
Abstract
New substitutional limited solid solution of the formula Yb8−xYxV2O17 and 0 < x < 4.0 was synthesised by the solid-state reaction from the mixtures of the compounds Yb8V2O17 and Y8V2O17. The monophasic samples containing Yb8−xYxV2O17 were characterised by powder XRD, DTA, IR, UV–Vis–DRS and SEM methods. The influence of the degree of Y3+ ion incorporation in the crystal lattice of Yb8V2O17 replacing Yb3+ ions, on the thermal stability, unit cell volume, band gap energies as well as on the position of the IR absorption bands in the spectrum of Yb8−xYxV2O17 was determined. The morphology of Yb8V2O17 and solid solution was analysed.
Similar content being viewed by others

Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
Introduction
Yttrium vanadates(V) doped with rare earth metals have been for a few decades the subject of interest because of their unique and attractive physicochemical properties and have been applied in many branches of industry [1–13]. Yttrium orthovanadates(V) doped with Nd3+, Er3+ or Yb3+ ions make interesting laser materials employed in lasers used in medicine or electronic industry [1–7]. YVO4 doped with Nd3+, Yb3+, Eu3+, Bi3+ or Er3+ ions has been applied as a luminophore or for production of plasma displays, electroluminescent diodes or fluorescence lamps [8–13].
Many scientific reports have been devoted to yttrium or ytterbium orthovanadates(V) [1–18], but much less attention has been paid to other vanadates(V) forming in the binary systems V2O5–Yb2O3 and V2O5–Y2O3, that is to the compounds Yb8V2O17 and Y8V2O17 [19–22]. The first of them Yb8V2O17 has been for the first time obtained by Brusset et al. [19] as a result of heating of a mixture of oxides Yb2O3/V2O5 at the molar ratio 4:1 to 1550 °C. This compound crystallises in the monoclinic system, and the parameters of its elementary cell are: a = 1.0397 nm, b = 0.8242 nm, c = 1.5891 nm, β = 98.47° [19]. The same authors [20] constructed the phase diagram of V2O5–Yb2O3, taking into account the formation of YbVO4 and Yb8V2O17.
According to literature data on the system V2O5–Y2O3 [21, 22], the compound Y8V2O17 has been obtained as a result of heating a mixture of oxides Y2O3/V2O5 at the molar ratio 4:1 at 1549 °C [21] and by the alkoxy method from VO(OC2H5)3 and YCl3 [22]. The authors of [22] have washed the sediment obtained many times with hot water and then after drying, subjected the sediment to calcination at 1500 °C. However, the X-ray structural data for the compound Y8V2O17 obtained by the above-mentioned two methods are significantly different [21, 22]. The authors of [21, 22] agree that Y8V2O17 occurs in two polymorphic varieties and that the low-temperature variety of Y8V2O17 undergoes a monotropic phase transition to the high-temperature variety at 1550 °C. Only Yamaguchi et al. [22] have reported that the low-temperature variety crystallises in tetragonal system, while the high-temperature one in monoclinic system and determined the parameters of their elementary cells.
The state of knowledge on Yb8V2O17 and Y8V2O17, and in particular the lack of information on their thermal stability and incomplete information on their structures, has prompted us to supplement the gap.
Taking into account the fact that the title pseudo-binary system Yb8V2O17–Y8V2O17 has not been studied yet, the general aim of our study was to which phases–if any–form as a result of reaction between the components of this system, determination of their thermal stability and basic physicochemical properties. These new phases can potentially find similar or wider application than the already known ones formed with yttrium orthovanadate(V) or orthovanadates of rare earth elements.
Experimental
The following reagents were used in our experiments: Yb2O3, a.p. (Alfa Aesar, Germany), Y2O3, a.p. (Alfa Aesar, Germany), V2O5, a.p. (POCh, Poland). The components of investigated system, i.e. Yb8V2O17 and Y8V2O17, were obtained by heating in air the mixtures of V2O5/Yb2O3 and V2O5/Y2O3 at the molar ratio of 1:4 in the temperature range 600–1300 °C [19, 21].
