
Abstract
The new hexadentate N2O4 Schiff base ligand from condensation of 2,4-hydroxybenzophenone and 1,3-propanediamine has been prepared in methanol and ethanol solutions. The Schiff base synthesized from EtOH contains half molecule of amine per one molecule of ligand in its structure. The complexes were synthesized from the direct reaction of this ligand and Mn(II), Co(II), Ni(II), Cu(II) and Zn(II) acetates. The Schiff base and its complexes were characterized by elemental analysis, X-ray crystallographic techniques, spectroscopic (UV–Vis, IR) and thermal (TG, TG-FTIR) methods, molar conductance and magnetic measurements. The complexes, except for Ni(II) compound, similar to the Schiff base are amorphous solids. The Ni(II) complex crystallizes in the monoclinic space group P21/c. The N2O2 coordination geometry around nickel(II) is slightly tetrahedrally distorted square planar. Variable temperature magnetic studies of the paramagnetic complexes show the existence of weak antiferromagnetic [for complexes of manganese(III) and cobalt(II)] and weak ferromagnetic [for copper(II)] interactions. The complexes are stable at ambient temperature. After heating at first they lose solvent molecules (water or methanol), and after that organic part undergoes defragmentation and combustion. Based on the obtained data from the TG-FTIR analysis, the main gaseous products resulting from thermal degradation in nitrogen atmosphere are CO2, CO, NH3, acetic acid, methane, C6H6, C6H5CN and C6H5CH3.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The metal complexes with polydentate ligands have been of great interest for the last few decades. It is well known that N and O donors play a key role in the coordination chemistry because they give the possibility of obtaining chelate compounds with interesting structures and properties [1–6]. The O,N-donor Schiff bases have often been used as chelating ligands in the field of coordination chemistry because they readily form stable complexes with most of the transition metals [4–7]. The Schiff base ligands and their complexes have numerous applications. The formation of stable complexes with metals gives the opportunity to design new systems selective to specific metallic ions. For this reason, they can be used in different extraction methods for preconcentration and separation of trace amount of metal ions or for the preparation of ion-selective electrodes for determination of anions and cations in analytical samples [8–10]. Metallic complexes of Schiff bases are efficient catalysts in oxidation and polymerization of organic compounds [8, 11–15]. Schiff base complexes are also noted for their significant biological activities such as antifungal, antibacterial, antimalarial, antitumor, antiproliferative, antiinflammatory, antiviral, antioxidant and antipyretic properties [6, 8, 11, 16, 17]. Special attention is paid to the aromatic Schiff bases with OH group in the ortho position to the amino group. This type of Schiff base compounds shows thermo- and photochromism providing potential applications as data storage, information processing, telecommunications and optical switching [10, 17–19].
As for numerous applications of Schiff base ligands and their metal complexes, there have been synthesized a salen-type Schiff base ligand derived from 2,4-dihydroxybenzophenone and 1,3-diaminopropane as well as its metal complexes of Mn(III), Co(II), Ni(II), Cu(II) and Zn(II). Hydroxybenzophenones (e.g. 2,4-dihydroxybenzophenone) have the ability to absorb and to dissipate UV light A, and therefore, they are used as UV stabilizers in plastic surface coatings on food packaging and sunscreens for humans [20]. The synthesized Schiff base and its metal complexes have been characterized on the basis of spectroscopic (IR, UV–Vis), X-ray crystallographic techniques and thermal (TG, TG-FTIR) methods as well as elemental analyses, molar conductance and magnetic measurements.
Experimental
Materials
The solvents (MeOH—methanol, EtOH—ethanol, DMF—N,N-dimethylformamide) and reagents (i.e. 1,3-propanediamine (1,3-PDA), 2,4-hydroxybenzophenone (K), acetates: Cu(CH3COO)2·H2O, Co(CH3COO)2·4H2O, Mn(CH3COO)2·4H2O, Ni(CH3COO)2·4H2O, Zn(CH3COO)2·2H2O) used for synthesis and studies were commercially available from Aldrich Chemical Company, Alfa Aesar Company and Polish Chemical Reagents. All solvents and chemicals were of reagent grade and used without further purification.
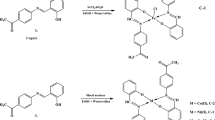
Preparation of 4,4′-{(1,3-propanediyl)bis[nitrilophenylmethylidene]}di(1,3-benzenodiol) (SchB)
The Schiff base ligand was synthesized (Scheme 1) using a similar method described in previous papers [8, 21, 22]. Mixtures of 10 mmol of 2,4-dihydroxybenzophenone and 5 mmol of 1,3-propanediamine in 40 mL of methanol or ethanol were refluxed for 2 h. The excess of the solvents (ca. 30 mL) was then evaporated. After cooling to 4 °C yellow solids were formed in both solvents. The products were collected by filtration, washed with methanol or ethanol and dried in air.

Synthesis of Schiff base in ethanol (1a) and methanol (1b)
Synthesis of complexes 2–6
The syntheses of complexes were performed in methanol solution with a ratio of metal(II) acetate to the Schiff base 2:1. To a stirred hot solution of the Schiff base ((1b), 0.43 mmol, 0.2 g) in MeOH (40 mL), 0.86 mmol of appropriate acetate dissolved in methanol (10 mL) was added. The obtained mixtures were refluxed for 2 h, and after this time about 30 mL of methanol was evaporated. In all the reaction mixtures with the exception of the nickel one, solid products were formed. Precipitates were collected by filtration and washed several times with methanol. The yields of the purified complexes are given in Table 1. In the case of nickel(II) complex, the reaction mixture was allowed to cool to room temperature. After 1 h the first crystal started to appear. The reaction mixture was left in the fridge and allowed to evaporate slowly until the volume was reduced to ca. 5 mL. Then, the solution was decanted from crystals and the crystals were washed several times with methanol.
Methods and physical measurements
Elemental analysis for C, H, N was performed using a PerkinElmer CHN 2400 analyser. Magnetic susceptibilities for the powdered samples were measured over the temperature range 1.8–300 K at the magnetic field 0.1 T using a Quantum Design SQUID-VSM magnetometer. Field dependences of magnetization were measured at 2 K in an applied field up to 5 T. The corrections were based on subtracting the sample—holder signal and contribution of χ D estimated from the Pascal’s constants [23]. The molar conductivities were measured at 25 °C using a PHYWE 13701.93 conductivity metre in the DMF solvent (except for 1a and 2, which were dissolved in DMSO). The molar conductivities reported in Table 1 are corrected values. The solvent conductivity was subtracted from that of the appropriate solution in order to obtain the real conductivity of the electrolyte. The infrared spectra were recorded on a Nicolet 6700 FTIR using the ATR accessory on the ZnSe crystal for pressed powder samples. The data were collected between 4000 and 600 cm−1, with a resolution of 4 cm−1 for 16 scans. The most important bands are given in Table 2, and all FTIR spectra are presented in Supplementary material (Figs. S1-S8). The UV–Vis spectra of Schiff base ligands (1a and 1b) and metal complexes were recorded in 10 × 10 mm quartz cells over the range 200–900 nm using a Genesys 10S UV–Vis spectrophotometer (DMF solution 2.5 × 10– 5 mol L−1, except for 1a and 2, which were dissolved in DMSO), see Table 3. Thermal stability and the decomposition of the analysed complexes were determined by means of a Setaram Setsys 16/18 derivatograph, recording TG, DTG and DSC curves. The samples (6.38–7.77 mg) were heated in a ceramic crucible in the temperature range of 30–700 (Cu), 30–800 (Ni, Zn, ligand 1b), 30–850 (ligand 1a) and 30–1000 °C (Co, Mn) under flowing air atmosphere (v = 1 dm3 h−1) with a heating rate of 10 °C min−1. TG-infrared spectrometry (TG-FTIR) of the title compounds was performed using a TG Q5000 analyser interfaced to the Nicolet 6700 FTIR spectrophotometer. About 11.53–24.13 mg of compounds was put in an open platinum crucible and heated from an ambient temperature (~22 to 23 °C) to 1000 °C. The analysis was made at a heating rate of 20 °C min−1 under nitrogen atmosphere at a flow rate of 25 mL min−1. In order to reduce the possibility of gases condensing along the transfer line, the temperature in the gas cell and transfer line was set to 250 and 240 °C, respectively. The FTIR spectra were recorded in the spectral range of 600–4000 cm−1 with a resolution of 4 cm−1 and 6 scans per spectrum.
