
Abstract
A series of potential biologically active 2-[3-(4-phenyl-1-piperazinyl)propyl]-3-(4-substituted-benzoyl)-4-hydroxy-2H-1,2-benzothiazine 1,1-dioxides was synthesized in a straightforward manner by condensation of respective 3-substituted-4-hydroxy-1,2-benzothiazine 1,1-dioxides with 1-(1-chloropropyl)-4-phenylpiperazine. The structures of all of the newly formed compounds were identified by elemental analysis, FTIR and 1H NMR. The synthesized compounds were subjected to preliminary evaluation using differential scanning calorimetry (DSC) to determine the existence of multiple crystal forms. The DSC scans for all compounds show more than one endothermic effect, which might suggest dynamic proton transfer between two or three possible tautomeric forms: O-keto/O-enol, O-enol/O-keto and O-keto/O-keto/CH2 in crystals 2-[3-(4-phenyl-1-piperazinyl)propyl]-3-(4-substituted-benzoyl)-4-hydroxy-2H-1,2-benzothiazine 1,1-dioxides.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sundry benzothiazine derivatives are known to possess a wide spectrum of biological activities like anti-inflammatory [1], analgesic and antipyretic [2], antimicrobial [3], antiviral [4], antitumor [5], antidiabetic [6] and Alzheimer’s and Parkinson’s disease treatment [7]. Among these, 1,2-benzothiazine 1,1-dioxide nucleus has got considerable attention after the discovery of 4-hydroxy-2-methyl-2H-1,2-benzothiazine-3-carboxamides 1,1-dioxides (known as oxicams) as potent anti-inflammatory and analgesic agents.
On the other hand, 1-arylpiperazines represent a potentially biologically active moiety of heterocyclic compounds. They have been studied as 5-HT1A, 5-HT2A, 5-HT2C, 5-HT3, 5-HT4, 5-HT7 receptors ligands [8–11], α1-adrenoreceptor antagonists [12] or D2, D3, D4 receptors ligands [13–15]. Some of the well-known antipsychotic drugs that contain 1-arylpiperazine moiety are already available in the market like buspirone [16], gepirone or ipsapirone [17].
Currently, much research and development is being focused on the discovery of highly selective dopamine D4 receptor ligands. The reason for the interest in this area derives from the possible involvement of D4 receptor in schizophrenia, Parkinson’s disease, bipolar disorder, additive behaviours, eating disorders such as anorexia nervosa, bulimia nervosa, binge eating or sexual disorders.
Keeping in view biological activity of benzothiazines in Alzheimers and Parkinsons diseases and D4 receptors as potentially target to 1-arylpiperazine derivatives, it was perceived that synergism of these two bioactive heterocycles may result in new molecules with accelerated activities.
For this purpose, as continuation our previous investigation on 1,2-benzothiazine derivatives, we have synthesized a series of novel 3-substituted-2H-2-{[3-(4-phenyl)-1-piperazinyl]-propyl}-4-hydroxy-1,2-benzothiazine 1,1-dioxides.
This new 1,2-benzothiazine 1,1-dioxide derivatives have some structural similarities with other D4 full or partial agonist like: WAY-100635 [18], A-412997 [19, 20], FAUC-316 and FAUC-299, ABT-724 and ABT-670 [21], which are in pharmacological screening tests.
On other hand solid form diversity of pharmaceutical substances may influence the efficacy and safety of drug products. Physical and chemical characterization of different solid phases that may occur during crystallization and pharmaceutical formulation processes, i.e. polymorphs, pseudopolymorphism, solvates, desolvated solvates and amorphous materials are advisable for both drugs and excipients. It is very important to control the crystal form of the drug during the various stages of drug development, because any phase changes and change in the degree of crystallinity can alter the bioavailability of the drug. Thermal analysis is one of the most frequently used instrumental techniques on pharmaceutical researches [22–31].
Experimental
Synthesis
Reagents and solvents were purchased from commercial suppliers and used as received. Dry solvent was obtained according to the standard procedure. Analytical silica gel 60 F254-coated TLC plates were obtained from Fluka Chemie Gmbh and were visualized with UV light. Flash column chromatographic purifications were performed using Sigma-Aldrich 60A silica gel 230–400 mesh. The proton nuclear magnetic resonance (1H NMR) spectra were measured on a Brucker 300 MHz NMR spectrometer. The samples were prepared by dissolving 5 mg of each form in 600 μl of CDCl3. Melting points were recorded using MEL-TEMP capillary melting point apparatus and were uncorrected. Elemental analyses were performed by Carlo Erba NA 1500 analyser and were within ±0.4 % of the theoretical value. FTIR spectra were run on a Perkin–Elmer spectrum two UATR FT-IR spectrometer. The samples were applied as solids.