Nine samples were prepared for the experiments. The samples were contained from 5.00 to 62.50 mol% of Y8V2O17 in mixtures with Yb8V2O17. The solid solution, Yb8−xYxV2O17, where 0 < x < 4, was prepared by the high-temperature solid-state reaction from the compounds: Yb8V2O17 and Y8V2O17 by the method described inter alia in [23–26]. Reagents weighed in suitable proportions were homogenised and calcined in air atmosphere in the temperature range 1400–1600 °C. The samples were heated in a tube furnace PRC 50/170/M (Czylok, Poland) equipped with a stationary optical pyrometer MARATHON MM (Raytek, Germany). The optical pyrometer was calibrated to the melting points of Ca2P2O7 (1353 °C), CaF2 (1420 °C) and Na3PO4 (1583 °C). After each heating stage, the samples were gradually cooled in the furnace to room temperature and analysed by the powder XRD or DTA methods too. The powder diffraction patterns of the samples obtained were recorded with the aid of the diffractometer EMPYREAN II (PANalytical, Netherlands) using CuKα with graphite monochromator. The phases were identified on the basis of XRD characteristics presented in the PDF cards [27]. The powder diffraction pattern of Yb8−xYxV2O17 was indexed in analogy to other phases [28], i.e. by means of the POWDER program [29]. The parameters of selected unit cell were refined using the REFINEMENT program of DHN/PDS package.
The DTA study was performed using an SDT 2960 thermoanalyser (TA Instruments, USA) under the air flow (110 mL min−1), at the heating rate of 10° min−1 in temperatures from the range 20–1450 °C. The DTA method for testing the thermal stability of the various phases forming in the multicomponent oxide systems was described, inter alia, in [30–33].
Selected samples were investigated also by means of an electron scanning microscope–SEM (JSM-6100, Jeol, Japan). The densities of Yb8−xYxV2O17 were determined in argon (5 N purity) with the help of an Ultrapyc 1200e ultrapycnometer (Quantachrome Instruments, USA). Initial mixtures and monophasic samples were examined by IR spectroscopy. The measurements were made within the wavenumber range of 1200–250 cm−1, using a spectrophotometer SPECORD M-80 (Carl Zeiss, Jena, Germany). A technique of pressing pellets with KBr at the mass ratio of 1:300 was applied. The UV–Vis–DR spectra were measured using a UV–Vis spectrometer V-670 (JASCO, Japan) equipped with a reflecting attachment for the solid-state investigation (integrating sphere attachment with horizontal sample platform PIV-756/(PIN-757). The spectra were recorded in the wavelength region of 200–750 nm at room temperature.
Results and discussion
The first stage of the study was to verify the literature crystallographic data [19–22] on the compounds Yb8V2O17 and Y8V2O17 and to evaluate their thermal stability. Analysis of XRD diffractograms of Yb8V2O17 and the low-temperature polymorph of Y8V2O17 synthesised by us revealed that only the diffractogram of Yb8V2O17 differed slightly from that reported by Brusset et al. [19]. Because of these differences including the presence of additional low-intensity XRD lines (d = 0.4675; 0.4492; 0.3911 nm), the Yb8V2O17 diffractogram was subjected to indexation using the program POWDER [29].
Indexation was performed for selected 20 reflections from the range of 2θ angles from 6 to 40° (CuKα). The best agreement with experimental data was obtained for the triclinic elementary cell model. The results of indexation of the powder diffractogram of Yb8V2O17 are presented in Table 1.
The elementary cell parameters of Yb8V2O17 were refined with the use of the program REFINEMENT (package DHN/PDS) to be:
-
a = 0.8932 nm, b = 0.9259 nm, c = 0.9794 nm,
-
α = 77.684°, β = 106.367°, γ = 116.348°
-
elementary cell volume V = 0.6928 nm3,
-
number of molecules in the elementary cell Z = 2.
The quality coefficient for this refined solution is F(20) = 12.37 (0.0192.84). The calculated density of Yb8V2O17 for the selected solution is equal to 8.42 g cm−3 and experimentally determined with a gas ultrapycnometer was 8.26 ± 0.05 g cm−3. The difference in densities is probably connected with porosity of obtained compound.