The crystallographic measurements were taken using an Oxford Diffraction Xcalibur CCD diffractometer with the graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). Data sets were collected at 100 K using the ω scan technique, with an angular scan width of 1.0°. The programs CrysAlis CCD [24] and CrysAlis Red [24] were used for data collection, cell refinement and data reduction. A multi-scan absorption correction was applied. The structures were solved by direct methods using SHELXL-2013 [25] and refined by the full-matrix least squares on F2 using SHELXL-97 [25] both operating under WinGX package program [26]. All non-H atoms were refined with anisotropic displacement parameters. Hydrogen atoms residing on carbon atoms were positioned geometrically and refined applying the riding model [with U iso(H) = 1.2 or 1.5 U eq(C)]. All remaining ones were found in the difference Fourier maps and refined with isotropic displacement parameters. The summary of crystal data, experimental details and refinement results is given in Table 4. The selected bond distances and angles are presented in Table S1 (Supplementary material). The molecular plots were drawn with ORTEP–3 for Windows [27, 28] and Mercury [29]. The geometrical calculations were performed using the PLATON program [30]. The CIF file for 4 refinement can be retrieved from the Cambridge Crystallographic Data Centre (CCDC) (deposition number CCDC 1450297).
Results and discussion
The synthesis of Schb was conducted from two different alcoholic solutions [ethanol (1a) or methanol (1b)]. In both cases solid, amorphous products differing in colour and composition were obtained (Table 1). The Schiff base synthesized from ethanol contains in its structure amine (Schb·(1.3-PDA) 0.5 ) which causes that the ligand 1a is less soluble than 1b. Therefore, the compound 1b was used in synthesis of complexes. The synthesized Schiff base has in its structure six possible coordination sites, i.e. two nitrogen atoms of azomethine groups and four oxygen atoms of the phenolic groups. For this reason, the synthesis of metal complexes was conducted through the reaction between metal ions and the ligand at 2:1 M:SchB ratio. The analysis of the power diffraction patterns of complexes (apart from 4) reveals that they, just like ligands 1a and 1b, are amorphous compounds. Despite several attempts, I was unable to obtain single crystal for them. The nickel(II) complex was obtained as crystalline solid. Compounds 1a–b and complexes 2–6 are stable in air in solid state. The molar conductivities of complexes were measured in DMF (3–6) or DMSO (2) solvents at the concentration 10−3 mol L−1. The obtained values are very low and indicate the non-electrolytic nature of complexes [31]. The elemental analyses and some physicochemical properties of the ligands (1a–b) and complexes (2–6) are shown in Table 1.
Infrared spectroscopy
The FTIR spectra of the free ligands (1a and 1b) and the complexes exhibit various bands in the 600–400 cm−1 region (see Supplementary material Figs. S1-S3). The most characteristic bands and their assignments are given in Table 2. The O–H stretching frequency of 1a and 1b is expected at 3500–3300 cm−1. In the o–hydroxy Schiff bases, this band is generally moved to lower frequencies (3200–2500 cm−1) due to the presence of strong intramolecular hydrogen bond O–H···N=C [6, 10, 11, 32]. In the studied Schiff base, the ν(O–H) vibration appears as broad bands at 2513 cm−1 for 1a and 2502 cm−1 for 1b which indicates the existence of a strong hydrogen bond between hydroxyl and azomethine groups. The medium peaks at 1339 cm−1 (1a) and 1337 cm−1 (1b) have been assigned to in-plane bending O–H vibration. Very strong bands observed at 1245 cm−1 (1a) and 1243 cm−1 (1b) are probably due to the C–O stretching vibration as reported for this type of compounds [6, 11–13, 16, 32]. The FTIR spectra of Schiff base also show a strong absorption band in the range 1690–1590 cm−1 which is characteristic of the C=N group [4, 10–12, 17]. In the spectrum of compound 1a this vibration is coupled with the ν(C=C) vibration and shifted towards lower wavelengths. For this reason, it is not observed as an independent band, but it appears as a shoulder peak at 1580 cm−1. In the case of 1b this band is better separated and observed at 1584 cm−1. In structure of 1a there is also one of the substrates, i.e. amine. The primary amines exhibit basically two stretching vibration bands due to the asymmetric and symmetric stretching vibration N–H situated in the 3500- to 3200-cm−1 region and one band due to in-plane bending vibration of N–H group at 1650–1550 cm−1. In the FTIR spectrum of 1a, the N–H stretching bands are not observed, but at 1671 cm−1 a week band is found. This band is shifted to higher values probably due to hydrogen bonding between 1,3-PDA and Schb.
The infrared spectra of complexes 2–5 exhibit a wide medium intense band in the range 3610–2500 cm−1, which points to the presence of hydrogen-bonded water or methanol molecules, and phenolic groups in their structures (Figs. S4-S8 in Supplementary material). In the case of 6 the broad band with the maximum at ~3056 cm−1 is also observed which is due to the O–H stretching vibrations of the phenolic groups involved in the hydrogen bonding between them as well as the phenolic group coordinated to zinc(II) ion. In the spectra of all complexes the band assigned to the vibration of azomethine groups, ν(C=N), is shifted to lower wavenumbers indicating participation of nitrogen atoms in coordination to metal ions. The ν(C–O) vibrations are shifted to higher frequencies (apart from 4) which suggests that coordination of metal ions takes place via phenolate and/or phenolic ortho-oxygen atoms as it is was observed for some transition metal complexes of o-hydroxy Schiff base [6, 11]. In the case of 4, the Ni(II) centre is also coordinated by phenoxide oxygen, but ν(C–O) vibration occurs at a bit lower wavenumber compared to the free ligand which is quite common for compounds of this type [4, 12, 13, 17]. In the structure of complexes 2, 3 and 6 also acetate ions occur. The bands characteristic of deprotonated carboxylic group, asymmetric and symmetric stretching vibration COO are observed at 1637–1565 and 1390–1483 cm−1, respectively. It is noteworthy that in the zinc complex spectrum the band derived from ν as(COO) is coupled with ν(C=N) vibration. In the IR spectrum of copper(II) compound at 1676 cm−1 a week band which is not found in the spectra of other complexes is observed. This IR region corresponds to the stretching C=O vibration indicating that acetic acid appears in the structure of 5. In general the C=O group of saturated aliphatic carboxylic acid absorbs in the region of 1740–1700 cm−1. The shift of this band to a lower wavelength may imply that probably carbonyl oxygen atom coordinates to copper(II) ion like in the molybdenum complexes reported by Bortoluzzi et al. [33]. It is also worth mentioning that the sharp, weak peak is observed at 3625 cm−1 which is assigned to stretching vibration of free O–H group [32]. This band can be due to the presence of free hydroxyl group of acetic acid which was observed for monomer of acetic acid by Maçôas et al. [34]. However, it cannot be ruled out that one of the phenolic groups in the para position may not be involved in any hydrogen bonds and also shows vibrations in this region [32, 35].