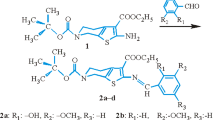
General procedure for the preparation of 2-[3-(4-phenyl-1-piperazinyl)propyl]-3-(4-substituted-benzoyl)-4-hydroxy-2H-1,2-benzothiazine 1,1-dioxide derivatives (Fig. 1, Scheme 1)
Procedure A. 2-(4-substituted-phenacyl)-2H-benzisothiazol-3-on 1,1-dioxides
A mixture of commercially available saccharine (0.92 g, 5 mmol) with 5 mmol of corresponding 2-bromoacetophenone 2a–2d [2-bromoacetophenone for obtaining 3a, 2-bromo-4′-methoxyacetophenone (3b), 2-bromo-4′-fluoroacetophenone (3c) and 2,4′-dibromoacetophenone (3d)] in 7 mL of N,N-dimethylformamide (DMF) and triethylamine (0.7 mL, 5 mmol) was stirred at room temperature for 10 h, then poured over ice cooled water (50 mL) resulted in the formation of a white solid, which was filtered and washed with cold water. The solid was dried and recrystallized from ethanol to give 2-(4-substituted-phenacyl)-2H-benzisothiazol-3-on 1,1-dioxides with 88.7–98.7 % yield;

Scheme for the preparation of 2-[3-(4-phenyl-1-piperazinyl)propyl]-3-(4-substituted-benzoyl)-4-hydroxy-2H-1,2-benzothiazine 1,1-dioxide derivatives
3a: Anal. C15H11NO4S (m.w. 301.32); m.p. 472–474 K (EtOH). 1H NMR δ: 5.15 s (2H, CH2), 7.49–7.97 m (9H, ArH). FT-IR: 1,739 and 1,694 (CO), 1,335 and 1,181 (SO2) cm−1.
3b: Anal. C16H13NO5S (m.w. 331.34); m.p. 434–436 K (EtOH). 1H NMR δ: 3.89 s (3H, OCH3), 5.11 s (2H, CH2), 6.96–7.97 m (8H, ArH). FT-IR: 1,738 and 1,693 (CO), 1,330 and 1,183 (SO2) cm−1.
3c: Anal. C15H10FNO4S (m.w. 319.31); m.p. 438–441 K (EtOH). 1H NMR δ: 5.11 s (2H, CH2), 7.16–8.01 m (8H, ArH). FT-IR: 1,738 and 1,693 (CO), 1,330 and 1,183 (SO2) cm−1.
3d: Anal. C15H10BrNO4S (m.w. 380.21); m.p. 462–464 K (EtOH). 1H NMR δ: 5.10 s (2H, CH2), 7.66–7.94 m (8H, ArH). FT-IR: 1,733 and 1,703 (CO), 1,328 and 1,179 (SO2) cm−1.
Procedure B. Rearrangement of benzisothiazolones 3a–3d to the corresponding derivatives of 3-(4-substituted-benzoyl)-4-hydroxy-2H-1,2-benzothiazines
Three millimoles of corresponding benzisothiazolone 1,1-dioxide 3a–3d (3a for obtaining 4a, 3b for 4b, 3c for 4c and 3d for 4d) were dissolved in 7.5 mL sodium ethoxide (2.3 %) at 313 K °C and stirred with heating to 328–333 K for 5–10 min. Colour changes from beige to deep red were observed. After this time and dissolving all of the substance, the mixture was rapidly cooled to 298 K and 7.5 mL HCl (9 %) was added. Colour changed from deep red to deep yellow and the product precipitated. The solid was filtered off, washed with cold water, dried and purified by crystallization from EtOH to give 4a–4d with 83.5–86.4 % yield.
4a: Anal. C15H11NO4S (m.w. 301.32); m.p. 434–436 K (EtOH). 1H NMR δ: 6.07 s (1H, NH), 7.44–8.23 m (9H, ArH), 15.71 s (1H, OH enolic); FT-IR: 3,262 (NH), 1,594 [C(OH)=C–C(O)], 1,301 and 1,176 (SO2) cm−1.
4b: Anal. C16H13NO5S (m.w. 331.34); m.p. 450–452 K (EtOH). 1H NMR δ: 3.90 s (3H, OCH3), 6.06 s (1H, NH), 7.27–8.21 m (8H, ArH), 16.02 s (1H, OH enolic). FT-IR: 3,258 (NH), 1,597 [C(OH)=C–C(O)], 1,310 and 1,179 (SO2) cm−1.
4c: Anal. C15H10FNO4S (m.w. 319.31); m.p. 466–468 K (EtOH). 1H NMR δ : 5.91 s (1H, NH), 7.13–8.20 m (8H, ArH), 15.80 s (1H, OH enolic). FT-IR: 3,151 (NH), 1,590 [C(OH)=C–C(O)], 1,280 and 1,158 (SO2) cm−1.