As the solution proposed by us is significantly different from that given in [19] from 1973, we decided to refine the Yb8V2O17 diffractogram known from literature with the use of program REFINEMENT. For the solution proposed by the authors of [19] which was the monoclinic system, the quality coefficient FM was very low, of only 2.24, which indicated a very small probability of crystallisation of this compound in the monoclinic system.
As thermal stability of the compounds Yb8V2O17 and Y8V2O17 has not been known, they were subjected to DTA–TG measurements in air atmosphere and in the temperature range 20–1450 °C. In this range no thermal effects were observed on both DTA and TG curves. This result means that these compounds undergo decomposition or melting at temperatures higher than 1450 °C. In view of this result, the study of reactivity between Yb8V2O17 and Y8V2O17 was started from a lower temperature of 1400 °C. The mixtures of Yb8V2O17 and Y8V2O17 of compositions specified in Table 2, were heated at the following stages: I:1400 °C (24 h) → II:1450 °C (24 h) → III:1500 °C (24 h) → IV:1550 °C (24 h).
As evidenced by XRD analysis, already after the first stage of heating the phase composition of all samples changed. The XRD diffractograms recorded for samples 1–7 did not reveal the reflections characteristic of Y8V2O17, and besides the lines characteristic of Yb8V2O17 slightly shifted towards smaller angles 2θ, it showed the lines undoubtedly evidencing the presence of YVO4 and Yb2O3. The subsequent two stages of the sample heating, at 1450 and 1500 °C, did not cause changes in the phase composition but changed the proportion of phases, i.e. the phases YVO4 and Yb2O3 were present in small amounts. The diffractograms recorded after the last stage of heating showed only the lines characteristic of Yb8V2O17. With increasing content of Y8V2O17 in the initial mixtures of reagents, the lines were increasingly shifted towards smaller 2θ angles that evidenced increasing interplanar distances dhkl relative to those in undoped Yb8V2O17 [19].
According to these results, in the system Yb8V2O17–Y8V2O17 in the concentration range studied, the reactions leading to formation of substitutive solid solution in which Y3+ replaced Yb3+ in the crystalline lattice of Yb8V2O17 took place. It is highly probable because of similar values of ionic rings of Yb3+ and Y3+ that can occur in the polyhedrons of NC = 6 (Yb3+-86.8 pm and Y3+-90.0 pm) as well as of NC = 8 (Yb3+-98.5 pm and Y3+-101.9 pm) [34]. The method for synthesis of this solid solution has been applied for patent protection in Poland [35].
According to phase composition determination in samples 8 and 9, representing the concentration ranges of the system components above 40.00 mol% Y8V2O17, after the last stage of heating (1550 °C) the samples were biphasic and besides the Yb8−xYxV2O17 contained the high-temperature polymorph of Y8V2O17 (Table 2).
The phase composition of all samples after the last stage of heating, presented in Table 2, shows that the solid solution formed in the system is substitutive, of limited solubility of components and general formula Yb8–xYxV2O17. The results collected in Table 2 additionally imply that the maximum degree of Y3+ ions incorporation into Yb8V2O17 reaches at least 40.00% mol (x = 3.2) and does not exceed 50.00% mol. The above-discussed results lead to the conclusion that in the reaction mixtures studied the following reaction took place:
Figure 1 presents a fragment of XRD diffractogram of Yb8V2O17 (Fig. 1a) to be compared with the analogous fragments of diffractograms of the new solid solution Yb8−xYxV2O17 for x = 0.8 (Fig. 1b), x = 2.8 (Fig. 1c) and x = 3.2 (Fig. 1d).

