
Abstract
Polyesters are a particularly interesting group of polymers because of their ester bonds in the main chain, which are sensitive to degradation. It has been shown that aromatic polyesters [eg. poly(ethylene terephthalate) (PET)] can be degraded when they are copolymerized with aliphatic polyesters. In this regard, the objective of our previous study was to obtain and investigate new copolymers with some fragments of PET and its hydrolytic degradation. In this work, the impact of ozone degradation on properties of bis-2-hydroxyethyl terephthalate-based copolymers was researched. Apart from the bis-2-hydroxyethyl terephthalate, the copolymers comprised oligomers of lactic acid and glycolic acid in different combination. During ozone degradation, samples were kept for 24 h in atmosphere containing 0.51 vol.% of ozone. Structure changes were determined by means of FTIR spectroscopy. In the IR spectra of ozonised samples, new bonds characteristic for ozonised polymers were observed. Thermal properties of copolymers before and after degradation process were reviewed based on differential scanning calorimetry (DSC) and thermogravimetry. DSC results revealed that melting point increased, especially for copolymers displaying higher quantity of PET units.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Poly(ethylene terephthalate) (PET) is one of the important, commercially available polymers with widespread applications and simultaneously extremely high resistance to atmospheric and biological agents. In order to improve its degradability, an incorporation of aliphatic units into structure of PET by copolymerization or reactive blending are commonly applied [1]. The study of degradation and stabilization of polymers and their copolymers is an extremely important area from the scientific and industrial point of view. It is expected that within the usability span a particular product will continue to display its key features and after the lapse of this period will gradually deteriorate forcing the consumer to purchase a new model of the product. The common methods of polymer degradation are biodegradation, pyrolysis, oxidative degradation, photodegradation and catalytic degradation [2–5]. In view of the high accumulation of ozone in the troposphere, it is important to determine the influence of this gas on the oxidative degradation and consequently, on the basic chemical and physical properties of polymers [6]. For this reason, the investigations of influence of ozone on the structure and thermal properties of new polymeric materials seem to be an important issue. Active oxygen forms, such as singlet oxygen and ozone, are responsible for oxidative degradation of a variety of materials, including polymers. Ozone-induced degradation of common polymers yields volatile radicals and peroxide species which may escape into the environment. Simultaneously, in the solid matter hydroxides, peroxides, or carbonyl groups are formed [7, 8]. Detailed studies concerning ozone degradation of the majority of commercial polymers are available in the literature [9]. On the other hand, there is relatively little data concerning ozone degradation of new materials such as conductive polymers [6, 10] or biodegradable polymers.
In our work, the impact of ozone degradation on properties of bis-2-hydroxyethyl terephthalate-based copolymers was studied. The synthesis and hydrolytic degradation of some analysed copolymers were described in our earlier works [11–13]. These copolymers contain bis-2-hydroxyethyl terephthalate (B), glycolic acid (G) and l-lactic acid (L) units, which are non-toxic and renewable raw materials. The composition of investigated copolymers was 80/20 wt% for samples BL1 and BG; 70/30—BL2, 60/40—BL3; 50/50—BL4 and 80/10/10 for sample BLG.
The degradation processes of the investigated copolyesters are an important characteristic determining their final applications. Each polymer can undergo degradation at different rates in different conditions. In our case, samples during ozone treatment were kept for 24 h in atmosphere containing 0.51 vol.% of ozone. Structure changes were determined by means of FTIR spectroscopy. Thermal properties of copolymer before and after ozone degradation process were investigated by means of differential scanning calorimetry (DSC) and thermogravimetry (TG).
Experimental
Materials and condition of the degradation tests
Polymeric material used in this work was a series of copolyesters synthesized by melt polycondensation based on bis-2-hydroxyethyl terephthalate, l-lactic acid and glycolic acid oligomers [11–13] (Fig. 1).

Scheme of investigated copolymers BL, BG and BLG
Samples which were used in this study contained 10, 20, 30, 40 and 50 wt% of lactic acid for samples BLG, BL1, BL2, BL3 and BL4. respectively, and 10, 20 wt% of glycolic acid for samples BLG and BG, respectively. Obtained polymer powder was pressed into pellets (0,8 g), applying 20–40 Bar pressure. On one side of the pellets, golden contacts were placed by vacuum evaporation technique. The ozonization of polymer samples has been performed in the test vessel used earlier for investigation of electronic conducting polymers [8]. Bach mode ozonization vessel containing polymer samples was filled with ozonized oxygen. The ozone gas was generated from analytical grade oxygen gas applying electric discharge ozone generator made by ELTO, Poland. The ozone content in the gas was about 0.51 vol.%. Ozonization was carried out for 24 h at room temperature.