4d: Anal. C15H10BrNO4S (m.w. 380.21); m.p. 511–514 K (EtOH). 1H NMR δ: 6.17 s (1H, NH), 7.27–8.26 m (8H, ArH), 15.73 s (1H, OH enolic). FT-IR: 3,225 (NH), 1,583 [C(OH)=C–C(O)], 1,287 and 1,179 (SO2) cm−1.
Procedure C. 1-(3-chloropropyl)-4-phenylpiperazine (Fig. 1, Scheme 2)
A commercially available 1-phenylpiperazine (6.48 g, 40 mmol) and 1-bromo-3-chloropropane (4.4 mL, 44 mmol) were dissolved in acetone (12 mL) with addition of 25 % NaOH (7 mL), stirred slowly in room temperature for 8 h and left overnight. After this time ether (80 mL) was added and stirred further for 30 min. Then, the mixture was divided in a funnel into two parts. The ether part was dried with MgSO4 and evaporated in vacuum. The residue was purified by flash silica gel chromatography, eluting with EtOAc to give 7.62 g (79.8 %) a pale yellow oil of 1-(3-chloropropyl)-4-phenylpiperazine 5; Anal. C13H19ClN2 (m.w. 238.76); 1H NMR δ: 1.95–2.09 m (2H, CH 2 CH 2 CH 2 ), 2.53–2.70 m [6H, CH2N(CH2)2], 3.19–3.22 m [4H, N(CH2)2], 3.61–3.65 t (2H, CH 2 CH 2 CH 2 N-piperazine, J = 6 Hz), 6.84–7.27 m (5H, ArH).
Procedure D. 3-(4-substituted-benzoyl)-2-[3-(4-phenyl-1-piperazinyl)propyl]-4-hydroxy-2H-1,2-benzothiazine 1,1-dioxide derivatives
To the stirred mixture of 5 mmol of corresponding 1,2-benzothiazine 4a–4d in 20 mL of anhydrous EtOH was added 5 mL of EtONa (prepared from 0.115 g of Na and 5 mL of anhydrous EtOH). Then, 5 mmol of 1-(3-chloropropyl)-4-phenylpiperazine 5 was added and refluxed with stirring for 10–12 h. When the reaction was ended, which was controlled on TLC plates, ethanol was distiled off, the residue was treated with 50 mL of CHCl3 and insoluble materials were filtered off. The filtrate was then evaporated and the residue was purified by crystallization from ethanol to give 6a–6d (yield: 34.0–54.0 %).
2-[3-(4-phenyl-1-piperazinyl)propyl]-3-benzoyl-4-hydroxy-2H-1,2-benzothiazine 1,1-dioxide ( 6a ). Anal. C28H29N3O4S (m.w. 503.61); m.p. 440–443 K (EtOH). 1H NMR δ: 1.21–1.26 m (2H, CH 2 CH 2 CH 2 ), 1.94–1.99 m (2H, CH 2 CH 2 CH 2 N-piperazine), 2.21–2.28 m [4H, N(CH2)2], 3.01–3.27 m [6H, CH2N(CH2)2], 6.82–8.22 m (14H, ArH), 15.71 s (1H, OH enolic); FT-IR: 1,598, 1,611 [C(OH)=C–C(O)], 1,331 and 1,173 (SO2) cm−1; Anal. Calcd: C, 66.78; H, 5.80; N, 8.34. Found: C, 66.66; H, 5.94; N, 8.39.
2-[3-(4-phenyl-1-piperazinyl)propyl]-3-(4-methoxybenzoyl)-4-hydroxy-2H-1,2-benzothiazine 1,1-dioxide ( 6b ). Anal. C29H31N3O5S (m.w. 533.64); m.p. 388–391 K (EtOH). 1H NMR δ: 1.22–1.31 m (2H, CH 2 CH 2 CH 2 ), 1.96–1.98 m (2H, CH 2 CH 2 CH 2 N-piperazine), 2.24–2.30 m [4H, N(CH2)2], 3.01–3.35 m [6H, CH2N(CH2)2], 3.90 s (3H, OCH3), 6.85–8.20 m (13H, ArH), 15.76 s (1H, OH enolic); FT-IR: 1,599, 1,612 [C(OH)=C–C(O)], 1,318 and 1,173 (SO2) cm−1; Anal. Calcd: C, 65.24; H, 5.85; N, 7.91. Found: C, 65.54; H, 5.90; N, 8.00.