Fragments of diffractograms of: a Yb8V2O17, b Yb7.2Y0.8V2O17, c Yb5.2Y2.8V2O17, d Yb4.8Y3.2V2O17
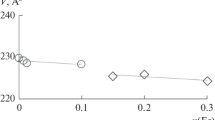
At the next stage of the study our aim was to confirm that the new solid solution Yb8−xYxV2O17 has the structure of the matrix, that is it crystallises in the triclinic system. Thus, the powder diffractograms of the new solution Yb8−xYxV2O17 for x = 0.80; 2.00; 2.80 and 3.20 were subjected to indexation. The elementary cell parameters were refined with the program REFINEMENT. The results confirmed that the new solid solution crystallises in the triclinic system and permitted calculation of the parameters of its elementary cell as a function of the degree of Yb3+ substitution with Y3+ in the crystal lattice of Yb8V2O17. Table 3 presents the elementary cell parameters, their volumes and densities for Yb8−xYxV2O17 with x = 0.00; 0.80; 2.00; 2.80 and 3.20.
According to the data presented in Table 3, with increasing x in Yb8−xYxV2O17, that is with increasing degree of Yb3+ substitution with Y3+, the crystal lattice expansion takes place; that is the elementary cell volume increases with respect to that of the matrix Yb8V2O17. Moreover, the density of the solid solution determined with the use of a gas ultrapycnometer is lower than the value calculated on the basis of parameters selected elementary cells.
With increasing x in Yb8−xYxV2O17 a change in its colour from orange to yellow was observed.
At the subsequent stage the compound Yb8V2O17 and the solid solution Yb8−xYxV2O17 with x = 3.2 were studied by scanning electron microscopy (SEM). The SEM images of the two polycrystalline samples are presented in Figs. 2 and 3.

SEM image of Yb8V2O17

SEM image of Yb8−xYxV2O17 (x = 3.2)
The morphology of the solid solution Yb8−xYxV2O17 crystallites is very similar to that of the matrix Yb8V2O17. They look like polygons of irregular shapes and different sizes, varying from 1 to 8 µm (Fig. 3). It should be pointed out that some crystallites of the matrix have ball-like shape of diameters close to ~6 µm (Fig. 2).
As the structures of Yb8V2O17 and the solid solution Yb8−xYxV2O17 were not fully resolved, their IR spectra were measured to get some preliminary information on the type of metal oxide polyhedrons in structures these phases. Figure 4 presents the IR spectra of Yb8V2O17 (Fig. 4a) and the newly obtained solid solution Yb8–xYxV2O17 with x = 0.80; 2.80 and 3.20 (Fig. 4b–d).

Fragments of the IR spectra of: a Yb8V2O17, b Yb7.2Y0.8V2O17, c Yb5.2Y2.8V2O17, d Yb4.8Y3.2V2O17
The IR spectrum of Yb8V2O17 (Fig. 4a) shows a few absorption bands of maxima appearing at 940, 820, 450 cm−1. The bands in the wavenumber range 970–620 cm−1 can be assigned to stretching vibrations of the symmetric and asymmetric V–O bonds in VO4 tetrahedrons and to the vibrations of Yb–O bonds in the YbO6 octahedrons [36–38]. The presence of YbO8 polyhedrons cannot be excluded [38]. On the basis of the IR spectrum of ytterbium vanadate(V) of the known structure [36–38] the absorption band in the range 650–300 cm−1 can be assigned to bending vibrations of V–O bonds in VO4 tetrahedrons and the band at ~630 cm−1 can be attributed to V–O–V bond. On the other hand a broad band with a maximum at 450 cm−1 can likely be attributed to the stretching vibrations of M–O bonds in the YbO6 octahedrons [36–38].
As follows from analysis of the IR spectrum of the new solid solution (Fig. 4b–d) the positions of absorption bands in this spectrum are very similar to those in the IR spectrum of Yb8V2O17. With increasing degree of Y3+ incorporation to replace Yb3+ in the crystal lattice of Yb8V2O17, the positions of absorption bands shift towards smaller wavenumbers or remain unchanged with respect to their positions in the IR spectrum of the matrix. The results of this part of study confirmed that the solid solution Yb8–xYxV2O17 shows the structure of Yb8V2O17, so contains VO4 tetrahedrons and MO6 polyhedrons (M=Y and Yb) or/and YbO8 [36–39]. It is not possible to conclude from IR spectra on the way the polyhedron is joined and therefore studies are only qualitative.
