
Abstract
A series of siloxane–silsesquioxane resins with Q8 structures, as network nodes containing reactive Si–H groups in the siloxane backbone, connecting silsesquioxane moieties, were prepared in a hydrolytic condensation process and successfully functionalized by hydrosilylation of allyl alcohol. The proposed material being an alternative to the well-defined silsesquioxanes has been characterized and used for preparation of a series of polyurethane (PU)-based composites by simple reaction of 3-hydroxypropyl groups of siloxane–silsesquioxane resin with 1,6-diisocyanatohexane and subsequently with 1,6-hexanediol to get polyurethane composite. A series of composites based on PU and octakis(3-hydroxypropyldimethylsiloxy)octasilsesquioxane were prepared in the same manner to investigate the influence of the filler amount (1, 3 or 5%) and its structure on thermal properties of the obtained materials.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The incorporation of inorganic or organometallic components into polymers has been recently explored to develop new materials of enhanced or tailored properties. Silsesquioxanes are widely used as components for the preparation of such materials [1,2,3,4,5,6,7,8,9,10]. Commonly reactive, functionalized polyhedral oligomeric silsesquioxanes of well-defined, 3D structure, bearing one or more functional groups were applied for this purpose [11,12,13,14,15,16]. The cage-like silsesquioxanes are known as nanoscale, organic–inorganic hybrids of approximately 1–3 nm diameter. Generally, the procedures for the synthesis of this group of compounds are based on the hydrolysis and condensation of trialkoxy- or trichlorosilanes, substitution reactions with retention of the siloxane cage, corner-capping reactions, and the functionalization of the obtained compounds with many catalytic or stoichiometric methods [13, 17,18,19,20]. Despite all the benefits arising from their use, their price often makes a barrier, which prevents their use on a large scale. An alternative and solution to this problem could be the use of cheaper, silsesquioxane resins which would retain the capability of functionalization and would have well-defined, 3D moieties in their structure. However, most of the cross-linked structures based on silsesquioxane cores do not have functional groups capable of producing chemical bonds to polymer, in contrast to the cage-like silsesquioxane derivatives [21,22,23,24,25,26,27,28,29,30,31,32]. The covalent incorporation of reactive silsesquioxane monomers into polymers is the greatest advantage of the proposed fillers. The formation of covalent bonds between the polymer matrix and the filler permits obtaining inseparable hybrid material based on organic, functional polymers (e.g., epoxides, polyamides, methacrylates, or polyurethanes) [33,34,35,36].
Polyurethanes (PUs) make a class of materials well known for several decades. They can be obtained by polyaddition reaction in solution or mass polyaddition process of diols (HO–R–OH) and diisocyanates (OCN–R–NCO). Polyurethanes may exist as liquid, soft solids, rubbery materials as well as rigid thermoplastic and thermoset materials. PUs have found extensive use in numerous commercial applications such as coatings, foams, adhesives, sealants, synthetic leathers, membranes, elastomers, as well as in many biomedical applications. They are one of the most useful commercial classes of polymers that have been widely exploited in the industry and in everyday life. More than 70% of the literature that deals with PUs are devoted to their thermal stability or flame retardancy [37,38,39,40,41]. In order to increase above-mentioned parameters of polyurethanes, different strategies can be applied. Thermal stability and flame retardancy of PU composites can be improved by incorporation of specific comonomers into the polymer backbone or by blending with fillers of different nature (silsesquioxanes, borax, graphite, carbon fillers, silicates, metals, graphene, phosphates, and many others) [42,43,44,45,46,47,48,49].
In this contribution, we present a new type of functional filler susceptible to modification and capable to form a covalent bonding with the polymer matrix as well as its exemplary application as a component for polyurethane-based hybrid material. To demonstrate the influence of the filler amount (1, 3 or 5%) and its structure on thermal properties of the obtained materials, the results are compared with those for the materials prepared with the use of well-defined silsesquioxane derivative bearing functional groups of the same type.
Experimental
Materials
Tetraethyl orthosilicate (98%), tetramethylammonium hydroxide (solution 25% in methanol) acquired from Sigma-Aldrich, Poland, methanol (99.8%) purchased from POCH, Poland, and redistilled water were used for octaanion solution according to the procedure described elsewhere [50]. Obtained octaanion solution, chlorodimethylsilane (98%), dichloromethylsilane (97%) from Sigma-Aldrich, Poland, and hexane (99%) methylene chloride (99.5%), and methanol (99.8%) purchased from POCH, Poland, were used for the synthesis of octakis(hydridodimethylsiloxy)octasilsesquioxane and a series of siloxane–silsesquioxane resins (SiHQ-2, SiHQ-4, and SiHQ-8). Allyl alcohol (99%), Karstedt complex (2% in xylenes) purchased from Sigma-Aldrich, Poland, and toluene (99.5%) from POCH, Poland, were used for octakis(hydridodimethylsiloxy)octasilsesquioxane and siloxane–silsesquioxane resins functionalization. Hexamethylene diisocyanate (HDI, 98%) and 1,6-hexanediol (HDO, 97%) purchased from Sigma-Aldrich, Poland, were used for polyurethane composites preparation containing synthesized octakis(3-hydroxypropyldimethylsiloxy)octasilsesquioxane and functionalized siloxane–silsesquioxane resins (R2, R4, and R8). All chemicals were used without any further purification. Water used in the experiments was redistilled immediately prior to use.