2-[3-(4-phenyl-1-piperazinyl)propyl]-3-(4-fluorobenzoyl)-4-hydroxy-2H-1,2-benzothiazine 1,1-dioxide ( 6c ). Anal. C28H28FN3O4S (m.w. 521.60); m.p. 376–378 K (EtOH). 1H NMR δ: 1.21–1.26 m (2H, CH 2 CH 2 CH 2 ), 1.95–2.00 m (2H, CH 2 CH 2 CH 2 N-piperazine), 2.26 s [4H, N(CH2)2], 3.04–3.30 m [6H, CH2N(CH2)2], 6.82–8.20 m (13H, ArH), 15.50 s (1H, OH enolic); FT-IR: 1,597, 1,609 [C(OH)=C–C(O)], 1,333 and 1,174 (SO2) cm−1; Anal. Calcd: C, 64.47; H, 5.41; N, 8.06. Found: C, 64.35; H, 5.44; N, 8.02.
2-[3-(4-phenyl-1-piperazinyl)propyl]-3-(4-bromobenzoyl)-4-hydroxy-2H-1,2-benzothiazine 1,1-dioxide ( 6d ). Anal. C28H28BrN3O4S (m.w. 582.51); m.p. 374–377 K (EtOH). 1H NMR δ: 1.22–1.26 m (2H, CH 2 CH 2 CH 2 ), 1.97–2.07 m (2H, CH 2 CH 2 CH 2 N-piperazine), 2.24–2.34 m [4H, N(CH2)2], 3.03–3.31 m [6H, CH2N(CH2)2], 6.86–8.19 m (13H, ArH), 15.70 s (1H, OH enolic); FT-IR: 1,599, 1,610 [C(OH)=C–C(O)], 1,328 and 1,173 (SO2) cm−1; Anal. Calcd: C, 57.71; H, 4.84; N, 7.24. Found: C, 57.32; H, 4.85; N, 7.22
Differential thermal analysis
The DSC traces were recorded on a Mettler Toledo DCS 25 measuring cell with TC15TA Controller, calibrated with indium to ensure the accuracy of the calorimetric scale. Samples weighing 6–10 mg (crystals form) were characterized in sealed 40 μl aluminium pans with lids and subjected to thermal analysis under a flowing argon atmosphere (30 cm3 min−1). Analysis was carried out from 303 K using heating rate 5 K min−1 (additionally 1 and 10 K min−1) with an identical empty sample pan as reference. For each compound several measurements were performed. Melting temperatures were determined by the onset because the onset temperature is independent of sample mass and scanning rate. Pans with lids and samples were weighed both before and after the experiment. In all cases, the mass was identical.
Computational methods
The primary task for the computational work was to determine the optimized geometry of the compounds. Calculations were carried out using the GAUSSIAN 09 software package [32]. Density functional theoretical (DFT) computations were performed using the closed shell Becke–Lee–Yang–Parr hybrid exchange–correlation three-parameter functional (B3LYP) [33–35] in combination with 6-311+G(d,p) basis set. Harmonic vibrational wavenumbers were calculated using analytic second derivatives to confirm the convergence to minima in the potential surface. The absence of imaginary frequency modes for the optimized structures confirm a true minimum on the potential energy surface. All calculations were carried out in vacuum.
Results and discussion
The DSC scans for benzisothiazolones 3a–3d are shown in Fig. 2. For all compounds only one endothermic effect, corresponds to the melting, is observed. The melting points were found as 470 K (3a), 440 K (3b), 439 K (3c), 466 K (3d). The peaks are sharp and symmetrical, characteristic for a pure substance.

DSC curves for compounds 3a–3d heated at 5 K min−1
The DSC scans for derivatives of 3-(4-substituted-benzoyl)-4-hydroxy-2H-1,2-benzothiazines 4a–4d are given in Fig. 3. For compound 4a two endothermic effects were detected. The DSC signal starts to fall below the baseline at 428 K. The second peak is symmetrical. The onset temperature is 428 K. For compound 4b the DSC trace shows a wide endothermic effect with a small bend at 452 K. For compound 4c and 4d one endothermic effect is recorded. The peak is wide and asymmetric. For compound 4c signal starts to fall below the baseline at 460 K and then rapidly at 460 K. For compound 4d a small bend at 504 K is observed. For these substances, DSC runs show the presence of overlapping peaks, corresponding probably to two thermal events, close to each other. In order to determine the overlapping peak positions and areas, a curve fitting analysis for DSC curves was carried out. Deconvolved signal was calculated using Exponentially Modified Gaussian function (EMG, r 2 = 0.991, 0.973, 0.982 for 4b, 4c, 4d, respectively). As a result two separate peaks were obtained (Fig. 3). The onset temperatures are: 450 and 453 K for 4b, 464 and 468 K for 4c, 499 and 505 K for 4d.