In order to assess the thermal stability of Yb8V2O17 and the solid solution Yb4.8Y3.2V2O17 obtained at 1550 °C, the samples containing these two phases were heated from 1450 to 1700 ± 20 °C in air atmosphere in a horizontal tube furnace equipped with an optical pyrometer.
The samples heated to 1600 °C did not show signs of melting, and their phase composition was unchanged. The diffractograms of the samples heated up to ~1650 °C show the set of XRD lines which, according to the PDF chart no. 00-035-0153, were assigned to the high-temperature polymorph of Yb8V2O17.
The lines in the diffractogram of Yb4.8Y3.2V2O17 corresponded to slightly greater interplanar distances, which suggest the presence of the solid solution Yb8–xYxV2O17, as the high-temperature polymorph of Yb8V2O17 is its matrix. The results obtained at this stage imply that at 1650 ± 20 °C the compound Yb8V2O17 undergoes monotropic polymorphous transformation.
Heating of the samples up to ~1700 °C did not result in their melting and did not cause changes in their phase composition. As we have no possibility to heat the samples above 1700 °C, we could only conclude that the high-temperature polymorph of Yb8V2O17 and the solid solution of the same structure are thermally stable in air at least to ~1700 °C. Figure 5 presents a fragment of XRD diffractogram of Yb8V2O17 (Fig. 5a) with the analogous fragment of diffractogram of Yb4.8Y3.2V2O17 (of the new solid solution Yb8−xYxV2O17 for x = 3.2), i.e. of samples which were heated at ~1700 °C (Fig. 5b).

Fragments of diffractograms of samples were heated at ~1700 °C: a Yb8V2O17 b Yb4.8Y3.2V2O17
Literature data [40] on the high-temperature polymorph of Yb8V2O17 are fragmentary so the studies should be continued to provide its physicochemical characterisation and detail structure.
In this study the physicochemical characterisation of Yb8V2O17 and the solid solution Yb8−xYxV2O17 was extended by the UV–Vis–DR measurements that helped evaluate the energy gap for these phases.
The reflectance spectra of Yb8V2O17 and the solid solution Yb8–xYxV2O17 for x = 0.80; 2.80; 3.20 after Kubelka–Munk transformation are shown in Fig. 6.

Kubelka–Munk transformation of reflectance spectra of Yb8V2O17 and the solid solution Yb8–xYxV2O17 for x = 0.80; 2.80; 3.20
The Kubelka–Munk transformation of measured reflectance performed using package Spectra Manager Version 2 (Spectra Analysis program) according to equation:
where K is reflectance transformed according to Kubelka–Munk and R is reflectance (%).
Then plotted the relationship (K·hν)2 = f(hν)—see Fig. 6, where on the ordinate variable (K/M) means (K·hν)2. The intersection between the linear fit and the photon energy axis gives the value to E g.
The energy gap (E g) obtained for the compound Yb8V2O17 is ~2.60 eV, while the energy gap for the solid solution Yb8–xYxV2O17 evaluated by UV–Vis–DR method decreases with increasing degree of substitution x from E g = ~ 2.52 eV for Yb7.2Y0.8V2O17 to ~2.44 eV for Yb4.8Y3.2V2O17. The above energy gap values mean that both the compound Yb8V2O17 and the solid solution Yb8−xYxV2O17 are semiconductors.
Conclusions
-
The substitutional, limited solid solution of the formula Yb8−xYxV2O17 and 0 < x < 4.0 is formed in the Yb8V2O17–Y8V2O17 system.
-
This phase has been obtained in air by high-temperature reaction from the specially synthesised compounds: Yb8V2O17 and Y8V2O17.
-
Yb8−xYxV2O17 crystalises in the triclinic system.
-
With increasing x in Yb8−xYxV2O17, the crystal lattice of solid solution expands.
-
The solid solution Yb8−xYxV2O17 is stable in air atmosphere up to ~1700 °C.
-
Yb8V2O17 and Yb8−xYxV2O17 belong to the group of semiconductors with the band gab energies from 2.60 to 2.44 eV.
References
O’Connor JR. Unusual crystal-field energy levels and efficient laser properties of YVO4:Nd. Appl Phys Lett. 1966;9:407–9.