Preparation of organosilicon fillers
Synthesis of octakis[(3-hydroxypropyl)dimethylsiloxy]octasilsesquioxane (SF)
Synthesized according to the procedure described by Filho et al. [50] octakis(hydridodimethylsiloxy)octasilsesquioxane (20 g, 19.6 mmol) together with 5% excess of allyl alcohol (11.2 mL, 165 mmol) and 200 mL of toluene as solvent was placed in a three-necked round-bottom flask equipped with a thermometer, a condenser, and a magnetic bar. Then Karstedt catalyst (100 mg, 1.02 × 10−6 mol Pt) was added at room temperature, and the solution was heated to 110 °C and kept at this temperature for 8 h. After the reaction mixture cooled down, the solvent and the excess of olefin were evaporated under vacuum, and the residue was filtered off to give a product as a wax (28.5 g, 98% of theoretical yield). The results of spectroscopic analysis of the product confirmed its structure.
1H NMR (CDCl3, 298 K, 300 MHz) δ [ppm] = 0.10 (OSiCH3); 0.58 (SiCH2); 1.60 (CH2); 3.55 (CH2OH); 3.64 (OH). 13C NMR (CDCl3, 298 K, 75.5 MHz) δ [ppm] = − 0.54 (SiCH3); 10.76 (SiCH2); 24.45 (CH2); 51.1 (CH2OH). 29Si NMR (CDCl3, 298 K, 59.6 MHz) δ [ppm] = 13.27 (OSi(CH3)2); − 108.85 (SiOSi).
Synthesis of siloxane–silsesquioxane resins (SiHQ-2, SiHQ-4, SiHQ-8)
Three types of silicone resins have been synthesized with differing in amount of SiH groups (and the length of polysiloxane chains connecting network nodes). In all cases, the reactions were carried out according to an identical procedure, changing only the amount of substrates [51].
To the solution of hexane (as solvent) and dichloromethylsilane placed in three-necked flask equipped with magnetic stirrer, reflux condenser, thermometer, and dropping funnel, and cooled down to 3 °C, a water–methanolic solution of octaanion (received in accordance with the procedure described by Filho et al. [50]) was introduced dropwise through the addition funnel. Resulting suspension was vigorously stirred for 2 h. After separation of the layers, the upper hexane layer was collected, and the solvent was evaporated under reduced pressure. Obtained a crude product was washed several times with a mixture of methanol and methylene chloride to remove possible by-products and unreacted substrates. Purified product as a white powder was dried under vacuum, and substrates quantities and obtained products yields are given in Table 1.
Functionalization of siloxane–silsesquioxane resins (R2, R4, R8)
The obtained siloxane–silsesquioxane resins with different amount of SiH bonds were functionalized with allyl alcohol in the hydrosilylation process carried in the presence of Karstedt complex as a catalyst.
Siloxane–silsesquioxane resin (SiHQ-2, SiHQ-4 or SiHQ-8) 5.0 g, together with 50 mL of toluene (as a solvent to improve mixing of the suspension) and 50 mL of allyl alcohol, was placed in a three-necked flask equipped with magnetic stirrer, heating bowl, condenser, and thermometer. The system was heated to 110 °C, and the Karstedt complex solution in xylene 150 µL was added. The reaction was carried out for 24 h. After reaction mixture cooled down, solvent and excess of olefin were evaporated under vacuum. Obtained functionalized resins R2, R4, and R8 (6.6, 7.2, and 7.5 g, respectively) as white powders were used for the preparation of PU composites.
Preparation of PU composites
Polyurethane composites containing silsesquioxane derivatives of different structures were synthesized according to the known procedure given by Janowski et al. with minor modifications [52]. Hexamethylene diisocyanate (HDI) 4.0 g was charged into a 50-mL three-necked round-bottomed flask equipped with a magnetic stirrer and nitrogen inlet and heated to 80 °C. Next organosilicon fillers in the desired amount was added in one portion to react 1, 3, or 5% of isocyanate groups present in HDI (substrates quantities are given in Table 2). The reaction was carried out under a nitrogen atmosphere at 80 °C for 2 h to form a polyurethane prepolymer. The NCO group content was determined titrimetrically, and the prepolymer was mixed with a suitable amount of 1,4-hexanediol (HDO). The resulting mixture was poured out into a glass vial and cured at 110 °C for a further 2 h and post-cured at 80 °C for the next 16 h.
Analytical techniques
Fourier transformed infrared spectroscopy (FTIR)
FTIR spectra were recorded on a Bruker Tensor 27 Fourier Transform spectrometer equipped with a SPECAC Golden Gate diamond ATR unit. For all spectra, 16 scans at a resolution of 2 cm−1 were collected. Samples were measured as obtained, without any preparatory steps.