DSC curves for compounds 4a–4d heated at 5 K min−1 (solid line) and deconvolved signal (dash line)
The DSC scans for 3-(4-substituted-benzoyl)-2-[3-(4-phenyl-1-piperazinyl)propyl]-4-hydroxy-2H-1,2-benzothiazine 1,1-dioxide derivatives 6a–6d are presented in Fig. 4. For compound 6a, the DSC trace shows a wide endothermic effect with a small bend at 434 K. After deconvolved two separate peaks were obtained. The onset temperatures are: 435 and 441 K (r 2 = 0.997). For compound 6b one endothermic effect was detected. It is wide, with a small step at temperature 385 K. This effect is better to observe on the curves with scanning rate 10 K min−1. The onset temperatures after deconvolved are 385 and 391 K (r 2 = 0.999). Two endothermic effects were observed during heating substance 6c. The first with maximum at 359 K starts at 352 K. The second one is wide and asymmetric. This effect shows the presence of overlapping peaks, corresponding to two thermal events. The onset temperatures after deconvolved are 377 and 383 K (r 2 = 0.971). Three thermal effects are detected heating substance 6d. Two endothermic peaks at 374 and 428 K and wide exothermic one. It starts at 398 K and end at 423 K. The second endothermic peak is sharp and symmetrical, characteristic for melting process. The first one is wide and it seems to be the presence of overlapping peaks. After deconvolved two separate peaks were obtained. The onset temperatures are: 372 and 377 K (r 2 = 0.981).

DSC curves for compounds 6a–6d heated at 5 K min−1 (solid line) and deconvolved signal (dash line)
The experiments for heating and cooling rates were also performed. In addition, second heating run, after rapid cooling, was carried out. For all experiments, thermal effects were not observed. This indicate that thermal events are irreversible. After the melting process, substances go into the amorphous state.
We suggest that the presence of overlapping peaks, close to each other, is the result of dynamic proton tautomerism in their crystal structures. Three isomers are possible: enol–keto, keto–enol and keto–keto form (Fig. 5). However, crystalline state may be stabilized by strong intermolecular O–H···O hydrogen bonding and keto–keto form, probably is not present. These conditions were detected in crystal structure similar compounds: 3-[hydroxy(phenyl)methyl]-2,5,7-trimethyl-2,3-dihydropyrido[3,2-e]-[1, 2]thiazin-4-one 1,1-dioxide and 3-(1-hydroxyethylidene)-2,5,7-trimethyl-2,3-dihydropyrido[3,2-e][1, 2]-thiazin-4-one 1,1-dioxide [36].

Three possible tautomeric forms in crystals 2-[3-(4-phenyl-1-piperazinyl)propyl]-3-(4-substituted-benzoyl)-4-hydroxy-2H-1,2-benzothiazine 1,1-dioxides (compound 6a–6d): O-enol/O-keto, O-keto/O-enol and O-keto/O-keto/CH2
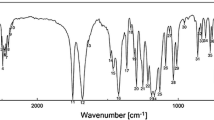
The FTIR spectra of compounds 6a–6d in the solid-state is shown in Fig. 6. In the region 3,900–1,800 cm−1, only a weak and broad bands are observed. It was assigned to stretching vibration of the hydroxyl group involved in the intra- and intermolecular hydrogen bond, which is superimposed on the C–H and Ar–H stretching vibration. A few small maximums were detected. By comparing FTIR spectra in this region, the sharpness and intensity of bands seem to indicate that H bonds are not as strong in 6c and 6d as they are in 6a and 6b. In addition, for substance 6c and 6d, weak and broad bands at 3,380, 3,670 cm−1 (6c) and 3,411, 3,723 cm−1 (6d) were detected. This can be probably assigned to intermolecular halogen bonding (interaction). In the region 1,800–450 cm−1, FTIR spectra for 6a–6d is similar. Single stretching band characteristic for C=O vibration is not observed. The band with a higher intensity near 1,600 cm−1. It was assigned to stretching vibration of C(OH)=C–C(O). It splits into two bands at 1,598, 1,611 cm−1 (6a), 1,599, 1,612 cm−1 (6b), 1,597, 1,609 cm−1 (6c), 1,599, 1,610 cm−1 (6d) since the C=O group is engaged in hydrogen bonding.

Experimental FTIR spectrum for compounds 6a–6d
The main absorption bands of FTIR theoretical and experimental were compared and shown in Table 1. The calculated vibrational wavenumbers are in good agreement with the experimental data expect vibration of C(OH)=C–C(O). But the calculations were performed for single molecules in the gas phase. The significant differences may indicate the hydrogen bonding in the solid-state.