Electronic absorption spectra
The electronic absorption spectra were recorded in DMF (for 1b and 3–6) or DMSO (for 1a and 2, because of their lower solubility in N,N-dimethylformamide) solutions. The data are summarized in Table 3 and presented at Fig. S9 (see Supplementary material). As can be seen from the table, the spectra of 1a and 1b exhibit four bands and according to the literature [9, 15–17] these bands can be assigned as follows. The absorption bands at 256 and 283 nm for 1a and 262 and 282 nm for 1b are most probably due to the π → π * transition of the aromatic rings. The bands at 310 nm (1a) and 312 nm (1b) are assigned to the π → π * transition of the azomethine group. The last bands at 381 and 385 nm (1a and 1b, respectively) have much lower molar extinction coefficients than the previous ones and are the n → π * transition involving the promotion of a lone pair electron on the nitrogen to the antibonding π-orbital associated with the azomethine group. It is worth mentioning that differences in the positions of ligands maxima are the result of used solvents. In the spectrum of a saturated solution of 1a in DMF, the bands occur at exactly the same wavelength as for the 1b.
The spectra of the complexes show changes in relation to the position and intensity of characteristic bands in comparison with the spectrum of free ligand. The new bands, although sometimes not well defined, are also present. The π → π * transition of the aromatic ring is observed for all complexes and occurs as one (for 2–4, at 260–273 nm) or two (for 5 and 6, at 266–267 and 284–296 nm) bands. In the case of complexes where π → π * transition of the aromatic ring is present as a single band, the shift of maxima towards shorter wavelengths is observed, while for 5 and 6 both absorption maxima show a negligible red shift. Moreover, the bands at 318, 312 and 320 nm, respectively, for the complexes of Mn(III), Ni(II) and Zn(II), can be assigned to π → π * transition of the azomethine group by analogy with related metal complexes of Schiff base [12, 15, 36]. In the electronic spectra of the transition metal bands associated with a charge transfer (CT) and d–d transitions are also observed. The charge transfer vibration is combined with n → π * /π → π * transition of (C=N) and observed as one bands at 346–430 nm for all complexes with exception of 4 [13, 16, 17, 37–41]. In the case of nickel(II) complex the CT transitions are observed at 343 and 409 nm [5, 15, 36]. The bands characteristic of the d–d transitions are not observed on spectra of 2–5 at the applied concentration due to their low intensity. In more concentrated solutions (10−3 mol L−1), the low intensity bands assigned to d–d transitions appear for 3 (575 nm), 4 (584 nm) and 5 (604 nm). The presence of only one d–d band at these wavelengths indicates that the coordination environment of copper(II) and nickel(II) ions in solution is square planar, while geometry of cobalt(II) is tetrahedral [7, 15, 36, 40, 42].
Structure of nickel(II) complex
Complex 4 was obtained as a crystalline substance which made it possible to carry out a single-crystal X-ray analysis. The molecular structure of complex 4 is displayed in Fig. 1. The crystallographic and refinement data for 2 are summarized in Table 4. The selected bond distances and angles are listed in Table S1, and the hydrogen bond parameters are given in Table S2 (see Supplementary material). The nickel(II) complex crystallizes in the monoclinic space group P21/c. In this complex, the Ni(II) metal centre is coordinated by a pair of phenolate O (O1 and O3) and imine N (N1 and N2) atoms from the Schiff base ligand. The square-planar N2O2 coordination geometry is a slightly tetrahedrally distorted as indicated by less than linear bond angles (171.97(10) O(1)–Ni1–N(2) and 171.78(9) O(3)–Ni1–N(1)) and the relatively small dihedral angle between the two N–Ni–O planes (θ = 9.09°). The Ni(II) atom lies practically in the plane of the N2O2 core (with a small rms deviation of 0.005(1) Å). In the coordination plane, Ni–O (1.834(2), 1.844(2) Å) and Ni–N (1.881(2), 1.885(2) Å) bond distances are similar to the corresponding distances observed in other Schiff base nickel(II) complexes with square-planar environment of metal centre [43–46]. It is worth mentioning that in the case of the increasing coordination number, the increase in Ni–O and Ni–N bond lengths is observed; e.g. the Ni–O and Ni–N bond distances 1.936(2)–1.963(3) Å and 1.953(2)–2.000(3) Å [47, 48] for a square pyramidal and 1.980(11)–2.039(2) Å and 2.032(15)–2.071(2) Å [5, 49, 50] for octahedral coordination geometries have been reported. Methanol molecules are linked to Ni(C29H24N2O4) molecule via intermolecular O–H···O hydrogen bonds with phenoxy and phenolic oxygen atoms as proton acceptors (Table S2 in Supplementary material). What is more, one methanol molecule is engaged in the formation of dimeric units through O2–H2P···O6 and O6–H6 M···O1 hydrogen bonds where it is both the donor and acceptor of proton, generating cyclic hydrogen-bonded motifs with the graph-set notation R 44 (16) [51], see Fig. S10 in Supplementary material. These dimeric units are further linked via another O4–H4P–O3i (symmetry code (i): x, −y + 1/2, z − 1/2) hydrogen bonds, forming a layer parallel to the bc plane (Figure S11). The structure is also stabilized by weak intermolecular C–H···O hydrogen bonds and C–H···π(ring) interactions. The presence of weak intermolecular C–H···O hydrogen bonds leads to the formation of a three-dimensional network, see Fig. S12 in Supplementary material. The intermolecular C–H···Cg contact (Table S2 given in Supplementary material) occurs between the centroid of the phenyl ring (Cg4, centroid of the phenyl formed by C1–C6) and phenyl hydrogen atom [C13-H13···Cg6iv, symmetry code (iv): −x, 1 − y, 1 − z] and stabilizes structure by creating other dimeric units.

ORTEP view with the atom numbering scheme of 4. Displacement ellipsoids are drawn at the 50 % probability level
Thermal analysis
Thermal analyses of studied compounds (1a–b) and complexes (2–6) were carried out by the TG-DSC (air) and TG-FTIR (nitrogen) techniques. The TG-DSC curves providing information about thermal properties of the compounds are shown in Figs. 2 and 3.