Fields RA, Birnbaum M, Fincher CL. Highly efficient Nd:YVO4 diode-laser end-pumped laser. Appl Phys Lett. 1987;51:1885–6.
Savitski VG, Malyarevich AM, Yumashev KV, Kalashnikov VL, Sinclair BD, Raaben H, Zhilin AA. Experiment and modeling of a diode-pumped 1.3 µm Nd:YVO4 laser passively Q-switched with PbS-doped glass. Appl Phys B. 2004;79:315–9.
Kisel VE, Troshin AE, Tostik NA, Shcherbitsky VG, Kuleshov NV, Matrosov VN, Matrosova TA, Kupchenko MI. Spectroscopy and continuous wave diode-pumped laser action of Yb3+:YVO4. Opt Lett. 2004;29:2491–3.
Orlova GYu, Vlasov VI, Zavartsev YuD, Zagumennyi AI, Kalashnikova II, Kutovoi SA, Naumov VS, Sirotkin AA. Effect of structural imperfections on lasing characteristics of diode-pumped YVO4, GdVO4 and mixed rare earth vanadate crystals. Quantum Electron. 2012;42:208–10.
Kalisky Y. New trends in lasers and laser crystals. Opt Mater. 1999;13:135–9.
Wu S, Wang G, Xiea J. Growth of high quality and large sized Nd3+:YVO4 single crystal. J Cryst Growth. 2004;266:496–9.
Pode RB, Band AM, Juneja HD, Dhoble SJ. Thermoluminescence studies YVO4:Yb3+. Phys Status Solidi. 1996;157:493–8.
Chen J, Guo F, Zhuang N, Lan J, Hu X, Gao S. A study on the growth of Yb:YVO4 single crystal. J Cryst Growth. 2002;243:450–5.
Buissette V, Huignard A, Gacoin T, Boilot JP, Aschehoug P, Viana B. Luminescence properties of YVO4: Ln (Ln = Nd, Yb and Yb–Er) nanoparticles. Surf Sci. 2003;532–535:444–9.
Xu HY, Wang H, Jin TN, Yan H. Rapid fabrication of luminescent Eu:YVO4 films by microwave-assisted chemical solution deposition. Nanotechnology. 2005;16:65–9.
Xia Z, Chen D, Yang M, Ying T. Synthesis and luminescence properties of YVO4:Eu3+, Bi3+ phosphor with enhanced photoluminescence by Bi3+ doping. J Phys Chem Solids. 2010;71:175–80.
Huignard A, Gacoin T, Boilot JP. Synthesis and luminescence properties of colloidal YVO4: Eu phosphors. Chem Mater. 2000;12:1090–4.
Huignard A, Buissette V, Laurent G, Gacoin T, Boilot JP. Synthesis and characterizations of YVO4: Eu colloids. Chem Mater. 2002;14:2264–9.
Riwotzki K, Haase M. Wet-chemical synthesis of doped colloidal nanoparticles YVO4: Ln (Ln = Eu, Sm, Dy). J Phys Chem B. 1998;102:10129–35.
Yu M, Lin J, Fang J. Silica spheres coated with YVO4:Eu3+ layers via sol-gel process: a simple method to obtain spherical core-shell phosphors. Chem Mater. 2005;17:1783–91.
Zhang H, Yu Y, Cheng Y, Liu J, Li H, Ge W, Cheng X, Xu X, Wang J, Jiang M. Growth of YbVO4 stoichiometric crystal. J Cryst Growth. 2005;283:438–43.
Liu FQ, Sun SQ, Gao ChY, Xu JQ. Optical properties of Nd:YbVO4 crystal. Opt Appl. 2015;XLV:63–70.
Brusset H, Mahe R, Laude JP. Studies of rare-earth vanadates with formula T8V2O17. Bull Soc Chim Fr. 1973;2:495–9.
Brusset H, Madaule-Aubry F, Blanck B, Deboichet A. On binary systems V2O5–Gd2O3 and V2O5–Er2O3. Bull Soc Chim Fr. 1969;1:15–6.
Levin EM. The system Y2O3–V2O5. J Am Ceram Soc. 1967;50:381–2.