FTIR
Infrared spectra of the polymer samples in KBr disc form (3 mg of copolymer/300 mg of KBr) have been recorded on Perkin-Elmer 2000 FTIR from 400 to 4,000 cm−1 before and after ozonization.
Thermogravimetric analysis (TG)
TA Instruments, SPT 2960 Simultaneous DSC-TGA was used for studying thermal behaviours of degraded and non-degraded samples which were in a form of powder. The thermogravimetric analyses were carried out under nitrogen flow in the 293–873 K temperature range at heating rates of 10 K min−1.
Differential scanning calorimetry (DSC)
The melting characteristics of copolyesters were determined by DSC (Polymer Laboratories, Epson, GB). The DSC analyses were carried out under nitrogen with flow rate about 1.5 mL min−1 heating rates of 10 K min−1 from room temperature to 523 K. All runs were carried out with a sample of 5–9 mg. The crucibles used during DSC analyses were not hermetic.
Results and discussion
FTIR
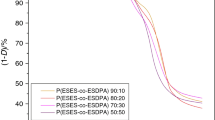
The spectroscopy in the infrared light is a valuable method in polymer chemistry. Careful analysis of spectra gives insight into a polymer chemical structure. The most important signals found in FTIR spectra of studied copolymers together with their assignments are listed in Table 1. Comparing the spectra of initial polymers with these of ozonized, allowed us to deduce the nature of chemical changes in the polymer. In this study, we have investigated a wide spectrum of samples consisting of different content of lactic acid, as well as glycolic acid. Despite all structural differences of the initial copolymers, the spectra of ozone-treated copolymers are fairly similar (see Fig. 2). Differences in the bands intensity were a subject of further investigations. In the spectra of ozonized polymer, several distinct bands were found. Dominating signals are related to oxygen bearing groups generated by the oxidation of polymer chains. In spite of the fact that most of the signals characteristic for the initial polymer are present in the spectra, their intensities are significantly decreased. One band was found to be missing in the treated polymers spectra i.e. band near 1,608 cm−1 which could be assigned to one of phenyl C–C in plane modes (see Figs. 2, 3). It is interesting that another phenyl C–C in plane mode appears at 1,506 cm−1 at a slightly decreased intensity [14]. The only reasonable explanation for this can be the formation of charge transfer complex between phenyl ring and ozone molecule resulting in the tightening of the ring structure, which avoid the specific vibrations. Moreover, ozone treatment causes considerable decrease of intensity of band at 1,720 cm−1 ascribed to ester C=O stretching. This indicates that the treatment results in the reduction of ester linkages in polymer chains [15]. The absence of extra bands that can be assigned to carbonyl moiety in spectra of ozonized polymers implies that in this case ozonization does not lead to carbonyl formation. This is understandable assuming that the reaction was still in the initial stage when the spectra were recoded. However, this is very unlikely. The alternative explanation is that the end product of the oxidation is mainly CO2, or resulting aldehydes, ketones and carboxylic acids are volatile and escape from the polymer matrix. During the analysis of variation of intensity of the strongest signals in the series of studied copolymers, we have found several interesting facts. The intensity of band about 1,269 cm−1, assigned to ester C–O–C stretching in ethylene terephthalate parts [14], is well correlated with the variation of intensity of most of the significant signals in the spectra of both initial and ozonized polymers. The only exceptions are the new peaks present in the spectra of ozone-treated polymers. These new bands i.e. 3,140 and 1,401 cm−1 together with the band at 3,016 which was substantially elevated due to the change in its background are highly correlated with each other. In addition, their intensities are not correlated with any other signal. Discussion concerning proper assignments of these bands is beyond the scope of this article, though, they typically occur in FTIR spectra of many ozonized polymers [8]. Together with the band at 1,269 cm−1 in the studied copolymers, there exists an ester C–O–C stretching band characteristic for lactic esters. It typically occurs at 1,195 cm−1, but in our case, it was found in the range between 2005 and 1,195 cm−1. In Table 2, the intensities of the two bands of C–O–C stretching are presented. Intensity of these bands is well correlated both before ozone treatment and after. Intensity decrease due to treatment is somewhat correlated with the intensity of band at 1,197 cm−1. In view of the above-mentioned facts, it can be stated that aliphatic fragments undergo oxidation by ozone easier than aromatic terephthalate fragments.