a TG, DTG and DSC curves of 1a in air, b TG, DTG and DSC curves of 1b in air, c TG curve of 1a in nitrogen, d FTIR spectra of gaseous products evolved during the decomposition of 1a

a–e TG, DTG and DSC curves of 2–6 in air, f TG curves of 2–6 in nitrogen
Ligands 1a and 1b
Compounds 1a and 1b have high thermal stability which depends on the alcohol used for the synthesis. In the case of compound 1a the first change on the TG curve recorded in air atmosphere has been found at 195 °C with an initial mass loss of 7.21 % which is in good agreement with the theoretical value of 7.36 % attributed to the loss of molecule 1,3-PDA, Fig. 2a. The deamination process is connected with an endothermic effect recorded on the DSC curve of 1a. The enthalpy of this process is equal to 57.39 ± 0.35 kJ mol−1. The same process is also observed on the TG curve recorded in nitrogen atmosphere (see Fig. 2c), but the initial mass loss is a bit higher (8.06 %) than the calculated one. This may suggest that in an inert atmosphere the partial decomposition of the Schiff base also appeared. The FTIR spectra of the evolved gas phase confirm that 1,3-PDA and its products as well as partial SchB decomposition (CO2, NH3 and H2O) are released in this stage (Fig. 2d). Further thermal decomposition of 1a in air proceeds in two stages. The first stage, the partial destruction and combustion of the Schiff base, starts at 265 °C, and intermediate, unstable products are formed. This step occurs together with the melting process. The DSC peak attributed to melting accompanied by the negligible mass loss is recorded at 264 °C, the melting point determined from the DSC curve is 268 °C, and the enthalpy of fusion is 30.90 ± 0.12 kJ mol−1. The second stage above 395 °C corresponds to the complete destruction and combustion of the remaining parts of the compound. Compound 1a fully decomposed at 678 °C. The thermal decomposition of 1a in nitrogen after the deamination process is a bit different from the one observed in air. The first step of compounds decomposition starts at 270 °C, and in this stage more than fifty per cent of mass loss is observed (see Fig. 2d; Table 5). Similar to thermal decomposition of 1a in air, this step is connected with formation of intermediate, unstable products. At the beginning of the first stage of decomposition process the main gaseous products emitted at around 245–330 °C are identified as: H2O (4000–3450 and 1950–1300 cm−1) [52–54], CO2 (peaks at 2450–2300 and 750–600 cm−1) [16, 17, 54] and NH3 (several bands with two characteristic maxima at 966 and 931 cm−1) [16, 17, 54], Table 6. These gaseous products indicate that probably this step can be associated with the pyrolysis of the Schiff base fragment containing amine and benzene rings with phenolic groups. At a higher temperature (~335 °C), the weak bands characteristic of benzonitrile are recorded, and the bands corresponding to C6H5CN are observed in the range 3100–3000 (ν(C–H)), 2275–2200 (ν(C≡N)) and 825–650 cm−1(δ(Car = Car), γ(CC≡N)) [17]. At the end of the first step of decomposition around 17 min (~350 °C) several bands characteristic of toluene were recorded. The bands assigned to toluene occur in the ranges 3200–2800 cm−1 (with four maxima at 3072, 3032, 2933 and 2885 cm−1) and 800–600 cm−1 (with the maxima at 728 and 692 cm−1). The other bands characteristic of toluene in the range of 2000–1000 cm−1 do not have sharp marked maxima because these vibrations are overlapped with those of water and ammonia molecules. As it was mentioned previously, the first step is connected with formation of unidentified, unstable product which immediately undergoes further thermal decomposition. The second stage of ligand pyrolysis starts around 430 °C, and initially it is associated with the release of similar products as they were observed at the end of the first stage, i.e. H2O, CO2, NH3 and toluene. At a higher temperature (ca. 485 °C) the peaks characteristic of CO (double peaks at 2275–2050 cm−1 [16, 17, 52, 54]) and benzene (in the ranges 3150–300, 1550–1450 and 1050–1000, and at 692, 672 and 653 cm−1 [52]) bands are recorded. With the increasing temperature, the intensity of the bands associated with the vibration of C6H6 and C6H5CH3 in the range 3200–2800 cm−1 decreases and the bands characteristic of methane are noted. The bands characteristic of CH4 are observed at ~525 °C in the range of 3175–2900 cm−1 with a characteristic maximum at 3016 cm−1 [16, 17, 54]. Probably methane is released at a lower temperature, but the vibrations are covered by the bands of benzene and toluene. At the end of this step (above 600 °C) in the FTIR spectra the bands characteristic of CO, CO2, H2O, NH3 and CH4 are only recorded, but ammonium and methane vibrations disappear below 800 °C. In contrast to the thermal decomposition in air atmosphere, here the decomposition process of 1a is not finished. The residue remaining after the analysis at 1000 °C is 32.71 %, and probably this is a mixture of carbon with hydrocarbons.
Thermal behaviour of 1b was studied in air atmosphere (see Fig. 2b). The TG-DSC curves show that the studied compound is thermally stable under air stream up to ~280 °C. The compound shows its first DSC peak (attributed to melting accompanied by negligible mass loss) at 282 °C. This endothermic peak is connected with melting process; the melting point and the enthalpy of fusion are 284 °C and 37.06 ± 0.23 kJ mol−1, respectively. This DSC peak is sharp which indicates that this is a pure substance. The decomposition of 1b in air is a two-step process. The first mass loss step is observed at the temperature 282 °C and occurred together with the melting process. This stage is connected with the partial destruction and combustion of 1b. Within this first step of 1b decomposition less than thirty per cent of mass loss is observed (Table 5), and an intermediate, unstable product is formed. The second stage above 430 °C corresponds to the complete destruction and combustion of the remaining parts of the compound. Compound 1b fully decomposed at a temperature of approximately 660 °C.
Complexes 2–6
The thermal degradation of manganese(III) complex in air atmosphere starts with an initial well-separated mass loss in the temperature range of 60–140 °C with an endothermic effect recorded on the DSC curve, see Fig. 3a. This stage corresponds to the desolvation process. On the basis of the TG-FTIR analysis, it was found that in this stage water molecules are released. A calculated mass loss due to removal of all water molecules is 5.86 %, while the measured mass loss is 5.37 %. The enthalpy of dehydration process is equal to 47.29 ± 0.42 kJ mol−1. The anhydrous compound is unstable and immediately undergoes thermal decomposition into Mn2O3 (found/calcd. overall mass losses: 87.17/87.15 %). At a high temperature (927–970 °C), this oxide is transformed into Mn3O4 as it can been observed in the previous work [55]. Thermal behaviour of 2 was also studied in nitrogen atmosphere together with the infrared spectroscopy analysis of gaseous decomposition products (Figs. 3f, 4a). The first step is analogous to that recorded in air atmosphere and corresponds to the loss of water molecules (found mass loss 5.25 %). As it has already been mentioned the FTIR spectra of the evolved gaseous products confirm the presence of water molecules in the composition of Mn(II) complex, see Fig. 4a. The characteristic valence and deformation vibration bands of water molecules appear at 45 °C. As in the case of thermal decomposition in air, anhydrous compound is immediately pyrolysed. At the beginning of the first stage of decomposition process in nitrogen the elimination of acetate anions as acid molecules is observed. The weak bands characteristic of acetic acid (ν(OH): 3595, 3580 and 3566 cm−1; ν(CH3): 3044, 2997 and 2969 cm−1; ν(C=O): 1797 and 1774 cm−1; δ(CH3): 1398 cm−1; ν(C–O): 1266 cm−1; δ(COH): 1177 cm−1; γ(CH3): 1087 and 999 cm−1; δ(OH): 979 cm−1; ν(C–C): 859 and 835 cm−1; τ(C–O): 656 and 642 cm−1 [56, 57]) appear in the FTIR spectra of the gaseous products at 110 °C. With the increasing temperature the intensity of the bands associated with the release of acid grows, but at 150 °C the bands characteristic of CO2 are also observed. The appearance of these vibrations with a simultaneous increase in the intensity of the bands derived from water molecules may be due to degradation of acetic acid as well as decomposition of the Schiff base. This is confirmed by the presence of bands characteristic of ammonia, and associated with defragmentation of SchB, which are visible when the intensity of the acetic acid bands decreases (~150 °C). At a higher temperature, the bands characteristic of benzonitrile (340 °C), toluene (380 °C) and methane (400 °C) are also recorded. It can be observed from the TG curve that the decomposition process was not completed in nitrogen atmosphere.