Solid-state nuclear magnetic resonance spectroscopy (MS-NMR)
The solid-state 29Si single-pulse/magic-angle spinning (SP/MAS) NMR spectra of the siloxane–silsesquioxane resins were obtained on a Bruker Ascend™ 600 MHz spectrometer with tetrakis(trimethylsilyl)silane as the reference.
Nuclear magnetic resonance spectroscopy (NMR)
1H NMR (300 MHz), 13C NMR (75 MHz), and 29Si NMR (59 MHz) spectra were recorded on a Varian XL 300 spectrometer at room temperature using CDCl3 as solvent.
X-ray diffraction analysis (XRD)
The X-ray diffraction patterns were measured by an XCALIBUR S2, Agilent, with a molybdenum lamp. The sample was placed in a soda-lime glass capillary (inner diameter 0.7 mm). The angular range for the measurement was 2θ (6–40°).
Scanning electron microscope, energy-dispersive X-ray spectroscopy (SEM, EDS)
The morphology of the fillers and PU composites was investigated by scanning electron microscopy (SEM) images using an FEI Quanta 250 FEG field emission scanning electron microscope equipped with EDAX EDS detector. Samples were prepared by gluing the polymer powder on the standard SEM carbon adhesive tape and were not covered by any layer. The microscope was operated at low- or high-vacuum mode. At low-vacuum mode (pressure of 50–100 Pa in the chamber) and 10 kV accelerating voltage, the signal was collected by an LFD detector type, while at high-vacuum mode (pressure of about 2.5 × 10−4 Pa in the chamber) accelerating voltage was 5 kV, and the signal was collected by an ETD detector type.
Textural characteristics
Low-temperature nitrogen sorption for unmodified siloxane resins was determined using an ASAP 2010 sorptometer (Micromeritics) at liquid nitrogen temperature (− 195.6 °C) under relative vacuum conditions in the range of 0.01–1. Masses of 0.2–0.3 g were degassed before testing at 300 °C for 3 h. The specific surface area was determined using the BET method. The area of the mesopores, the size distribution of the mesopores, and their volume were determined from the nitrogen adsorption–desorption isotherms, using the BJH method.
Thermogravimetric analysis (TGA)
Thermal stabilities of the samples prepared were measured on a Q50-TGA thermogravimetric analyzer (TA Instruments, Inc.) under an air flow of 60 mL min−1. Samples (10–15 mg) loaded in a platinum pan were heated from RT to 700 °C at a rate of 10 °C min−1.
Differential scanning calorimetry (DSC)
DSC measurements of prepared PU composites samples placed in a 40-µL aluminum pans with a pierced lid were carried out in N2 atmosphere at a flow rate of 25 mL min−1 in a temperature range from 25 to 230 °C at a heating/cooling rate of 10 °C min−1 using a Mettler Toledo DSC-1 differential scanning calorimeter performed for the determination of their melting and crystallization temperatures.
Results and discussion
Prepared polyurethane composites containing various amounts of octakis(3-hydroxypropyldimethylsiloxy)octasilsesquioxane or functionalized siloxane–silsesquioxane resins of different structures (Fig. 1) as fillers were subjected to the spectroscopic, morphological, and thermal characterization.

Structures of organosilicon compounds used
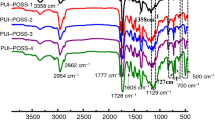
FTIR analysis of the obtained resins clearly showed that they differed depending on the reagents stoichiometry applied. The most significant differences can be observed in the intensities of bands at 2170 and 950–650 cm−1, characteristic of Si–H bond (Fig. 2). A significant increase in the intensity of these bands correlates with the rising amount of –OSi(CH3)H– units present in siloxane chains connecting Q8 silsesquioxane nodes. A similar tendency can be observed for 2970 and 1262 cm−1 bands characteristic for C–H and Si–C bonds, respectively.

FTIR spectra of SiHQ-2, SiHQ-4, and SiHQ-8 siloxane–silsesquioxane resins
FTIR analysis of the functionalized resins confirmed total conversion of the reactive Si–H by the disappearance of characteristic bands at 2170 and 950–650 cm−1. Also the appearance of new bands at 2934 and 2877 cm−1, characteristic for C–H bonds present in the introduced alkyl chain and a broad band at 3331 cm−1 characteristic for the terminal OH, confirm the complete resins functionalization (Fig. 3). Moreover, similarly as in the spectra of the starting resins, also in the spectrum of functionalized resins, a good correlation between the intensity of bands characteristic for hydroxypropyl groups (2934, 2877 and 3331 cm−1) and the reagents stoichiometry applied for the synthesis of the starting materials can be clearly observed. The intensity of the mentioned bands increases with the growing number of –OSi(CH3)H– units present in siloxane chains connecting Q8 silsesquioxane nodes.