Collection of FTIR spectra of all copolymers after ozone treatment

FTIR spectra region between 1,800 and 1,500 cm−1 of BL3 before and after ozone treatment. The feature is representative for all studied polymers
Thermal properties
The DSC thermograms were registered to establish the impact of the degradation process of each of the copolymers on their melt temperatures and enthalpy.
In Table 3, melt temperatures and enthalpies before and after the ozone degradation process of crystalline copolymers BL1, BL2 and BG copolymer were depicted. Depending on the aliphatic ingredient in the copolymer, a different kind of influence the ozone degradation has got on researched structures can be observed.
BL1 and BG copolymers once subjected to ozone degradation display an increase of melt temperature while the BL1 structure displays simultaneous reduction in the melt enthalpy. Higher melt temperature of both copolymers could be attributed to mer degradation derived from aliphatic fragments positioned on the ends of the chains which have got a significantly lower melt temperature compared to the aromatic part originating form PET [16, 17].
In the amorfic parts, the polymer segments are positioned in a less confined way and can be easily affected by ozone resulting in the breaking of the ester bond [18]. In spite of the above mentioned in the case of BL1 and BL2 copolymers, the disrupted chains were not able to improve the mobility of segments which therefore prevented the regrouping of regularity, as inferred from the reduction in phase transition enthalpy [15, 19].
In the case of BL2 copolymer, where the load of aliphatic fragments was at 30 wt% the melt enthalpy reduction was drastic, i.e. 23 J g−1 as well as the decrease in melt temperature—by 298 K. This confirms the fact that the polymers were degraded once subjected to ozone.
The other technique which was used to measure the thermal stability of copolymers, the before and after degradation process was TG. The main criteria to indicate the thermal stability of the copolymers were the percentage mass loss as a function of temperature. T 10, T 50 (temperature for 10 % and 50 % mass loss, respectively).
The results obtained for the BL copolymer series as well as the BG and BLG polymers were presented in Table 4. The impact of the ozone degradation process on thermal stability of copolyesters is apparent mainly in the shift in temperatures in the case of 10 % mass loss (T 10%) oriented at higher temperatures in comparison with the original compounds.
There was an increase in decomposition temperatures in the case of all researched compounds independent of the kind and quality of the present aliphatic groups [20]. As shown in the Table 2, the most significant increase in thermal resistance was displayed by copolymers containing small amount of lactic acid, i.e. BL1 and BLG with 12 and 10 K increase, respectively. The same rule does not apply to glycolic acid.
It could be easily observed that thermal stability of researched copolymers decreases with the increase in the load of aliphatic parts. The phenomenon is related to the loss of copolymer aliphatic fragments in the hydrolytic degradation process which results in the increase in the percentage of ET in the chain [21, 22]. The ET fragment has got a higher decomposition temperature and therefore the increase in its load in the copolymer’s structure results in the increase in degradation temperatures [19, 23, 24].
Conclusions
-
The degradation process was studied by FTIR, DSC and TG. The thermal behaviour suggested the degradation of the investigated copolyesters.
-
In the case of BL1 and BG copolymers, there was an increase in melt temperature which could be attributed to the degradation of aliphatic groups on the ends of chains.
-
Melt temperature of the BL2 copolymer decreased which could indicate the degradation of amorfic regions and the decomposition of polymers into fragments of lower molecular mass.
-
Temperatures of decomposition of the researched compounds increased independent of the kind and the load of used aliphatic ingredient whereas the most significant increase was observed in the case of compounds containing 20 wt% of lactic acid.
References
Patel MR, Patel JV, Sinha VK. Polymeric precursors from PET waste and their application in polyurethane coatings. Polym Degrad Stab. 2005;90:111–5.
Sivalingam G, Madras G. Thermal degradation of poly(3-caprolactone). Polym Degrad Stab. 2003;80:11–6.
Lee SR, Park HM, Lim H, Kang T, Li X, Cho WJ, Ha ChS. Microstructure, tensile properties, and biodegradability of aliphatic polyester/clay nanocomposites. Polymer. 2002;43:2495–500.