a FTIR spectra of gaseous products evolved during the decomposition of 2, b FTIR spectra of gaseous products evolved during the decomposition of 6
The complex of cobalt(II) is stable at room temperature. The first change of mass on the TG curve of 3 recorded in air is found above 49 °C, see Fig. 3b. The observed mass loss for this step is 6.69 % and is connected with an endothermic peak on a DSC curve (88.23 ± 0.45 kJ mol−1). Similar to the previously described complex this stage is related to the dehydration process. In this stage three molecules of water are lost (calcd. value 6.48 %). The remaining molecule of water is eliminated above 137 °C (found/calcd. mass losses: 1.88/2.16 %). The anhydrous compound is very unstable and immediately undergoes degradation into Co3O4 (found/calcd. overall mass losses: 80.90/80.74 %). Within this stage on the TG curve around 236 °C there can be seen an inflection point which corresponds to the loss of 9.11 %. This can be associated mainly with the partial decomposition of acetate anion (calcd. mass loss: 7.09, 14.17 and 21.25 % for a loss of 1, 2 and 3 acetate molecules, respectively). Above 907 °C Co3O4 transforms into CoO. Thermal behaviour of 3 in nitrogen begins, as in the air, with the two-step dehydration. The first stage is recorded at 38–110 °C, the second one at 120–150 °C. On the FTIR spectra of the evolved gaseous products up to 150 °C the bands characteristic of water molecules are only observed. The anhydrous complex is unstable and immediately decomposes. The thermal decomposition proceeds in two steps, and this process is not completed under measurement conditions (Fig. 4f). The first stage of pyrolysis is connected with the elimination of acetate anions as the acid whose vibrations are noted on the FTIR spectra together with trace products of its decomposition, i.e. H2O and CO2. Here, in contrast to thermal decomposition in air, this step is better marked on the TG curve and a greater part of acetate ions is eliminated (found mass loss: 15.65 %). The second step of thermal degradation of complex starts above 318 °C and initially is related to the removal of the remaining parts of acetate ion. The first bands indicating the Schiff base degradation, i.e. the bands of NH3 and CH4 molecules, appear on the FTIR spectra above 360 °C. Further heating causes the increase in the emission of these gaseous species, and new products are observed, i.e. benzene, benzonitrile and phenol. The most characteristic, qualitative bands for C6H5OH are observed at 3700–3600, 3150–3000, 1650–1100 and 850–650 cm−1 [17]. Above 560 °C only CO2, H2O and CO molecules are present in gaseous products.
Decomposition of nickel(II) complex in air is a two-step process as visible on the TG curves, see Fig. 3c. Thermal degradation of compound 4 starts with an initial well-separated mass loss of 5.60 %, between 50 and 143 °C and corresponding to the loss of one methanol molecule per formula unit (calcd. 5.45 %). The desolvation process is connected with an endothermic effect recorded on the DSC curve, and the enthalpy of this process is equal to 52.50 ± 0.41 kJ mol−1. The last methanol molecule present in the structure of 4 is removed at a higher temperature (297 °C) together with the degradation and combustion of the Schb ligand. The final product was a grey-black powder which indicates that Ni2O3 was formed (found/calcd. overall mass losses: 86.01/85.92 %) [58, 59]. The thermal behaviour of 4 in nitrogen together with the analysis of gaseous products was also investigated. As can be seen in Fig. 3f, the first stage starts at a slightly higher temperature than in the air and takes longer to 250° C. What is more, a greater loss in mass, 7.12 %, is registered on the TG curve. However, this value is still lower than those calculated for the loss of two molecules of methanol, i.e. 10.45 %. The next stage on the TG curve is observed in the range 330–975 °C and corresponds to removal of the remaining part of CH3OH from the crystal structure as well as destruction of the organic ligand. The desolvation process at such a high temperature was confirmed by the FTIR analysis of gaseous products. The traces of methanol as well as its decomposition products are recorded on the FTIR spectra up to about 350 °C. The characteristic vibration bands of CH3OH are observed in the wavenumber ranges 3750–3600, 3150–2750 and 1100–950 cm−1 [16, 17]. The beginning of the second stage is also associated with the release of ammonia molecules. The most characteristic, qualitative bands for NH3 are already observed at 320 °C. The FTIR spectra of the gas phase products indicate that further decomposition of 4 is connected with release of H2O, CO2, CO, toluene, benzene, benzonitrile and methane molecules as a consequence of ligand defragmentation, see Table 6. The final stable product has not been identified. It is formed at 975 °C, and this can be a mixture of nickel phenolates. The residue left after decomposition is 44.13 %, while the values calculated for the individual compounds are as follows: 20.19 %—NiCO3, 30.75 %—Ni(HCO3)2, 41.65 %—Ni(C6H5O)2 and 47.09 %—Ni(HOC6H4O)2.
The copper(II) complex has comparable thermal stability as the other hydrates. The dehydration process starts around 49 °C and occurs in one step during which all water molecules are lost (found/calcd. mass loss: 6.12/6.06 %). This process is connected with an endothermic effect recorded on the DSC curve of 5, Fig. 5d. The enthalpy of this process is equal to 237.64 ± 0.42 kJ mol−1. Further thermal decomposition takes place in two stages. The first step is observed at 246–370 °C. At the beginning, this is probably related to elimination of acetate ion from the structure (calcd. mass loss: 4.96 %), but with the increasing temperature, combustion and decomposition of the Schiff base ligand are also observed (found mass loss: 13.98 %), as evidenced by two maxima on the DTA curve. The second stage starts at 373 °C and corresponds to a complete degradation and combustion of the Schiff base ligand. The final product CuO is formed at 537 °C (found/calcd. total mass losses: 86.93/86.96 %). The first change in the mass on the TG curve recorded in nitrogen is found above 37 °C, and similar to the thermal behaviour of 5 in air, it is related to the dehydration process. The FTIR spectra of the evolved gas phase confirm that water is the only product released in this stage. Further decomposition of 5 in nitrogen is observed at a lower temperature (190 °C) compared to decomposition in air (248 °C). On the basis of the FTIR spectra analysis of the evolved gas phase, it was found that in this stage part of acetic acid molecules and its decomposition products (CO2 and H2O) are released. The found mass loss is 1.13 %, and this is about 25 % of total amount of acetic acid present in structure of 5. The next stage is observed at 270 °C, and at the beginning it is connected with further elimination of remaining parts of acid. The degradation of the Schiff base is observed above 300 °C. The gaseous products, which are released and recorded on the FTIR spectra during decomposition of Schb, are as follows: CO2, H2O, NH3, benzonitrile, benzene, phenol, CO and CH4. Most of these gases disappear below ~550 °C, and on the FTIR spectra of evolved volatile pyrolysis products mainly bands of carbon oxide (up to ~700 °C), methane (up to ~720 °C), water and carbon dioxide were recorded (Table 6). Similarly to the other complexes, thermal decomposition is incomplete under measurement conditions; the found total mass loss at 1000 °C is 59.86 %. The zinc(II) complex is stable at room temperature, Fig. 3e. In air atmosphere, the decomposition process of 6 proceeds in one step and starts at 260 °C. The final product is ZnO (found/calcd. overall mass loss: 86.19/86.21 %). Thermal stability of 6 in nitrogen is similar to that in air; i.e. the decomposition process starts at the same temperature. The first stage begins with the release of molecule of acetic acid which is the only degradation product of the complex up to 330 °C, Fig. 4b. Above this temperature on the FTIR spectra bands characteristic of ammonia and associated with the thermal decomposition of Schiff base can be observed. The beginning of the fragmentation of the Schiff base is also associated with the release of carbon dioxide (340 °C), water (340 °C), carbon monoxide (365 °C) and benzonitrile (365 °C). At the end of this step (above 455 °C) in FTIR spectra the bands characteristic of toluene and benzene are also recorded. On the FTIR spectra of gaseous decomposition products of 6, contrary to the other compounds, bands characteristic of methane were not observed. However, it cannot be definitely stated that there is no methane in the evolved gas because its vibrations may be overlapped by C6H6, C6H5CN and C6H5CH3 bands.