Dynamic proton transfer may cause small differences in molecular packing and possibility co-existence in a crystalline state two tautomeric forms: keto–enol cannot be precluded. Thus, on the DSC curves, overlapping peaks, close to each are observed. Our theoretical calculations show small differences in the energy between both forms of 1.46–1.58 kJ mol−1 (4a–4d) and 1.31–1.42 kJ mol−1 (6a–6d). Thus, the populations of the tautomers keto–enol in vacuum estimated using Boltzmann distribution are in the approximate ratio 0.55:0.45 for 4a–4d and 0.52:0.48 for 6a–6d. The results of theoretical calculations show also that third possible tautomeric form, keto–keto, is the most stable form. However, as already mentioned, ability to form hydrogen bonds stabilize the crystal structure.
The molecular electrostatic potential (MEP) is helpful in identifying sites for intra- and intermolecular interactions [37]. In drug-receptor, it is a very useful descriptor in understanding sites for electrophilic and nucleophilic reactions as well as hydrogen bonding interactions [38]. The calculated MEP for compound 4c is shown in Fig. 7. Similar results were obtained for other molecules. Negative regions (red and yellow) are found around the S=O groups, –F atom in benzene ring and also close to O atom at –OH and –CO group. Our theoretical calculations for the dimer interaction through the hydrogen bond show a stabilizing effect. The energy of interaction is higher for molecule 6a and 6b. Energy value obtained for molecule 6c and 6d are lower by 2 kJ mol−1. It is worth noting that the enthalpy of melting is also the highest for substance 6a and the lower for substance 6d.

3D plots of the molecular electrostatic potential map of the molecule 4c
The DSC traces for compound 6c and 6d are different from the trace for compound 6a and 6b. An additional thermal effect is observed. The presence of the –Br and –F substituent in the benzene ring causes a strong inductive effect. Electrons are withdrawn from the ring through a σ bond. This may facilitate intermolecular O–H···F bond formation. It also seems that an important factor affecting the strong of intermolecular O–H···O–S hydrogen bond is a propyl-4-phenylpiperazine group (spherical barrier). As a consequence, co-existence in crystalline state forms with differences in molecular packing is possible (three peaks on DSC traces for 6c). On other hand a resonance effect (donation of electrons through a π bond) increases π electron density. It can influence the van der Waal’s interactions between benzene rings. Compound 6d shows two melting peaks with a recrystallization step. On heating, the substance is not directly transformed from crystalline state to an isotropic melt. The second melting peak corresponds to other modification.
Conclusions
Four new 2-[3-(4-phenyl-1-piperazinyl)propyl]-3-(4-substituted-benzoyl)-4-hydroxy-2H-1,2-benzothiazine 1,1-dioxides were synthesized. The studied compounds are interesting materials due to their potential biologically active. Thermal analysis may be very useful for monitoring and identification substances during organic synthesis. In pharmaceutical industry, it is very important to control the purity of crystal form. The important aspect of our investigation was to determine the existence of multiple crystal forms. The DSC scans for all compounds show more than one endothermic effect (or overlapping peaks), which might suggest dynamic proton transfer between two or three possible tautomeric forms: O-keto/O-enol, O-enol/O-keto and O-keto/O-keto/CH2 in crystals 2-[3-(4-phenyl-1-piperazinyl)propyl]-3-(4-substituted-benzoyl)-4-hydroxy-2H-1,2-benzothiazine 1,1-dioxides. The study further demonstrates the utility of deconvolution software for separating overlapping DSC endotherms. Our theoretical calculations show small differences in the energy between keto–enol tautomeric forms. The FTIR spectra analysis and theoretical calculations suggest that the crystal state is stabilized by a strong intermolecular hydrogen bonding. The presence of the –Br and –F substituent in the benzene ring causes an inductive effect. As a consequence, co-existence in crystalline state forms with differences in molecular packing is possible.
References
Lombardino JG, Wiseman ED. Sudoxicam and related N-heterocyclic carboxamides of 4-hydroxy-2H-1,2-benzothiazine 1,1-dioxide. Potent nonsteroidal anti-inflammatory agents. J Med Chem. 1972;15:848–9.
Gupta SK, Bansal P, Bhardwaj RK, Jaiswal J, Velpandian T. Comparison of analgesic and anti-inflammatory activity of meloxicam gel with diclofenac and piroxicam gels in animal models: pharmacokinetic parameters after topical application. Skin Pharmacol Appl Skin Physiol. 2002;15:105–11.
Ziaurrehman M, Choudary JA, Ahmad S, Siddiqui HL. Synthesis of potential biologically active 1,2-benzothiazin-3-yl-quinazolin-4(3H)-ones. Chem Pharm Bull. 2006;54:1175–8.
Malagu K, Boustie J, David MD, Sauleau J, Amoros M, Girre R, Sauleau A. Synthesis and antiviral activity of new 1, 4-benzothiazines: sulphoxides and sulphone derivatives. Pharm Pharmacol Commun. 1998;4:57–60.