Yavuz H, Babac C, Tuzlakoglu K, Piskin E. Preparation and degradation of l-lactide and 3-caprolactone homo and copolymer films. Polym Degrad Stab. 2002;75:431–7.
Tomita K, Tsuji H, Nakajima T, Kikuchi Y, Ikarashi K, Ikeda N. Degradation of poly(d-lactic acid) by a thermophile. Polym Degrad Stab. 2003;81:167–71.
Cataldo F. Ozone interaction with conjugated polymers-I. Polyacetylene. Polym Degrad Stab. 1998;60:223–31.
Czerwinski W, Nowaczyk J, Kania K. Ozonization of electronic conducting polymers I. Copolymers based on poly[3-nonylthiophene]. Polym Degrad Stab. 2003;80:93–101.
Nowaczyk J, Czewinski W, Olewnik E. Ozonization of electronic conducting polymers: II. Degradation or doping. Polym Degrad Stab. 2006;91:2022–9.
Razumovskii SD, Zaikov GE. Ozone and its reactions with organic compounds. Elsevier; 1984.
Cataldo F. On the action of ozone on undoped and doped alkyl and N-alkyl-substituted polyanilines. Polym Degrad Stab. 2002;75:99–106.
Olewnik E, Czerwinski W, Nowaczyk J, Sepulchre MO, Tessier M, Salhi S, Fradet A. Synthesis and structural study of copolymers of l-lactic acid and bis(2-hydroxyethyl terephthalate). Eur Polym J. 2007;43:1009–19.
Olewnik E, Czerwinski W, Nowaczyk J. Hydrolytic degradation of copolymers based on l-lactic acid and bis-2-hydroxyethyl terephthalate. Polym Degrad Stab. 2007;92:24–31.
Olewnik E, Czerwinski W. Synthesis, structural study and hydrolytic degradation of copolymer based on glycolic acid and bis-2-hydroxyethyl terephthalate. Polym Degrad Stab. 2009;94:221–6.
Daniels WW, Kitson RE. Infrared spectroscopy of poly(ethylene terephthalate). J Polym Sci. 1958;33:161–70.
Murphy SH, Leeke GA, Jenkins MJ. Comparison of the use of FTIR spectroscopy with DSC in the characterisation of melting and crystallisation in polycaprolactone. J Therm Anal Calorim. 2012;107:669–74.
Kong Y, Hay JN. The measurement of the crystallinity of polymers by DSC. Polymer. 2002;43:3873–8.
Hsieh YT, Kuo NT, Woo EM. Thermal analysis on phase behavior of poly(l-lactic acid) interacting with aliphatic polyesters. J Therm Anal Calorim. 2012;107:745–56.
Cao X, Mohamed A, Gordon SH, Willett JL, Sessa DJ. DSC study of biodegradable poly(lactic acid) and poly(hydroxy ester ether) blends. Thermochim Acta. 2003;406:115–27.
Dekker M. In: Basel NY, editor. Handbook of polymer degradation. New York: Marcel and Decker; 2000.
Wunderlich B. Methodology of interpreting thermal analysis of polymers. J Therm Anal Calorim. 2011;106:85–91.
Galcera T, Fradet A, Marechal E. Synthesis and study of thermotropic block copolyesters containing aliphatic unsaturated units. Eur Polym J. 1995;31:733–9.
Smith ChG, Nyquist RA, Smith PB, Pasztor AJ, Martin SJ. Analysis of synthetic polymers. Anal Chem. 1991;63:11R–32R.
Yuan X, Mak AFT, Yao K. Comparative observation of accelerated degradation of poly(l-lactic acid) fibres in phosphate buffered saline and a dilute alkaline solution. Polym Degrad Stab. 2002;75:45–53.
Kiziltas A, Gardner DJ, Han Y, Yang HS. Thermal properties of microcrystalline cellulose-filled PET–PTT blend polymer composites. J Therm Anal Calorim. 2011;103:163–70.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Olewnik, E., Nowaczyk, J., Garman, K. et al. Influence of ozone treatment on structure and thermal properties of bis-2-hydroxyethyl terephthalate-based copolymers. J Therm Anal Calorim 112, 697–702 (2013). https://doi.org/10.1007/s10973-012-2608-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-012-2608-8