Temperature dependence of the magnetic moment for 2 (circle), 3 (square) and 5 (triangle)
Magnetic properties
The magnetic behaviours of paramagnetic complexes, i.e. Mn(III), Co(II) and Cu(II), are shown in Fig. 5. Temperature-dependent molar susceptibility measurements of powdered samples 2, 3 and 5 were taken in an applied magnetic field of 0.1 T over the temperature range 1.8–300 K. The plot of reciprocal magnetic moment against temperature of 2 is shown in Fig. 5. At room temperature the magnetic moment is 5.94 μB, and it is higher than the expected value for manganese(III) (4.90 μB), but such height values were observed before [60–62]. As the temperature was reduced, the magnetic moment was roughly constant ca. 180 K, then decreased slowly to 5.58 μB (70 K) and after that drop abruptly reached the value 3.73 μB at 1.8 K. The decrease of µ eff at low temperature is due to the zero-field splitting term of manganese(III) ion and/or an intermolecular antiferromagnetic interactions [60–62]. The χ M T value keeps decreasing, while the temperature decreases from 300 to 1.8 K which indicates that a possible weak antiferromagnetic interaction exists between Mn(III) ions. The corresponding χ versus T curve is Curie–Weiss like in shape, with an inflection occurring at ca. 7 K. The Weiss and Curie constants are θ = −12 K and C = 4.60 cm3 K mol−1. The negative value of the Weiss constant θ confirms the antiferromagnetic interactions between Mn(III) ions [63]. The magnetic moment of 3 at room temperature is 4.55 μ B (300 K). This value is characteristic of high-spin complex of Co(II) and decreases smoothly up to 3.84 μ B (1.8 K). The magnetic moment value of 3 at room temperature differs slightly from the spin-only moment which is observed for tetrahedral symmetry. The octahedral complexes typically have a moment of 4.7–5.2 μB [63]. The χ M T value decreases slowly with the decrease of temperature from 300 to 1.8 K which indicates the existence of antiferromagnetic coupling. The negative value of Weiss constant θ (−5.9 K) confirms the antiferromagnetic interactions between Co(II) ions [63]. The temperature dependence of the magnetic moment of 5 is shown in Fig. 5. The μ eff value at room temperature (300 K) is 1.90 μB, and it is a little higher than the expected, spin-only value for uncoupled copper(II) (1.73 μB) ions and is indicative of the presence of ferromagnetic intradimer interactions [63–65]. Moreover, as the temperature was decreased, the magnetic moment increased slowly to a maximum of 2.26 at 12 K and then decreased slightly to 2.19 at 1.8 K which confirms weak ferromagnetic coupling [64]. Compound 5 shows also behaviour typical of ferromagnetic coupling, with the χ M T versus T (K) profiles which involves an increase in the χ M T value as temperature is lowered, followed by a rapid drop at low temperatures (~7 K) [63, 65].
Conclusions
In summary, this paper deals with the synthesis and characterization of the Schiff base ligand derived from 2,4-benzophenone and their manganese(III), cobalt(II), nickel(II), copper(II) and zinc(II) complexes. The compounds were characterized by using different physicochemical techniques. The ligands (1a and 1b) and complexes (2–6) are stable at room temperature. The degradation of the Schiff base occurs in consecutive reactions where probably the destruction of azomethine double bonds firstly takes place which is confirmed by the TG-FTIR analysis; the ammonia bands appear as the first on the FTIR spectra of evolved gases. In the case of complexes (2–5), the first step is connected with the desolvation process. The TG-FTIR analysis confirms the presence of water (for 2, 3 and 5) or methanol (for 4) molecules in their structures. Next, the acetate ions (2, 3 and 6) or acetic acid (5) is removed from the structures of complexes which is confirmed by the presence of acetic acid bands in the FTIR spectra of gaseous products. At the end the Schiff base undergoes defragmentation and combustion (air) or pyrolysis (nitrogen). The heating in nitrogen resulted in evolution of: CO2, CO, NH3, acetic acid, methane, benzene, acetonitrile and toluene/phenol molecules as volatile products of Schiff base ligand thermal decomposition. Variable temperature magnetic studies of the paramagnetic complexes exhibit the existence of antiferromagnetic (for 2 and 3) or ferromagnetic (for 5) interactions.
References
Wichmann O, Sillanpää R, Lehtonen A. Structural properties and applications of multidentate [O, N, O, X′] aminobisphenolate metal complexes. Coord Chem Rev. 2012;256:371–92.
Zayed EM, Mohamed GG, Hindy AMM. Transition metal complexes of novel Schiff base. J Therm Anal Calorim. 2015;120:893–903.
Poddel’sky AI, Cherkasov VK, Abakumov GA. Transition metal complexes with bulky 4,6-di-tert-butyl-N-aryl(alkyl)-o-iminobenzoquinonato ligands: structure, EPR and magnetism. Coord Chem Rev. 2009;253:291–324.
Cristóvão B, Miroslaw B. Propeller-like heterotrinuclear CuII–LnIII–CuII compounds—physicochemical properties. Inorg Chim Acta. 2013;401:50–7.
Cristóvão B, Kłak J, Miroslaw B. Synthesis, crystal structures and magnetic behavior of NiII–4f–NiII compounds. Polyhedron. 2012;43:47–54.
Abdel Aziz AA, Salem ANM, Sayed MA, Aboaly MM. Synthesis, structural characterization, thermal studies, catalytic efficiency and antimicrobial activity of some M(II) complexes with ONO tridentate Schiff base N-salicylidene-o-aminophenol (saphH2). J Mol Struct. 2012;1010:130–8.
Anthonysamy A, Balasubramanian S, Usha G, Velmurugan D. Synthesis, spectroscopic, structural and electrochemical studies of nickel(II) and copper(II) complexes derived from 2-hydroxy-4-methacryloyloxybenzaldehyde. Transit Met Chem. 2008;33:681–90.