FTIR spectra of R2, R4 and R8 siloxane–silsesquioxane resins modified with allyl alcohol
The solid-state 29Si single-pulse/magic-angle spinning (SP/MAS) NMR spectra of obtained for raw siloxane–silsesquioxane and allyl alcohol functionalized siloxane–silsesquioxane resins (Fig. 4) also confirmed their structures and successful functionalization. The NMR spectra of all resins appear very similar in terms of a number of signals and their chemical shifts, but they differ remarkably in their relative intensities. For all resins, three signals at − 34.9, − 65.2, and − 110.2 ppm can be observed. They can be attributed to the presence of D (SiO2), T (SiO3), and Q (SiO4) units in the obtained resins structure, respectively. The presence of T units can be explained by the occurrence of a possible side reaction of one of the Cl atoms from dichloromethylsilane with methanol and subsequent condensation of methoxy group formed with Si–H bond with a formation of T structure. It can be observed that the intensity of D units signal at − 34.9 ppm increases relative to the intensity of Q units (− 110.2 ppm) which remains in good correlation with the reagents stoichiometry applied for the synthesis of investigated samples. For all the functionalized resins, the signals attributed to D units are shifted from − 34.9 to − 21.9 ppm which is related to the addition of hydroxypropyl group to the –OSi(CH3)H– segments (Fig. 4). Simultaneously, the shift of the signal at − 65 (T units) to − 55 ppm is observed explained by the change in the nature of adjacent groups. The Q unit signals at − 110.0 ppm are unchanged, which confirms the retention of the silsesquioxane moieties structures in the products.

Solid-state 29Si NMR spectra of SiHQ-2, SiHQ-4, SiHQ-8 and R2, R4 R8 resins
The reaction stoichiometry applied for the synthesis of siloxane–silsesquioxane resins strongly affected their structure as described above and in consequence influenced their morphological properties.
The crystalline structures of the siloxane–silsesquioxane resins verified by XRD analysis, shown in Fig. 5, were quite different depending on the lengths of siloxane chains incorporated into their structures. For the SiHQ-2 resin, three major characteristic diffraction peaks at c.a. 24°, 28°, and 41° were observed, indicating a relatively high proportion of crystalline phase in its structure. For SiHQ-4 resin, the above-mentioned signals are much less pronounced, while for SiHQ-8 resin, they are not observed at all, which indicates the amorphous nature of this material.

XRD analysis of SiHQ siloxane–silsesquioxane resins
The morphological effect of different lengths of siloxane chains linking the Q8 cores can be also observed on the scanning electron microscopy images of siloxane–silsesquioxane resins, especially for SiHQ-8 one (Fig. 6). For SiHQ-2 and SiHQ-4 resins, no major changes resulting from the number of siloxane units in the chains are observed, and their structure is built mainly with small particles growing on one another. In SiHQ-4 also terrace structures can be observed. SiHQ-8 resin differs significantly in morphology from SiHQ-2 and SiHQ-4 ones. The sample is characterized by a monolithic structure with no crystallite phase present.

SEM micrographs of siloxane–silsesquioxane resins. a SiHQ-2, b SiHQ-4 and c SiHQ-8
Scanning electron microscopy (SEM) was also used to investigate changes in the prepared PU composites surface morphology, related to the type and amount of organosilicon compounds used. For the samples prepared with the use of octakis(3-hydroxypropyldimethylsiloxy)octasilsesquioxane, numerous crystalline domains can be observed on the composite surface. The number as well as the size of the domains increased with growing silsesquioxane content and was the most noticeable for the sample PU-SF-5% containing 5 mass% of the filler. For the composite samples containing R2, R4, or R8 functionalized resins, no similar effect was observed even for the highest (5 mass%) filler loadings (Fig. 7).

SEM micrographs of PU composites containing 5 wt.% of a silsesquioxane, b R2, c R4, and d R8 resins
Samples of polyurethane hybrid materials were also analyzed by the energy-dispersive X-ray spectroscopy (EDS) to identify the degree of fillers dispersion in the polymer matrix. The results obtained for polyurethane composites containing octakis(3-hydroxypropyldimethylsiloxy)octasilsesquioxane reveal the homogenous filler distribution in PU matrix regardless of its content. No formation of agglomerates is observed. For PU-R2, R4, and R8 composites, the formation of agglomerates is observed (Fig. 8). It was also observed that the homogeneity of the filler dispersion increases with increase in siloxane chain length and number of reactive hydroxypropyl groups present in the resin used (from R2 to R8).

