
Abstract
Substitutional, continuous solid solution of the general formula Y2–xYbxO3 was obtained from the mixture of Y2O3 and Yb2O3 oxides, for the first time by the mechanochemical method in a high-energy ball milling. The monophasic samples of nanocrystalline solid solution for x > 0.00 and x < 2.00 were examined by the methods: XRD, DTA, SEM, IR and UV–Vis–DR. As follows from the results, the solid solution crystallizes in cubic system and is isostructural with Y2O3 and Yb2O3. The solution is stable in the air atmosphere up to at least 900°C, and its decomposition temperature decreases with the increase in x, that is, with decreasing number of Yb3+ ions replacing Y3+ ions in the crystal lattice of Y2O3. The energy band gap estimated for the solid solution varies from ~ 5.30 eV for x = 0.50 to ~ 4.90 eV for x = 1.50, which means that it is an insulator.
Similar content being viewed by others


Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
Introduction
Binary and multicomponent systems of different oxides have been for a few decades the subjects of comprehensive studies. The knowledge gained from the studies is used for devising advanced technologies for the production of new materials of target chemical, electric, magnetic, optical or catalytic properties. The new materials not only should be synthesized by environmentally friendly methods, but also should contribute to limitation of its pollution. In view of growing depletion of natural resources and increasing cost of energy, rationalization of production processes has become an urgency [1,2,3,4]. A method for production of new materials proposed as an alternative to high-temperature treatment, sol–gel, hydrothermal, sonochemical or Pechini processes is mechanochemical synthesis [1,2,3,4,5,6,7,8,9,10,11]. Its main advantage is simplicity as the method is based on milling of all kinds of reagents in high-energy mills [1, 2, 4, 8, 9]. The mechanical energy generated upon milling is supplied to the substrates and accumulated in them in the form of all kinds of lattice stresses which can increase their reactivity. The process of high-energy milling can lead to very fine comminution of the product, often to nanosize particles [1, 4, 9]. Moreover, the mechanochemical method can lead to materials whose syntheses by other methods have failed [9]. Such materials include solid solutions. According to the literature data, the solid solutions obtained by mechanochemical method often show a greater range of homogeneity than those obtained by the solid-state reactions [8, 9].
Formation of solid solution in the system of V2O5–Yb2O3, obtained by mechanochemical method, has been described in the literature [9]. It is a substitutional solution of limited range of solubility of vanadium(V) oxide in the crystal lattice of ytterbium oxide and of the general formula Yb2−5x□2xV3xO3, where 0.00 < x <0.1667 [9]. It is stable in the air at least up to ~ 800°C (for x = 0.1176), and its thermal decomposition point decreases with increase in x, so with increasing degree of V5+ replacement of Yb3+ in the crystal lattice of Yb2O3 [9].
According to the literature data on the oxide system Y2O3–Yb2O3 [10, 11], in this system, a solid solution of the general formula Y2−xYbxO3 is formed. It has been obtained by high-temperature (1400 °C) treatment of mixtures of oxides containing Y2O3 and Yb2O3, of defined compositions (solid-state reactions), for a few days [10]. The same authors have resolved full crystalline structure of this solution by the Rietveld method and examined its magnetic properties [10]. Kimmel et al. [11] have also reported obtaining the same phase by the sol–gel method. The xerogel obtained was heated in the air atmosphere in temperatures from 100 to 1400 °C for 3 h.
Taking into account the hitherto knowledge of the phases forming in the binary system of Y2O3–Yb2O3, in particular no information on the obtaining of the phases by mechanochemical synthesis, the main aim of our studies was to identify the compounds and/or phases of solid solution type that would form in this system, in the entire range of concentrations of its components, as a result of processes induced by high-energy ball milling. If some compounds and/or solid solutions formed as a result of this procedure, the studies would be undertaken to establish basic properties of these phases, mainly their structures, thermal stability and optical properties.
Experimental
The following reagents were used in the experiments:
Y2O3, a.p. (Sigma-Aldrich, USA) and Yb2O3, a.p. (Sigma-Aldrich, USA). Nine samples were prepared for the experiments (Tables 1, 2). The samples were synthesized by mechanochemical method using a laboratory planetary ball mill Pulverisette (Fritsch GmbH, Germany) with vessel and balls made of zirconia, rpm = 250, BPR = 1:20 and time = 3.0 h, under air atmosphere.