Yamaguchi O, Mukaida Y, Shigeta H, Takemura H, Yamashita M. Preparation of Alkoxy-Derived Yttrium Vanadate. J Electrochem Soc. 1989;136:1557–60.
Filipek E, Pacześna A, Piz M. Sr2InV3O11—new ceramic compound in Sr2V2O7–InVO4 system and its characteristic. Ceram Int. 2016;42:14148–54.
Dabrowska G, Filipek E, Piz M. A new ceramic continuous solid solution in the CrSnSbO6–FeSnSbO6 system and some of its properties. Ceram Int. 2015;41:12560–7.
Bosacka M, Filipek E, Paczesna A. Unknown phase equilibria in the ternary oxide V2O5–CuO–In2O3 system in subsolidus area. J Therm Anal Calorim. 2016;125:1161–70.
Walczak J, Filipek E, Tabero P. CrVMoO7 and phase equilibria in the V9Mo6O40–CrVMoO7 system. Thermochim Acta. 1992;206:279–84.
Powder Diffraction File, International Center for Diffraction Data, Swarthmore (USA). 1989. (PDF-4 +), File Nos.: 00-043-1037, 00-041-1105, 04-008-4555, 00-025-1249, 00-022-1001, 00-035-0153.
Piątkowska M, Tomaszewicz E. Synthesis, structure and thermal stability of new scheelite type Pb1–3x□xGd2x(MoO4)1–3x(WO4)3x ceramic materials. J Therm Anal Calorim. 2016;126:111–9.
Taupin D. A powder-diagram automatic-indexing routine. J Appl Cryst. 1973;6:380–5.
Tabero P, Frąckowiak A. Synthesis of Fe8V10W16O85 by a solution method. J Therm Anal Calorim. 2016;125:1445–51.
Gorodylova N, Šulcova P, Bosacka M, Filipek E. DTA–TG and XRD study on the reaction between ZrOCl2. 8H2O and (NH4)2HPO4 for synthesis of ZrP2O7. J Therm Anal Calorim. 2014;118:1095–100.
Olszak-Humienik M, Jablonski M. Thermal behavior of natural dolomite. J Therm Anal Calorim. 2015;119:2239–48.
Checmanowski J, Pelczarska AJ, Szczygiel I, Szczygiel B. Influence of ceria and yttria on the protective properties of SiO2–Al2O3 coatings deposited by sol–gel method on FeCrAl alloy. J Therm Anal Calorim. 2016;126:371–80.
Shannon RD, Prewitt CT. Effective ionic radii in oxides and fluorides. Acta Cryst. 1969;B25:925–46.
Piz M, Filipek E. Patent application RP. 2016; No. P 417368, Poland.
Garg AB, Rao R, Sakuntala T, Wani BN, Vijayakumar V. Phase stability of YbVO4 under pressure: in situ X-ray and Raman spectroscopic investigations. J App Phys. 2009;106:063513.
Panchal V, Lopez-Moreno S, Santamaria-Perez D, Errandonea D, Manjon FJ, Rodriguez-Hernandez P, Munoz A, Achary SN, Tyagi AK. Zircon to monazite phase transition in CeVO4: X-ray diffraction and Raman-scattering measurements. Phys Rev B. 2011;84:024111.
Jindal R, Sinha MM, Gupta HC. Lattice vibrations of AVO4 crystals (A = Lu, Yb, Dy, Tb, Ce). Spectrochim Acta, Part A. 2013;113:286–90.
Rast HE, Caspers HH, Miller SA. Infrared spectral emittance and optical properties of yttrium vanadate. Phys Rev. 1968;169:705–9.
Ustalova O, Rykova G, Skorykov W, Ianaev I. Oksyvanadaty redkosemielnykh elementov ittrevoy podgruppy. Russ J Inorg Chem. 1980;25:1223–8.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Piz, M., Filipek, E. Synthesis and homogeneity range of Yb8−xYxV2O17 in the Yb8V2O17–Y8V2O17 system. J Therm Anal Calorim 130, 277–283 (2017). https://doi.org/10.1007/s10973-017-6379-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-017-6379-0