Maps of silicon distribution in PU composites containing 5 mass% of a silsesquioxane, b R2, c R4, and d R8 resins
The siloxane chain lengths, degree of crystallinity, and resins morphology influenced also their textural properties such as the specific surface area, average pore diameter, or pore volumes determined by the adsorption–desorption nitrogen isotherms of investigated samples. The obtained results are summarized in Table 3. The shape of measured isotherms reflects the mesoporous nature of the tested materials. Most similar in shape to type IV isotherm (according to classification IUPAC characteristic of mesoporous adsorbents) is the isotherm recorded for SiHQ-8 sample. The isotherms recorded for SiHQ-2 and SiHQ-4 can be defined as deformed type IV, because the adsorption hysteresis pulses are not closed even for the small values of relative pressure p p0−1. The observation of not closing the hysteresis is difficult to explain. For no reason, IUPAC can be referenced in the systems studied (it is the stiffening of the non-rigid porous structure, irreversible adsorption of adsorbent particles in pores with a diameter close to the particle size of the adsorbent molecule, and strong chemical interaction of adsorbate with adsorbent) [53]. Loops of adsorption hysteresis of SiHQ-8 sample represent type H2 according to IUPAC classification (or E, according to de Boer classification [54]). The hysteresis of SiHQ-2 and SiHQ-4 samples can be specified as Type H3 or B. It should be noted that there is a difference between the hysteresis of the last two samples and the shape of the classical H3 loop, in the fact that the hysteresis does not close. Clearly, different textural properties of individual samples are determined by the length of the siloxane chains linking the silsesquioxane Q8 units.
The surface area and the pore volume are clearly decreasing with increase in the number of siloxane units in the chain. The average pore diameter indicates the presence of mesopores (it is 2–50 nm according to the IUPAC classification) [53]. This demonstrates that the characteristic mesoporous character of silsesquioxane units has been retained in all resin samples. The largest surface area and pore volume (293 m2 g−1 and 0.387 cm3 g−1) were observed for SiHQ-2 resin, in which the lengths of the siloxane chains connecting the network nodes were statistically the smallest. The dramatic decrease in the surface area and the pore volume values was observed with increase in siloxane chains length in samples SiHQ-4 and SiHQ-8.
As mentioned above, the obtained and characterized hydroxypropyl functionalized resins were subsequently used as reactive fillers for the preparation of series of PU-based hybrid materials containing different amounts of inorganic phase (1, 3 or 5 mass%). The influence of the type of the filler used and its amount on the thermal properties of the obtained composites was evaluated on the basis of TG analysis and DSC measurements. It was found that the thermal degradation process in air atmosphere occurs generally in two distinct stages with a maximum mass loss at about 350–400 and 450–600 °C. The effects of the fillers content were described in the terms of 1, 5, and 10% mass loss temperatures and ΔT values for each sample series. All data obtained are presented in Table 4.
It can be observed that the influence of the amount of filler on the thermal properties of each polyurethane composite series is slightly different for each filler. The analysis of TG curves for polyurethane composite series obtained with the addition of octakis(3-hydroxypropyldimethylsiloxy)octasilsesquioxane showed that 1 and 5% filler addition drastically reduced the materials’ thermal stability. However, 3 mass% content of silsesquioxane increased its 1, 5 and 10% mass loss temperatures by about 15 °C. The analysis of TG curves obtained for R2, R4, and R8 resins based PU composites showed that the thermal stability was in all samples improved, regardless of the amount of the filler used. However, the difference between the 1, 5, and 10% mass loss temperatures observed for pure PU and the composites containing siloxane–silsesquioxane resins R2, R4, and R8 were the highest for 1% of filler loading and decreased with its increase.
Results of DTG curves analysis presented in Table 5 lead to the conclusion that, in average, the highest temperatures of maximum mass loss rate Tmax were observed for R4 resin composite samples. Simultaneously measured for R4 resin composite samples, maximum mass loss rate dmax and mass loss d at Tmax values were the lowest at all. The highest values of dmax and d at Tmax were observed for composites based on R8 resin.
A comparison of the TG and DTG curves for pure PU and the composites containing 1% of octakis(3-hydroxypropyldimethylsiloxy)octasilsesquioxane, R2, R4 and R8 resins is presented in Fig. 9. Based on the results of TG and DTG curves analysis, it is easy to observe that the composites of the highest thermal stability were those obtained with the use of R4 resin and the lowest thermal stability was observed for R8 resin containing composites. This stems from the difference in the structure of both resins which influences not only their morphological and textural properties but also the polymer–filler interactions and composite properties. Lower thermal stability of R8 resin-based composites in comparison with R4 ones should be attributed to the presence of statistically twice as long siloxane chains linking silsesquioxane Q8 nodes in its structure and lower network density. Increased siloxane chain length favor the thermo-oxidative mechanism of their decomposition and decreases composites thermal stability [55].

TG (a) and DTG (b) curves of modified PU with 1 wt.% fillers content
The thermogravimetric analysis was followed by the DSC measurements. Analysis of the DSC curves of second heating run presented in Fig. 10 lead to an observation that the addition of 1 and 3% of octakis(3-hydroxypropyldimethylsiloxy)octasilsesquioxane does not affect significantly the melting temperatures (Tm) of the PU composites in comparison to those of the neat polyurethane. Only for 5% silsesquioxane loading, a considerable reduction (almost 12 °C) of the melting point was observed. For all composites based on R2, R4, or R8 siloxane–silsesquioxane resins, no trends in changes in melting temperatures were observed regardless of the amount of filler used. The observed variations in the melting points were not greater than ± 3 °C.