The powder diffraction patterns of the samples obtained were recorded on a diffractometer EMPYREAN II (PANalytical, Netherlands) using CuKα with graphite monochromator. The crystallite size of the phases was evaluated using the High Score Plus PANalytical.
The phases were identified on the basis of XRD characteristics contained in the PDF4 + cards [12]. The parameters of selected unit cell solid solutions were refined using the REFINEMENT program of DHN/PDS package.
Monophasic samples were examined by IR spectroscopy. The measurements were made within the wavenumber range of 1200–250 cm−1, using a spectrophotometer SPECORD M-80 (Carl Zeiss, Jena, Germany). A technique of pressing pellets with KBr at the mass ratio of 1:300 was applied.
The monophasic samples were examined using the following methods: scanning electron microscopy using FE-SEM Hitachi SU–70 microscope and EDS X-ray microanalysis using NORAN™ System 7 of Thermo Fisher Scientific (UltraDry X-ray detector). SEM analyses were performed at accelerating voltage of 15 kV, and secondary electron images were acquired. The densities of the resulting samples were determined in argon (5 N purity) with the help of an Ultrapyc 1200e ultrapycnometer (Quantachrome Instruments, USA).
Selected samples were also subjected to examination by the DTA method using a SDT 2960 apparatus, (TA Instruments Company, USA). The measurements were taken in air atmosphere, within the temperature range 20–1400 °C, at the heating rate of 10 deg/min. The tests were conducted in corundum crucibles. The mass of the samples was ~ 20 mg.
The UV–Vis–DR spectra were recorded using a UV–Vis spectrometer V-670 (JASCO, Japan) equipped with a reflecting attachment for the solid-state investigation, integrating sphere attachment with horizontal sample platform PIV-756/(PIN-757). The spectra were recorded in the wavelength region of 200–750 nm at room temperature.
Results and discussion
The study of the oxide system Y2O3–Yb2O3 was begun with preparation of nine mixtures of Y2O3 and Yb2O3 of the compositions specified in Table 1, selected to represent different ranges of concentration of the system’s components. The mixtures of isostructural oxides Y2O3 and Yb2O3 [13,14,15,16,17,18,19,20,21,22,23,24,25,26], after preliminary homogenization in an agate mortar, were subjected to mechanochemical synthesis in a high-energy ball milling, under air atmosphere, in a few 3-h long stages. According to XRD studies of all samples, only samples no. from 5 to 9, that is those containing over 40% mol of Yb2O3, changed their initial composition after the first stage of milling. The lines appearing on the XRD diffractograms of the samples corresponded to the interplanar distances, dhkl, whose values varied from those typical of pure Y2O3 to those typical of pure Yb2O3 (PDF Cards No. 00-043-1037 and 01-083-5700). As follows from the XRD results, the samples were monophasic and contained only the substitutional solid solution of the structure of Y2O3 or Yb2O3. The XRD diffractograms of the other samples no. 1–4, containing Yb2O3 in the amount not exceeding 40% mol, after the first 3-h stage of milling showed the same set of lines as in the diffractograms of the initial mixtures. Differences were only noted in the intensity of XRD lines. For samples no. 1–4, the same results of phase analysis as for samples 5–9 after 3-h milling were obtained after eight subsequent 3-h milling stages.
The results showed that by the method of high-energy ball milling of the mixtures of oxides Y2O3 and Yb2O3, a solid solution was obtained in the entire range of concentrations of the system components. Thus, it is a continuous substitutional solid solution whose general formula can be written as Y2−xYbxO3(s.s.) as well as YzYb2−zO3(s.s.). The time of synthesis of the solid solution by this method varied from 3 to 30 h and depended on the initial composition of the sample. The time of synthesis was much longer for the samples containing Yb2O3 in concentrations lower than 50% mol in the mixtures with Y2O3, but still this method of synthesis was much shorter and less complex than other methods [10, 11].