Bartyzel A, Cukrowska EM. Solid phase extraction method for the separation and determination of chromium(III) in the presence of chromium(VI) using silica gel modified by N,N′-bis-(alpha-methylsalicylidene)-2,2-dimethyl-1,3-propanediimine. Anal Chim Acta. 2011;707:204–9.
Galic N, Cimerman Z, Tomisic V. Spectrometric study of tautomeric and protonation equilibria of o-vanillin Schiff base derivatives and their complexes with Cu(II). Spectrochim Acta A Mol Biomol Spectrosc. 2008;71:1274–80.
Tozzo É, Romera S, Santos MP, Muraro M, Santos RHA, Lião LM, Vizotto L, Dockal ER. Synthesis, spectral studies and X-ray crystal structure of N,N′-(±)-trans-1,2-cyclohexylenebis(3-ethoxysalicylideneamine) H-2(t-3-EtOsalchxn). J Mol Struct. 2008;876:110–20.
Ambrozini B, Dockal E, Cavalheiro É. Thermal behavior of tetradentate Schiff base chromium(III) complexes. J Therm Anal Calorim. 2014;115:979–86.
Olar R, Badea M, Ferbinteanu M, Stanica N, Alan I. Spectral, magnetic and thermal characterization of new Ni(II), Cu(II), Zn(II) and Cd(II) complexes with a bischelate Schiff base. J Therm Anal Calorim. 2016;. doi:10.1007/s10973-016-5433-7.
Ebrahimi HP, Hadi JS, Abdulnabi ZA, Bolandnazar Z. Spectroscopic, thermal analysis and DFT computational studies of salen-type Schiff base complexes. Spectrochim Acta A Mol Biomol Spectrosc. 2014;117:485–92.
Cozzi PG. Metal-Salen Schiff base complexes in catalysis: practical aspects. Chem Soc Rev. 2004;33:410–21.
Glaser T, Heidemeier M, Fröhlich R, Hildebrandt P, Bothe E, Bill E. Trinuclear nickel complexes with triplesalen ligands: simultaneous occurrence of mixed valence and valence tautomerism in the oxidized species. Inorg Chem. 2005;44:5467–82.
Bartyzel A, Kaczor A. The formation of a neutral manganese(III) complex containing a tetradentate Schiff base and a ketone—synthesis and characterization. J Coord Chem. 2015;68:3701–17.
Bartyzel A. Synthesis, crystal structure and characterization of manganese(III) complex containing a tetradentate Schiff base. J Coord Chem. 2013;66:4292–303.
Hadjoudis E, Mavridis IM. Photochromism and thermochromism of Schiff bases in the solid state: structural aspects. Chem Soc Rev. 2004;33:579–88.
Minkin VI, Tsukanov AV, Dubonosov AD, Bren VA. Tautomeric Schiff bases: iono-, solvato-, thermo- and photochromism. J Mol Struct. 2011;998:179–91.
Suzuki T, Kitamura S, Khota R, Sugihara K, Fujimoto N, Ohta S. Estrogenic and antiandrogenic activities of 17 benzophenone derivatives used as UV stabilizers and sunscreens. Toxicol Appl Pharmacol. 2005;203:9–17.
Black R, Billing D, Bartyzel A, Cukrowska E. 2,2′-[(Propane-1,3-diyldinitrilo)bis(phenylmethylidyne)]diphenol. Acta Crystallogr Sect E. 2010;66:o1002–3.
Black R, Billing D, Bartyzel A, Cukrowska E. N,N′-Bis[(2-hydroxyphenyl)(phenyl)methylidene]propane-1,2-diamine. Acta Crystallogr Sect E. 2010;66:o1256–7.
Bain GA, Berry JF. Diamagnetic corrections and Pascal’s constants. J Chem Educ. 2008;85:532.
CrysAlis PRO, Agilent Technologies Ltd., Yarnton, Oxfordshire, UK;2013.
Sheldrick GM. A short history of SHELX. Acta Crystallogr Sect A. 2008;64:112–22.
Farrugia LJ. WinGX suite for small-molecule single-crystal crystallography. J Appl Crystallogr. 1999;32:837–8.
Farrugia LJ. ORTEP-3 for Windows-a version of ORTEP-III with a graphical user interface (GUI). J Appl Crystallogr. 1997;30:565.
Farrugia LJ. WinGX and ORTEP for windows: an update. J Appl Cryst. 2012;45:849–54.
Macrae CF, Edgington PR, McCabe P, Pidcock E, Shields GP, Taylor R, Towler M, van de Streek J. Mercury: visualization and analysis of crystal structures. J Appl Crystallogr. 2006;39:453–7.
Spek A. Single-crystal structure validation with the program PLATON. J Appl Crystallogr. 2003;36:7–13.
Geary WJ. Use of conductivity measurements in organic solvents for characterisation of coordination compounds. Coord Chem Rev. 1971;7:81–122.
Socrates G. Infrared and Raman characteristic group frequencies: tables and charts. Chichester: Wiley; 2004.
Bortoluzzi M, Marchetti F, Pampaloni G, Zacchini SY. Oxido-molybdenum complexes obtained by Cl/O interchange between MoCl5 and carboxylic acids: a crystallographic, spectroscopic and computational study. Dalton Trans. 2014;16416–16423.
Maçôas EMS, Khriachtchev L, Pettersson M, Fausto R, Räsänen M. Rotational isomerism in acetic acid: the first experimental observation of the high-energy conformer. J Am Chem Soc. 2003;125:16188–9.
Keresztury G, Billes F, Kubinyi M, Sundius T. A density functional, infrared linear dichroism, and normal coordinate study of phenol and its deuterated derivatives: revised interpretation of the vibrational spectra. J Phys Chem A. 1998;102:1371–80.
Bosnich B. An interpretation of the circular dichroism and electronic spectra of salicylaldimine complexes of square-coplanar diamagnetic nickel(II). J Am Chem Soc. 1968;90:627–32.
Gupta SK, Hitchcock PB, Kushwah YS, Argal GS. Synthesis, structure and DNA binding studies of a mononuclear cobalt(III) complex with a NNO donor Schiff base derived from 4-methyl-2,6-dibenzoylphenol and ethane-1,2-diamine. Inorg Chim Acta. 2007;360:2145–52.
Amirnasr M, Mahmoudkhani AH, Gorji A, Dehghanpour S, Bijanzadeh HR. Cobalt(II), nickel(II), and zinc(II) complexes with bidentate N,N′-bis(β-phenylcinnamaldehyde)-1,2-diiminoethane Schiff base: synthesis and structures. Polyhedron. 2002;21:2733–42.
Şenol C, Hayvali Z, Dal H, Hökelek T. Syntheses, characterizations and structures of NO donor Schiff base ligands and nickel(II) and copper(II) complexes. J Mol Struct. 2011;997:53–9.
Mustafa IM, Hapipah MA, Abdulla MA, Ward TR. Synthesis, structural characterization, and anti-ulcerogenic activity of schiff base ligands derived from tryptamine and 5-chloro, 5-nitro, 3,5-ditertiarybutyl salicylaldehyde and their nickel(II), copper(II), and zinc(II) complexes. Polyhedron. 2009;28:3993–8.
Back DF, de Oliveira GM, Fontana LA, Ramão BF, Roman D, Iglesias BA. One-pot synthesis, structural characterization, UV–Vis and electrochemical analyses of new Schiff base complexes of Fe(III), Ni(II) and Cu(II). J Mol Struct. 2015;1100:264–71.