DSC curves of the second heating run
Similarly, as it can be observed in Fig. 11 presenting DSC curves of second cooling run, no simple correlation between the amount and type of filler and the crystallization temperatures was observed except for the PU-R8 composites. For all PU-R8 composites, regardless of the filler content, the increase in crystallization temperatures was observed by more than 10 °C.

DSC curves of the second cooling run
This phenomenon, similarly to the results of XRD, SEM, and TG analyses, is straightly related to the structure of the fillers used and the length of siloxane chains linking the silsesquioxane Q8 units present in their structures. The siloxane chains linking silsesquioxane nodes in the R8 resin are statistically twice as long as those in R4 and four times longer from those in R2 resin. The mobility of Si–O–Si bonds of the siloxane backbone as well as loosening of the resin network strongly affects the properties of filler and subsequently composites containing it by enabling polyurethane chains motion, align for nucleation and crystal lattice formation which is manifested in the case in the increase of the crystallization temperatures observed for PU-R8 composite samples [56]. The results of DSC analysis are summarized in Table 6.
Conclusions
The synthesis of a novel hybrid polyurethane composite has been studied. To get the desired composites, a series of siloxane–silsesquioxane resins bearing reactive Si–H groups were synthesized (SiHQ-2, -4, -8) and functionalized with hydroxypropyl groups in the allyl alcohol hydrosilylation process to get a series of reactive fillers (R2, R4, R8) capable of formation of covalent bonding to polyurethane matrix. The prepared reactive fillers and polyurethane composites were characterized with spectroscopic techniques, scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDS), thermogravimetric analysis (TGA), and differential scanning calorimetry (DSC). The results obtained confirmed strong dependence between the structure of resins, their morphology, textural and thermal properties. FTIR and NMR analyses confirmed successful functionalization of starting SiHQ resins with different amounts of hydroxypropyl groups. XRD analysis showed that the crystallinity of synthesized SiHQ resins strongly depends on their chemical structures. The significant influence of the chemical structures of the synthesized materials on specific surface and pore volumes, as well as their morphology, has been also observed based on sorption and microscopic analyses. Mentioned above parameters affected also the thermal properties of prepared PU composites. The most significant was the impact of R4 resin series on the improvement on PU composites thermal stability, while R8 resins caused notable reduction in composites stability, what is related to the different siloxane chains length present in their structure. The structure–property dependency was also clearly observed in case of crystallization temperatures determined by DSC measurements for PU composites.
It has been proven that the proposed materials can be a valuable alternative for well-defined silsesquioxane derivatives and their use permits the synthesis of polyurethane composites of improved thermal stability even at low filler contribution.
References
Barczewski M, Dobrzyńska-Mizera M, Dutkiewicz M, Szołyga M. Novel polypropylene β-nucleating agent with polyhedral oligomeric silsesquioxane core: synthesis and application. Polym Int. 2016;65(9):1080–8.
Dutkiewicz M, Szołyga M, Maciejewski H, Marciniec B. Thiirane functional spherosilicate as epoxy resin modifier. J Therm Anal Calorim. 2014;117(1):259–64.
Lee SH, Yu S, Shahzad F, Hong JP, Kim WN, Park C, Hong SM, Koo CM. Crystallization derivation of amine functionalized T12 polyhedral oligomeric silsesquioxane-conjugated poly(ethylene terephthalate). Compos Sci Technol. 2017;146:42–8.
Groch P, Dziubek K, Czaja K, Dudziec B, Marciniec B. Copolymers of ethylene with monoalkenyl- and monoalkenyl(siloxy)silsesquioxane (POSS) comonomers—synthesis and characterization. Eur Polym J. 2017;90:368–82.
Zhang W, Camino G, Yang R. Polymer/polyhedral oligomeric silsesquioxane (POSS) nanocomposites: an overview of fire retardance. Prog Polym Sci. 2017;67:77–125.
Chen HL, Jiao XN, Zhou JT. The research progress of polyhedral oligomeric silsesquioxane (POSS) applied to electrical energy storage elements. Funct Mater Lett. 2017;10(2):1730001.
Wang Z, Ma H, Chu B, Hsiao BS. Super-hydrophobic polyurethane sponges for oil absorption. Sep Sci Technol (Philadelphia). 2017;52(2):221–7.
Gu P, Yang G, Lee SC, Lee JK. Thermal characterization of epoxy nanocomposites containing polyhedral oligomeric silsesquioxane: glass transition temperature and chemical conversion. Fibers Polym. 2017;18(1):131–9.
Dobrzyńska-Mizera M, Dutkiewicz M, Sterzyński T, Di Lorenzo ML. Isotactic polypropylene modified with sorbitol-based derivative and siloxane–silsesquioxane resin. Eur Polym J. 2016;85:62–71.
Czarnecka-Komorowska D, Sterzynski T, Dutkiewicz M. Polyoxymethylene/polyhedral oligomeric silsesquioxane composites: processing, crystallization, morphology and thermo-mechanical behavior. Int Polym Proc. 2016;31(5):598–606.
Raftopoulos KN, Pielichowski K. Segmental dynamics in hybrid polymer/POSS nanomaterials. Prog Polym Sci. 2016;52:136–87.