Henceforth, for the clarity of presentation, we assume that the matrix of the continuous substitutional solid solution is the oxide Y2O3. The solid solution is formed by incorporation of Yb3+ ions in the crystal lattice of Y2O3, replacing Y3+ ions (Yb3+—86.8 pm, Y3+—90.0 pm) which can be described by the following reaction formula:
According to the XRD diffractograms of monophasic samples, Table 1, containing solid solution Y2−xYbxO3, with increasing concentration of Yb2O3 in the initial mixtures of the reagents (that is with the increase in x in the solution formula), the XRD lines characterizing this phase Y2−xYbxO3(s.s.) are shifted towards lower 2Θ angles, and so correspond to longer interplanar distances dhkl relative to those in the pure oxide Y2O3.
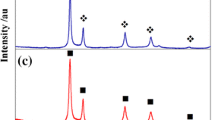
Figure 1 shows fragments of XRD diffractograms of the commercial oxide Y2O3 (Fig. 1a) and the oxide Yb2O3 (Fig. 1e) in comparison with the fragments of the diffractograms of the solid solution Y2−xYbxO3 obtained for the first time by the mechanochemical method, for x = 0.5 (Fig. 1b), x = 1.0 (Fig. 1c) and x = 1.5 (Fig. 1d).

Fragments of diffractograms of a Y2O3, b Y1.5Yb0.5O3, c YYbO33, d Y0.5Yb1.5O3 and e Yb2O3
The next task was to confirm that the solid solution Y2−xYbxO3 obtained by the mechanochemical method has the structure of Y2O3 and Yb2O3, i.e. it crystallizes in the cubic system. The powder diffractograms of the solution Y2−xYbxO3 for x = 0.5, 1.0 and 1.5 were subjected to refinement by the program REFINEMENT. The results confirmed that the obtained solid solution crystallized in the cubic system, and the parameters of its elementary cells were calculated as a function of concentration of Yb3+ incorporated in the place of Y3+ in the crystal lattice of Y2O3. Table 2 presents the elementary cell parameters and volumes as well as X-ray and experimental density for Y2−xYbxO3, with x = 0.5, 1.0 and 1.5 and the corresponding data for the pure oxides Y2O3 and Yb2O3.
As can be concluded from Table 2, with the increase in x in the system Y2−xYbxO3, and with increasing concentration of Yb3+ ions replacing Y3+ ones, the crystal lattice contraction is observed. The volume of unit cells decreases from the value corresponding to pure Y2O3 to that characteristic of pure Yb2O3.
The density of the solid solution Y2−xYbxO3 measured using a gas ultrapycnometer increases with the increase in x, and its value ranges from 4.99 g cm−3 for pure Y2O3 to 9.26 g cm−3 for Yb2O3. The increase in density is mainly due to the fact that the ion mass Yb3+ (173.054 u) is significantly larger than the mass of the ion Y3+ (88.906 u) and that the increase in the concentration of ions Yb3+ in the crystal lattice Y2O3 results in a decrease in the volume of the elementary cell. Additionally, experimental density of solid solution is in a good agreement with the calculated X-ray density, which confirms that the solutions adapted were correct.
The oxides Y2O3 and Yb2O3 as well as the solid solution Y2–xYbxO3 for x = 1.0 (YYbO3) were subjected to scanning electron microscopy (SEM/EDS). The obtained images of polycrystalline samples are presented in Figs. 2, 3 and 4.

SEM image of Y2O3

SEM image of Yb2O3

SEM image of YYbO3 (x = 1.0)
The crystallites of solid solution Y2−xYbxO3x = 1.0 (Fig. 4) have shapes very similar to that of the crystallites of Y2O3 and Yb2O3 (Fig. 2, 3). They resemble flattened polyhedrons of different sizes varying from ~ 35 to 150 nm (Fig. 4). According to their size distribution analysis, over 80% of them are nanosize, so are smaller than 100 nm. X-ray microanalysis (EDS) of the contents of elements in the crystallites of the solid solution YYbO3 for x = 1.0 evidenced the presence of ytterbium, yttrium and oxygen in the averaged amounts of Yb—56.6% mas., Y—32.8% mas. and O—10.6% mas. The calculated contents of these elements in the solid solution are Yb—55.8% mas., Y—28.7% mas. and O—15.5% mas. Taking into account that the mean error in determination of oxygen as a light element reaches over 10%, it can be concluded that the results obtained by SEM/EDS method are in good agreement with the values calculated from the formula YYbO3 for x = 1.0, so they confirm the correctness of the formula.