Calu L, Badea M, Chifiriuc M, Bleotu C, David G-I, Ioniţă G, Măruţescu L, Lazăr V, Stanică N, Soponaru I, Marinescu D, Olar R. Synthesis, spectral, thermal, magnetic and biological characterization of Co(II), Ni(II), Cu(II) and Zn(II) complexes with a Schiff base bearing a 1,2,4-triazole pharmacophore. J Therm Anal Calorim. 2015;120:375–86.
Allen FH. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr B. 2002;58:380–8.
Kargar H, Kia R, Moghadam M, Froozandeh F, Tahir MN. {2,2′-[(2,2-Dimethylpropane-1,3-diyldinitrilo)bis(phenylmethylidyne)]diphenolato}nickel(II). Acta Crystallogr Sect E. 2011;67:m1173.
Akine S, Varadi Z, Nabeshima T. Synthesis of planar metal complexes and the stacking abilities of naphthalenediol-based acyclic and macrocyclic salen-type ligands. Eur J Inorg Chem. 2013;2013:5987–98.
Habibi MH, Mokhtari R, Harrington RW, Clegg W. [N,N′-Bis(6-methoxysalicylidene)-1,3-diaminopropane]nickel(II). Acta Crystallogr Sect E. 2007;63:m2304.
Guo Z, Li L, Xu T, Li J, Wang D. Aqua{6,6′-dimethoxy-2,2′-[ethane-1,2-diylbis(nitrilomethylidyne)]diphenolato}nickel(II). Acta Crystallogr Sect E. 2009;65:m1158–9.
Mukherjee T, Costa Pessoa J, Kumar A, Sarkar AR. Synthesis, structure, magnetic properties and biological activity of supramolecular copper(II) and nickel(II) complexes with a Schiff base ligand derived from vitamin B6. Dalton Trans. 2013;42:2594–607.
Gomes L, Sousa C, Freire C, Castro BD. Diaqua{6,6′-dimethoxy-2,2′-[propane-1,3-diylbis(nitrilomethylidyne-N)]diphenolato-O, O’}nickel(II). Acta Crystallogr Sect C. 2000;56:1201–3.
Datta A, Machura B, Huang J-H, Sheu S-C. Diaqua{2,2′-dimethoxy-6,6′-[(1E,1′E)-propane-1,3-diylbis(azanylylidene)bis(methanylylidene)]diphenolato}nickel(II). Acta Crystallogr Sect E. 2013;69:m399.
Bernstein J, Davis RE, Shimoni L, Chang N-L. Patterns in hydrogen bonding: functionality and graph set analysis in crystals. Angew Chem Int Ed Engl. 1995;34:1555–73.
Łyszczek R, Ostasz A, Bartyzel A, Lipke A. Thermal, spectroscopic and luminescence investigations of lanthanide(III) coordination polymers based on V-shaped 4,4′-sulfonyldibenzoic acid. J Anal Appl Pyrolysis. 2015;115:370–8.
Zong G, Ren N, Zhang J, Qi X, Gao J. Lanthanide complexes with 3-bromine-4-methyl benzoic acid and 1,10-phenanthroline. J Therm Anal Calorim. 2016;123:105–16.
Bartyzel A, Sztanke M, Sztanke K. Thermal studies of analgesic active 8-aryl-2,6,7,8-tetrahydroimidazo[2,1-c][1,2,4]triazine-3,4-diones. J Therm Anal Calorim. 2016;123:2053–60.
Rzaczyńska Z, Bartyzel A, Glowiak T. Synthesis and characterization of triaquabis(1,1-cyclobutanedicarboxylato-O,O′,O″,O′′′)dimanganese(II). Polyhedron. 2003;22:2595–9.
Maçôas EMS, Khriachtchev L, Fausto R, Räsänen M. Photochemistry and vibrational spectroscopy of the trans and cis conformers of acetic acid in solid Ar. J Phys Chem A. 2004;108:3380–9.
Taghizadeh MT, Yeganeh N, Rezaei M. Kinetic analysis of the complex process of poly(vinyl alcohol) pyrolysis using a new coupled peak deconvolution method. J Therm Anal Calorim. 2014;118:1733–46.
Brockner W, Ehrhardt C, Gjikaj M. Thermal decomposition of nickel nitrate hexahydrate, Ni(NO3)2·6H2O, in comparison to Co(NO3)2·6H2O and Ca(NO3)2·4H2O. Thermochim Acta. 2007;456:64–8.
Masoud MS, Ali AE, Shaker MA, Elasala GS. Synthesis, computational, spectroscopic, thermal and antimicrobial activity studies on some metal–urate complexes. Spectrochim Acta A Mol Biomol Spectrosc. 2012;90:93–108.
Kennedy BJ, Murray KS. Magnetic properties and zero-field splitting in high-spin manganese(III) complexes. 1. Mononuclear and polynuclear Schiff-base chelates. Inorg Chem. 1985;24:1552–7.
Batten SR, Murray KS. Structure and magnetism of coordination polymers containing dicyanamide and tricyanomethanide. Coord Chem Rev. 2003;246:103–30.
Batten SR, Jensen P, Kepert CJ, Kurmoo M, Moubaraki B, Murray KS, Price DJ. Syntheses, structures and magnetism of α-Mn(dca)2, [Mn(dca)2(H2O)2]·H2O, [Mn(dca)2(C2H5OH)2]·(CH3)2CO, [Fe(dca)2(CH3OH)2] and [Mn(dca)2(L)2], where L = pyridine, CH3OH or DMF and dca– = dicyanamide, N(CN)2–. J Chem Soc Dalton Trans. 1999;2987–2997.
Carlin RL. Magnetochemistry. Heidelberg: Springer; 1986.
Ismayilov RH, Wang W-Z, Lee G-H, Peng S-M. One-, two- and three-dimensional Cu(II) complexes built via new oligopyrazinediamine ligands: from antiferromagnetic to ferromagnetic coupling. Dalton Trans. 2006;478–491.
Tandon SS, Thompson LK, Manuel ME, Bridson JN. Magnetostructural correlations in μ2-1,1-N3 bridged, dinuclear copper(II) complexes. 1. ferromagnetic and antiferromagnetic coupling associated with the azide bridge. X-ray crystal structures of [Cu2(DMPTD)(μ2-N3)(μ2-Cl)Cl2]·CH3CN, [Cu2(DMPTD)(μ2-N3)2(N3)2], [Cu2(DIP)(μ2-N3)(μ2-Cl)Cl2]·0.5CH3OH, [Cu2(PAP46Me-H)(μ2-N3)(N3)2]·0.33H2O, [Cu2(PAP)(μ2-N3)Cl3]·CH2Cl2, [Cu2(PAP)(μ2-N3)(N3)(NO3)(CH3OH)](NO3)·CH3OH, [Cu2(PPD3Me)(μ2-N3)Cl3(H2O)1.5], and [Cu2(PPD)(μ2-N3)(NO3)3(H2O)1.6]. Inorg Chem. 1994;33:5555–70.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bartyzel, A. Synthesis, thermal study and some properties of N2O4—donor Schiff base and its Mn(III), Co(II), Ni(II), Cu(II) and Zn(II) complexes. J Therm Anal Calorim 127, 2133–2147 (2017). https://doi.org/10.1007/s10973-016-5804-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-016-5804-0