Kuo SW, Chang FC. POSS related polymer nanocomposites. Prog Polym Sci. 2011;36:1649.
Cordes DB, Lickiss PD, Rataboul F. Recent developments in the chemistry of cubic polyhedral oligosilsesquioxanes. Chem Rev. 2010;110(4):2081–173.
Tanaka K, Chujo Y. Advanced functional materials based on polyhedral oligomeric silsesquioxane (POSS). J Mater Chem. 2012;22(5):1733–46.
Kuo SW, Chang FC. POSS related polymer nanocomposites. Progr Polym Sci (Oxford). 2011;36(12):1649–96.
Tanaka K, Chujo Y. Chemicals-inspired biomaterials: developing biomaterials inspired by material science based on POSS. Bull Chem Soc Jpn. 2013;86(11):1231–9.
Zhang W, Müller AHE. Architecture, self-assembly and properties of well-defined hybrid polymers based on polyhedral oligomeric silsequioxane (POSS). Prog Polym Sci. 2013;38(8):1121–62.
Blanco I, Abate L, Bottino FA. Synthesis and thermal characterization of new dumbbell-shaped cyclopentyl-substituted POSSs linked by aliphatic and aromatic bridges. J Therm Anal Calorim. 2015;121(3):1039–48.
Zhao M, Feng Y, Li Y, Li G, Wang Y, Han Y, Sun X, Tan X. Preparation and performance of phenyl-vinyl-POSS/addition-type curable silicone rubber hybrid material. J Macromol Sci Part A Pure Appl Chem. 2014;51(8):639–45.
Liu N, Li L, Wang L, Zheng S. Organic–inorganic polybenzoxazine copolymers with double decker silsesquioxanes in the main chains: synthesis and thermally activated ring-opening polymerization behavior. Polymer (UK). 2017;109:254–65.
Zhang C, Babonneau F, Bonhomme C, Laine RM, Soles CL, Hristov HA, Yee AF. Highly porous polyhedral silsesquioxane polymers. Synthesis and characterization. J Am Chem Soc. 1998;120(33):8380–91.
Naga N, Oda E, Toyota A, Furukawa H. Mesh size control of organic–inorganic hybrid gels by means of a hydrosilylation co-gelation of siloxane or silsesquioxane and α, ω-non-conjugated dienes. Macromol Chem Phys. 2007;208(21):2331–8.
Naga N, Nagino H, Furukawa H. Synthesis of organic–inorganic hybrid gels by means of thiol-ene and azide–alkene reactions. J Polym Sci Part A Polym Chem. 2016;54(14):2229–38.
Guo S, Okubo T, Kuroda K, Shimojima A. A photoresponsive azobenzene-bridged cubic silsesquioxane network. J Sol-Gel Sci Technol. 2016;79(2):262–9.
Zhang A, Gao H, Li W, Bai H, Wu S, Zeng Y, Zhou X, Li L. Hybrid microporous polymers from double-decker-shaped silsesquioxane building blocks via Friedel–Crafts reaction. Polymer (UK). 2016;101:388–94.
Liu C, Li Z, Wang Y. Novel fluorescent terphenyl bridged crystalline silsesquioxane through self-directed assembly. J Sol-Gel Sci Technol. 2017;81(2):593–9.
Li H, Zhang J, Xu R, Yu D. Direct synthesis and characterization of crosslinked polysiloxanes via anionic ring-opening copolymerization with octaisobutyl–polyhedral ohgomeric silsesquioxane and octamethylcyclotetrasiloxane. J Appl Polym Sci. 2006;102(4):3848–56.
Gunji T, Shioda T, Tsuchihira K, Seki H, Kajiwara T, Abe Y. Preparation and properties of polyhedral oligomeric silsesquioxane–polysiloxane copolymers. Appl Organomet Chem. 2010;24(8):545–50.
Pawlak T, Kowalewska A, Zgardzińska B, Potrzebowski MJ. Structure, dynamics, and host–guest interactions in POSS functionalized cross-linked nanoporous hybrid organic–inorganic polymers. J Phys Chem C. 2015;119(47):26575–87.
Zhou Y, Huang F, Du L, Liang G. Synthesis and properties of silicon-containing arylacetylene resins with polyhedral oligomeric silsesquioxane. Polym Eng Sci. 2015;55(2):316–21.
Handke M, Kowalewska A. Siloxane and silsesquioxane molecules—precursors for silicate materials. Spectrochim Acta Part A Mol Biomol Spectrosc. 2011;79(4):749–57.
Arsalani N, Akbari A, Amini M, Jabbari E, Gautam S, Chae KH. POSS-based covalent networks: supporting and stabilizing Pd for heck reaction in aqueous media. Catal Lett. 2017;147(4):1086–94.
Pramudya I, Rico CG, Lee C, Chung H. POSS-containing bioinspired adhesives with enhanced mechanical and optical properties for biomedical applications. Biomacromol. 2016;17(12):3853–61.