The subsequent task in our research work was to preliminarily confirm whether the solid solution obtained has the structure of Y2O3. In order to check this, selected monophasic samples of the solid solution were subjected to IR study. Figure 5 presents the IR spectrum of the commercial oxides: Y2O3, Yb2O3 (Fig. 5a, e) and the IR spectra of the solid solution Y2−xYbxO3 for x = 0.5, x = 1.0 and x = 1.5 (Fig. 5, spectra b, c and d).

Fragments of IR spectra of: a Y2O3, b Y1.5Yb0.5O3, c YYbO3, d Y0.5Yb1.5O3 and e Yb2O3
The IR spectrum of Y2O3 (Fig. 5a) shows three sharp bands of clear extremes appearing at ~ 308, 344 and 560 cm−1 and one broad band in the wavenumber range 540–356 cm−1 with an inflection point at ~ 500 cm−1. These bands can be assigned to the stretching vibrations of Y–O in the deformed YO6 octahedra [17,18,19,20,21,22,23,24,25,26]. In the IR spectra of the obtained solid solution Y2−xYbxO3 (Fig. 5b-d), the absorption bands appear in almost the same ranges of wavenumbers, from 650 to 300 cm−1, but they are broadened and their maxima are poorly marked at 564, 416 and 344 cm−1 (for x = 0.5), 568, 428 and 344 cm−1 (for x = 1.0) and at 576, 432 and 344 cm−1 (for x = 1.5). The bands can be assigned to the stretching vibrations of Yb–O and Y–O bonds, either symmetric or asymmetric, in the octahedra of YbO6 and YO6 [13,14,15,16,17,18,19,20,21,22,23,24,25,26]. With increasing number of Yb3+ replacing Y3+ in the crystal lattice of Y2O3, the absorption bands in IR spectra are shifted towards higher wavenumbers relative to those recorded in the IR spectrum of the matrix. These results confirmed that the solid solution Y2−xYbxO3 shows the Y2O3 structure and is built of the octahedra of YbO6 and YO6.
As thermal stability of the solid solution Y2−xYbxO3 (for 0.0 < x < 2.0) has not been known, monophase samples were subjected to DTA–TG measurements in air atmosphere and in the temperature range 20–1450°C. In this range, no thermal effects were observed on both DTA and TG curves. This result means that this phase undergoes decomposition or melting at temperatures higher than 1450°C.
In order to evaluate the thermal stability of the solid solution Y2−xYbxO3 obtained for the first time by the mechanochemical method, the monophasic samples 1–9 (Table 1) containing this solid solution were heated in 3-h stages, in the range from 600 to 1650 ± 10°C, in air atmosphere in a horizontal tube furnace equipped with an optical pyrometer. The samples heated at 600, 700, 800 or 900°C did not show signs of melting and their phase composition did not change. Subsequent heating of samples at ~ 1000°C (3 h) resulted in a change in phase composition of sample no. 5 that contains YYbO3. The diffractogram of this not-molten sample did not show the diffraction lines characteristic of the solid solution Y2−xYbxO3 (x = 1.0), but showed the XRD lines assigned to the oxides Y2O3 and Yb2O3, according to the PDF Cards No. 00-043-1037 and 01-083-5700. The other samples showed the same phase composition only after their heating at ~ 1600°C (3 h). After this stage of heating, the samples still did not show signs of melting. As follows from these results, the solid solution Y2−xYbxO3 obtained by the mechanochemical method is thermally stable in air atmosphere up to at least 900°C for x = 1.0 and up to ~ 1500°C for 1 > x >0 and 2 > x >1. Above these temperatures, the solid solution decomposes to Y2O3 and Yb2O3. Explanation of the minimum of the thermal stability temperature determined for the solid solution Y2−xYbxO3 for x = 1 needs separate studies beyond the scope of this paper.
