia. 1988;25:99–106.
Rogulska M. The synthesis, structure and properties of new nonsegmented and segmented polyurethanes with diphenylethane unit. Doctoral dissertation (in Polish), Maria Curie-Skłodowska University, Lublin, Poland, 2004.
Rogulska M, Kultys A, Pikus S. Studies on thermoplastic polyurethanes based on new diphenylethane-derivative diols. III. The effect of molecular weight and structure of soft segment on some properties of segmented polyurethanes. J Appl Polym Sci. 2008;110:1677–89.
Niemczyk A, Piegat A, Sonseca Olalla Á, El Fray M. New approach to evaluate microphase separation in segmented polyurethanes containing carbonate macrodiol. Eur Polym J. 2017;93:182–91.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rogulska, M., Maciejewska, M. & Olszewska, E. New thermoplastic poly(carbonate-urethane)s based on diphenylethane derivative chain extender. J Therm Anal Calorim 139, 1049–1068 (2020). https://doi.org/10.1007/s10973-019-08433-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-019-08433-z