Barczewski M, Czarnecka-Komorowska D, Andrzejewski J, Sterzyński T, Dutkiewicz M, Dudziec B. Processing properties of thermoplastic polymers modified by polyhedral oligomeric silsesquioxanes (POSS). Polimery/Polymers. 2013;58(10):805–15.
Heneczkowski M, Oleksy M, Oliwa R, Dutkiewicz M, Maciejewski H, Galina H. Application of silsesquioxanes for modification of epoxy resins. Polimery/Polymers. 2013;58(10):759–65.
Prządka D, Jęczalik J, Andrzejewska E, Marciniec B, Dutkiewicz M, Szłapka M. Novel hybrid polyurethane/POSS materials via bulk polymerization. React Funct Polym. 2013;73(1):114–21.
Oprea S. Synthesis and properties of polyurethane elastomers with castor oil as crosslinker. J Am Oil Chem Soc. 2010;87(3):313–20.
Kim H, Miura Y, MacOsko CW. Graphene/polyurethane nanocomposites for improved gas barrier and electrical conductivity. Chem Mater. 2010;22(11):3441–50.
Chattopadhyay DK, Webster DC. Thermal stability and flame retardancy of polyurethanes. Progr Polym Sci (Oxford). 2007;32(3):352–418.
Chattopadhyay DK, Webster DC. Thermal stability and flame retardancy of polyurethanes. Progr Polym Sci (Oxford). 2009;34(10):1068–133.
Delebecq E, Pascault JP, Boutevin B, Ganachaud F. On the versatility of urethane/urea bonds: reversibility, blocked isocyanate, and non-isocyanate polyurethane. Chem Rev. 2013;113(1):80–118.
Kong W, Lei Y, Jiang Y, Lei J. Preparation and thermal performance of polyurethane/PEG as novel form-stable phase change materials for thermal energy storage. J Therm Anal Calorim. 2017;130(2):1011–9.
Datta J, Kasprzyk P, Błażek K, Włoch M. Synthesis, structure and properties of poly(ester-urethane)s obtained using bio-based and petrochemical 1,3-propanediol and 1,4-butanediol. J Therm Anal Calorim. 2017;130(1):261–76.
Michałowski S, Hebda E, Pielichowski K. Thermal stability and flammability of polyurethane foams chemically reinforced with POSS. J Therm Anal Calorim. 2017;130(1):155–63.
Chao C, Gao M, Chen S. Expanded graphite: borax synergism in the flame-retardant flexible polyurethane foams. J Therm Anal Calorim. 2018;131(1):71–9.
Ciecierska E, Jurczyk-Kowalska M, Bazarnik P, et al. The influence of carbon fillers on the thermal properties of polyurethane foam. J Therm Anal Calorim. 2016;123(1):283–91.
Cheng J-J, Zhou F-B. Flame-retardant properties of sodium silicate/polyisocyanate organic–inorganic hybrid material. J Therm Anal Calorim. 2016;125(2):913–8.
Chen X, Ma C, Jiao C. Synergistic effects between iron–graphene and ammonium polyphosphate in flame-retardant thermoplastic polyurethane. J Therm Anal Calorim. 2016;126(2):633–42.
Liu L, Zhao X, Ma C, et al. Smoke suppression properties of carbon black on flame retardant thermoplastic polyurethane based on ammonium polyphosphate. J Therm Anal Calorim. 2016;126(3):1821–30.
Filho NLD, de Aquino HA, Pires G, Caetano L. Relationship between the dielectric and mechanical properties and the ratio of epoxy resin to hardener of the hybrid thermosetting polymers. J Braz Chem Soc. 2006;17:533–41.
Szołyga M, Dutkiewicz M, Marciniec B, Maciejewski H. Synthesis of reactive siloxane–silsesquioxane resins [Synteza reaktywnych żywic siloksanowo-silseskwioksanowych]. Polimery/Polymers. 2013;58(10):766–71.
Janowski B, Pielichowski K. A kinetic analysis of the thermo-oxidative degradation of PU/POSS nanohybrid elastomers. Silicon. 2016;8(1):65–74.
Sing KSW, Everett DH, Haul RAW, Moscou L, Pierotti RA, Rouquérol J, Siemieniewska T. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity. Pure Appl Chem. 1985;57(4):603–19.
de Boer JH. The structure and properties of porous materials. Londyn: Butterworth; 1958.
Camino C, Lomakin SM, Lazzari M. Polydimethylsiloxane thermal degradation part 1. Kinetic aspects. Polymer. 2001;42(6):2395–402.
Shalaby WS, Bair HE. Block copolymers and polyblends. In: Turi E, editor. Thermal characterization of polymeric materials. London: Academic Press; 1981. p. 366–408.
Acknowledgements
The authors gratefully acknowledge the financial support from the National Centre for Research and Development in Poland (Grant No. PBS3/A1/16/2015).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Szołyga, M., Dutkiewicz, M. & Marciniec, B. Polyurethane composites based on silsesquioxane derivatives of different structures. J Therm Anal Calorim 132, 1693–1706 (2018). https://doi.org/10.1007/s10973-018-7096-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-018-7096-z