Gunning WT, Kramer PM, Lubet RA, Steele VE, Pereira MA. Chemoprevention of vinyl carbamate-induced lung tumors in strain A mice. Exp Lung Res. 2000;26:757–72.
Tawada H, Sugiyama Y, Ikeda H, Yamamoto Y, Meguro K. Studies on antidiabetic agents. IX. A new aldose reductase inhibitor, AD-5467, and related 1,4-benzoxazine and 1,4-benzothiazine derivatives: synthesis and biological activity. Chem Pharm Bull. 1990;38:1238–45.
Shen XM, Dryhurst G. Influence of glutathione on the oxidation chemistry of 5-S-cysteinyldopamine: potentially neuroprotective reactions of relevance to Parkinson’s disease. Tetrahedron. 2001;57(2):393–405.
Heinrich T, Bottcher H, Gericke R, Bartoszyk GD, Anzali S, Seyfried CA, Greiner HE, Van Amsterdam C. Synthesis and structure–activity relationship in a class of indolebutylpiperazines as dual 5-HT1A receptor agonists and serotonin reuptake inhibitors. J Med Chem. 2004;47:4684–92.
Seo HJ, Park E-J, Kim MJ, Kang SY, Lee SH, Kim HJ, Lee KN, Jung ME, Lee MW, Kim MS, Son EJ, Park WK, Kim J, Lee J. Design and synthesis of novel arylpiperazine derivatives containing the imidazole core targeting 5-HT2A receptor and 5-HT transporter. J Med Chem. 2011;54:6305–18.
Cappelli A, Anzini M, Vomero S, Mennuni L, Makovec F, Doucet E, Hamon M, Bruni G, Romeo MR, Menziani C, Benedetti PG, Langer T. Novel potent and selective central 5-HT3 receptor ligands provided with different intrinsic efficacy. 1. Mapping the central 5-HT3 receptor binding site by arylpiperazine derivatives. J Med Chem. 1998;41:728–41.
Vermeulen ES, Smeden M, Schmidt AW, Sprouse JS, Wikstrom HV, Grol C. Novel 5-HT7 receptor inverse agonists. Synthesis and molecular modeling of arylpiperazine- and 1,2,3,4-tetrahydroisoquinoline-based arylsulfonamides. J Med Chem. 2004;47:5451–66.
Betti L, Corelli F, Floridi M, Giannaccini G, Maccari L, Manetti F, Strappaghetti G, Botta M. Structural optimization of pyridazinone-arylpiperazines. Study of the influence on affinity and selectivity of cyclic substituents at the pyridazinone ring and alkoxy groups at the arylpiperazine moiety. J Med Chem. 2003;46:3555–8.
Wustrow DJ, Smith WJ III, Corbin AE, Davis MD, Georgic LM, Pugsley TA, Whetzel SZ, Heffner TG, Wise LD. 3-[[(4-Aryl-1-piperazinyl)alkyl]cyclohexyl]-1H-indoles as dopamine D2 partial agonists and autoreceptor agonists. J Med Chem. 1997;40:250–9.
Leopoldo M, Berardi F, Colabufo NA, De Giorgio P, Lacivita E, Perrone R, Tortorella V. Structure–affinity relationship study on N-[4-(4-arylpiperazin-1-yl)butyl]arylcarboxamides as potent and selective dopamine D3 receptor ligands. J Med Chem. 2002;45:5727–35.
Perrone R, Berardi F, Colafubo NA, Leopoldo M, Tortorella V. 1-(2-methoxyphenyl)-4-alkylpiperazines: effect of the N-4 substituent on the affinity and selectivity for dopamine D4 receptor. Bioorganic Med Chem Lett. 1997;7:1327–30.
Goa KI, Ward A. Buspirone. A preliminary review of its pharmacological properties and therapeutic efficacy as an anxiolytic. Drugs. 1986;32:114–29.
Becker OM, Dhanoa DS, Marantz Y, Chen D, Shacham S, Cheruku S, Heifetz A, Mohanty P, Fichman M, Sharadendu A, Nudelman R, Kauffman M, Noiman S. An integrated in silico 3D model-driven discovery of a novel, potent, and selective amidosulfonamide 5-HT1A agonist (PRX-00023) for the treatment of anxiety and depression. J Med Chem. 2006;49:3116–35.
Chemel BR, Roth BL, Armbruster B, Watts VJ, Nichols DE. WAY-100635 is a potent dopamine D4 receptor agonist. Psychopharmacology. 2006;188:244–51.