The oxides Y2O3, Yb2O3 and the solid solution obtained Y2−xYbxO3 for x = 0.5, 1.0 and 1.5 were also subjected to UV–Vis–DRS study to estimate the energy gap of these phases. Figure 6 presents the dependencies K2 = f(E), where K is a constant following from the Kubelka–Munk transformation [5, 6, 9, 27] and on the plot is marked as K/M. The energy values of the forbidden bands (Eg) for the phases studied were read off as the coordinate of the point of intersection of the tangent to an appropriate curve with the x-axis.

Kubelka–Munk transformation of the reflection spectra of Y2O3, Yb2O3 and the solid solution Y2−xYbxO3 for x = 0.5, 1.0 and 1.5
The energy gap (Eg) determined for Y2O3 is ~ 5.50 eV, for Yb2O3 is ~ 5.00 eV, while the energy gap established for the solid solution decreases with increasing number of Yb3+ ions replacing Y3+ ions in the crystal lattice of Y2O3, from Eg = ~ 5.30 eV for Y1.5Yb0.5O3 to ~ 4.90 eV for Y0.5Yb1.5O3. The above results prove that the solid solution obtained Y2−xYbxO3 belongs to insulators and can be used as ceramic insulator or dielectric.
Conclusions
-
The substitutional continuous solid solution of the formula Y2−xYbxO3,x > 0.00 and x < 2.00 is formed in the Y2O3–Yb2O3 system,
-
The solid solution has been obtained in air for the first time by high-energy ball milling from the oxides: Y2O3 and Yb2O3,
-
The Y2−xYbxO3 crystallizes in the cubic system,
-
With the increase in x in Y2−xYbxO3, the crystal lattice of solid solution is contracted,
-
The solid solution Y2−xYbxO3 for x = 1 is stable in air atmosphere up to ~ 900°C and for 1 > x > 0 and 2 > x > 1 up to ~ 1500°C and
-
Y2−xYbxO3 belongs to the group of insulators with the band gab energies from 5.30 to 4.90 eV.
References
Baláž P, Achimovičová M, Baláž M, Billik P, Cherkezova-Zheleva Z, Manuel Criado J, Delogu F, Dutková E, Gaffet E, Jose Gotor F, Kumar R, Mitov I, Rojac T, Senna M, Streleckii A, Wieczorek-Ciurowa K. Hallmarks of mechanochemistry: from nanoparticles to technology. Chem Soc Rev. 2013;42:7571–637.
James SL, Adams ChJ, Bolm C, Braga D, Collier P, Friščić T, Grepioni F, Harris KDM, Hyett G, Jones W, Krebs A, Mack J, Maini L, Orpen G, Parkin IP, Shearouse WC, Steed JW, Waddell DC. Mechanochemistry: opportunities for new and cleaner synthesis. Chem Soc Rev. 2012;41:413–47.
Garay AL, Pichon A, James SL. Solvent-free synthesis of metal complexes. Chem Soc Rev. 2007;36(6):846–55.
Dulian P, Garbarz-Glos B, Bak W, Antonova M, Kajtoch C, Wieczorek-Ciurowa K, Noga H. Dielectric behavior of BaTi1-xZrxO3 ceramics obtained by means of a solid state and mechanochemical synthesis. Ferroelectric. 2016;497(1):62–8.
Piz M, Filipek E. Synthesis and homogeneity range of Yb8-xYxV2O17 in the Yb8V2O17–Y8V2O17 system. J Therm Anal Calorim. 2017;130:277–83.
Dabrowska G, Filipek E, Piz M. A new ceramic continuous solid solution in the CrSnSbO6–FeSnSbO6 system and some of its properties. Ceram Int. 2015;41:12560–7.
Walczak J, Filipek E. Phase-equilibria in the Cr2V4O13-CrVMoO7 system. Therm Acta. 1990;161(2):239–45.
Filipek E, Wieczorek-Ciurowa K. Comparison between the synthesis in molybdenum and antimony oxides system by high-temperature treatment and high-energy ball milling. J Therm Anal Calorim. 2009;97:105–10.
Piz M, Dulian P, Filipek E, Wieczorek-Ciurowa K, Kochmanski P. Characterization of phases in the V2O5-Yb2O3 system obtained by high-energy ball milling and high-temperature treatment. J Mater Sci. 2018;53:13491–500.
Mitric M, Antic B, Balanda M, Rodic D, Napijalo MLJ. An X-ray diffraction and magnetic susceptibility study of YbxY2−xO3. J Phys Condens Mater. 1997;9:4103–11.
Kimmel G, Zabicky J, Goncharov E, Mogilyanski D, Venkert A, Bruckental Y, Yeshurun Y. Formation and characterization of nanocrystalline binary oxides of yttrium and rare earths metals. J Alloys Compd. 2006;42:102–6.
Powder Diffraction File (1989) International Center for Diffraction Data, Swarthmore USA (PDF-4+).
Atkinson SC. Crystal structures and phase transitions in the rare earth oxides. Salford: School of Computing, Science and Engineering University of Salford; 2013.
Fotiev AA, Slobodin BV, Khodos MY, Pletnev RN. Vanadates: composition, synthesis, structure, properties. Moskwa: Izd. Nauka; 1988.
Vasundhara K, Achary SN, Patwe SJ, Sahu AK, Manoj N, Tyagi AK. Structural and oxide ion conductivity studies on Yb1-xBixO1.5 (0.00 ≤ x ≤ 0.50) composites. J Alloys Compd. 2014;596:151–7.
Heiba ZK, Akin Y, Sigmund W, Hascicek YS. X-ray structure and microstructure determination of the mixed sesquioxides (Eu1−xYbx)2O3 prepared by a sol-gel proces. J Appl Crystallogr. 2003;36:1411–6.
Baldinozzi G, Béear JF, Calvarin-Amiri G. Rietveld refinement of two-phase Zr-doped Y2O3. Mater Sci Forum. 1998;278:680–5.
Antic B, Önnerud P, Rodic D, Tellgren R. The structure characteristics of the diluted magnetic semiconductor Y2−xDyxO3. Powder Diffr. 1993;8(4):216–20.
Mills SJ, Kartashov PM, Ma Ch, Rossman GR, Novgorodova I, Kampf R, Raudsepp M. Yttriaite-(Y): the natural occurence of Y2O3 from the Bol’shaya Pol’ya River, Subpolar Urals, Russia. Am Mineral. 2011;96:1166–70.
Santos C, Daguano JKMF, Rodrigues D Jr, Suzuki PA, Santos FA, Silva OMM. Synthesis of a mixed-rare-earth aluminium garnet solid solution. Mater Lett. 2013;91:283–6.
Som S, Dutta S, Chowdhury M, Kumar V, Kumar V, Swart HC, Sharma SK. A comparative investigation on ion impact parameters and TL response Y2O3:Tb3+ nanophosphor exposed to swift heavy ions for space dosimetry. J Alloys Compd. 2014;589:5–18.
Ishibashi H, Shimomoto K, Nakahigashi K. Electron density distribution and chemical bonding of Ln2O3 (Ln = Y, Tm, Yb) from powder X-ray diffraction data by the maximum-entropy method. J Phys Chem Solids. 1994;55(9):809–14.
Som S, Sharma SK, Lochab SP. Swift heavy ion induced structural and luminescence characterization of Y2O3:Eu3+ phosphor: a comparative study. Luminescence. 2014;29:480–91.
Ambrosini A, Duarte A, Poeppelmeier R, Lane M, Kannewurf CR, Mason T. Electrical, optical and structural properties of tin-doped In2O3–M2O3 solid solutions (M = Y, Sc). J Solid State Chem. 2000;153:41–7.
Leng J, Yu Z, Li Y, Zhang D, Liao X, Xue W. Optical and electrical properties of Y2O3 thin films prepared by ion beam assisted deposition. Appl Surf Sci. 2010;256(20):5832–6.
Basak D, Sen SK. Electrical, dielectric and optical properties of M/Y2O3/M devices. Thin Solid Films. 1995;254:181–6.
Tabero P, Frackowiak A. Reinvestigations of the Li2O-WO3 system. J Therm Anal Calorim. 2017;130:311–8.
Acknowledgements
This work was partly supported by National Science Centre No. 2017/01/X/ST8/01114.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Piz, M., Filipek, E. & Jablonski, M. Mechanochemical synthesis and thermal stability of phases in the Y2O3–Yb2O3 system. J Therm Anal Calorim 138, 4313–4319 (2019). https://doi.org/10.1007/s10973-019-08407-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-019-08407-1