Moreland RB, Patel M, Hsieh GC, Wetter JM, Marsh K, Brioni JD. A-412997 is a selective dopamine D4 receptor agonist in rats. Pharmacol Biochem Behav. 2005;82:140–7.
Browman KE, Curzon P, Bao Pan J, Molesky AL, Komater VA, Decker MW, Brioni JD, Moreland RB, Fox GB. A-412997, a selective dopamine D4 agonist, improves cognitive performance in rats. Pharmacol Biochem Behav. 2005;82:148–55.
Patel MV, Kolasa T, Mortell K, Matulenko MA, Hakeem AA, Rohde JJ, Nelson SL, Cowart MD, Nakane M, Miller LN, Uchic ME, Terranova MA, El-Kouhen OF, Donnelly-Roberts DL, Namovic MT, Hollingsworth PR, Chang R, Martino BR, Wetter JM, Marsh KC, Martin R, Darbyshire JF, Gintant G, Hsieh GC, Moreland RB, Sullivan JP, Brioni JD, Stewart AO. Discovery of 3-methyl-N-(1-oxy-3¢,4¢,5¢,6¢-tetrahydro-2¢H-[2,4¢-bipyridine]-1¢-ylmethyl)benzamide(ABT-670), an orally bioavailable dopamine D4 agonist for the treatment of erectile dysfunction. J Med Chem. 2006;49:7450–65.
Ford JL, Timmins P. Pharmaceutical thermal analysis: techniques and applications. New York: Wiley; 1989.
Haines PJ. Thermal methods of analysis—principles, applications and problems. London: Blackie Academic and Professional; 1995.
Charsley EL, Warrington SB. Thermal analysis: techniques and applications. Cambridge: Royal Society of Chemistry; 1992.
Craig Duncan QM, Reading M. Thermal analysis of pharmaceuticals. Boca Raton: CRC Press; 2007.
Giron D. Applications of thermal analysis and coupled techniques in pharmaceutical industry. J Therm Anal Cal. 2002;68(2):335–57.
Zayed MA, Hawash MF, Fahmey MA, Ali El-Gizouli MM. Investigation of ibuprofen drug using mass spectrometry, thermal analyses, and semi-empirical molecular orbital calculation. J Therm Anal Cal. 2012;108(1):315–22.
Kohutová A, Honcová P, Svoboda L, Bezdička P, Maříková M. Structural characterization and thermal behaviour of biological hydroxyapatite. J Therm Anal Cal. 2012;108(1):167–73.
Pani NR, Nath LK, Acharya S, Bhuniya B. Application of DSC, IST, and FTIR study in the compatibility testing of nateglinide with different pharmaceutical excipients. J Therm Anal Cal. 2012;108(1):219–26.
El-Gamel Nadia EA, Hawash MF, Fahmey MA. Structure characterization and spectroscopic investigation of ciprofloxacin drug. J Therm Anal Cal. 2012;108(1):253–62.
Thakur SS, Maheswaram MPK, Mantheni DR, Kaza L, Perara I, Ball DW, Moran J, Riga AT. Solid-state mechanical properties of crystalline drugs and excipients. J Therm Anal Cal. 2012;108(1):283–7.
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03. Pittsburgh: Gaussian Inc.; 2003.
Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98:5648–53.
Lee C, Yang W, Parr RG. Development of the Colle–Salvetti correlation energy formula into a functional of the electron density. Phys Rev. 1988;B37:785–9.
Perdew JP, Wang Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys Rev. 1992;B45:13244–9.
Karczmarzyk Z. Keto–enol tautomerism in crystals of 3-[hydroxy(phenyl)methyl]-2,5,7 trimethyl-2,3-dihydropyrido[3,2-e]-[1, 2]thiazin-4-one 1,1-dioxide and 3-(1-hydroxyethylidene)-2,5,7-trimethyl-2,3-dihydropyrido[3,2-e][1, 2]-thiazin-4-one 1,1-dioxide. Acta Crystallogr. 2008;C64:590–4.
Roohi H, Nowroozi AR, Anjomshoa E. H-bonded complexes of uracil with parent nitrosamine: a quantum chemical study. Comput Theor Chem. 2011;965:211–20.
Munshi P, Guru Row TN. Intra- and intermolecular interactions in small bioactive molecules: cooperative features from experimental and theoretical charge–density analysis. Acta Crystallogr. 2006;B62:612–26.
Acknowledgments
Calculations have been carried out in Wroclaw Centre for Networking and Supercomputing (http://www.wcss.wroc.pl).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Krzyżak, E., Szczęśniak-Sięga, B. & Malinka, W. Synthesis and thermal behaviour of new benzo-1,2-thiazine long-chain aryl-piperazine derivatives. J Therm Anal Calorim 115, 793–802 (2014). https://doi.org/10.1007/s10973-013-3185-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-013-3185-1