
Abstract
A new “in situ” sol–gel synthesis procedure was exploited to produce silica/epoxy nanocomposites with 6 wt.% maximum silica content. 3-Aminopropyltriethoxysilane (APTS) was used as a coupling agent. The experimental results (fourier-transform infrared spectroscopy, FTIR, small-angle X-ray scattering, SAXS, transmission electron microscopy, TEM, nuclear magnetic resonance, NMR, and dynamic mechanical analysis, DMA) support that the structure consists of nanosized silica particles (maximum 1.25 nm in size) embedded in a hybrid co-continuous network. A post cure non-isothermal heating from 15 to 100 °C (beyond the Tg of the neat epoxy) caused Tg and storage modulus to increase. The fire behavior, that, owing to severe regulations (i.e., in aerospace engineering), often prevents composites applications, was also studied. The formed silica domains prevented melt dripping phenomena during vertical flame spread tests. Cone calorimetry tests showed a remarkable decrease of the heat release rate (HRR) for all the hybrid systems with respect to the neat cured resin, even at very low silica loadings (i.e., 2 wt.%). This decrease was much more pronounced for the hybrid structures that were not subjected to the post cure thermal treatment. The use of multiple structural investigation techniques allowed to choose among multiple hypothesis and conclude that nanoparticles clustering is the main reason of the effects of the post curing treatments.

SAXS of the samples before (EPO) and after (EPO_t) post cure thermal treatment in the glass transformation range. Effect of the post cure on the glass transformation temperature (Tg) and heat release rate (HRR) Photos of samples EPO and EPO 2%Si after vertical flame spread tests
Highlights
-
A hybrid co-continuous network embedding nanoparticles (maximum 1.25 nm in size) was obtained;
-
A post-cure was performed above the glass transformation temperature, Tg, of neat epoxy;
-
The post cure makes Tg, storage modulus and Heat Release Rate (HRR) to increase;
-
No dripping in flame test and up to 40% HRR reduction for only 2 wt.% silica content is observed;
-
The post cure effects depend on nanoparticles clustering in the glass transformation range.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
It is well-known that polymeric materials cannot be used for high-performance applications because of their limited properties. This limitation can be overcome by using organic/inorganic composites. Epoxy/silica systems represent one of the most widely utilized organic/inorganic systems. Because of heat, moisture, and chemical resistance and good adhesion to many substrates, epoxy resins are mostly exploited in the field of coatings, adhesives, casting, potting, composites, laminates, and for encapsulating semiconductor devices [1, 2].
Recently, organic–inorganic polymer hybrids raised great attention. They require nano-level molecular design but are expected to have unique properties that are not simply the sum of those of the composite components: unprecedented materials may be created with the hybrid strategy [3]. Aerogels may be strengthened through proper organic–inorganic hybridization strategies [4]. Supercapacitors may be created [5]. Organic-inorganic hybrid perovskites (e.g., CH3NH3PbI3), with advantages of easy processing, tunable bandgaps, and superior charge-transfer properties, have emerged as a new class of revolutionary optoelectronic semiconductors promising for various applications [6].
In this paper, the attention is focused on materials for linings in the aerospace field. In this case, the requirements related to the mechanical properties are often limited. On the contrary, severe regulations about the fire resistance must be respected in order use the materials [7]. As it is known, traditional halogen-based flame retardants are persistent organic pollutants of global concern and generate corrosive/toxic combustion products [8]. In this prospect, polymer nanocomposites, i.e., polymer matrices filled with specific, finely dispersed nanofillers, are considered to pave the way for future materials combining physico-chemical and thermo-mechanical performances with enhanced flame retardant behavior [9]. However, most of the literature on these materials is qualitative, and often points to conflicting/misleading suggestions from the perspectives of short-term and long-term fire exposure tests [8]. Hence, there is a renewed need to fundamentally understand the fire response of such materials [8]. These findings justify the topic proposed in this paper, which investigates the structure and fire behavior of new in-situ generated silica/epoxy hybrid materials.
The organic/inorganic hybrid materials usually consist of nanostructured domains of the inorganic filler, homogeneously dispersed in the polymer matrix [10]. Epoxy/silica hybrids may be prepared by simply dispersing preformed silica particles or (better) by promoting the “in-situ” formation of a silicate phase through sol–gel chemistry. The “in-situ” method allows obtaining a wide range of morphologies ranging from the particles dispersion to the formation of co-continuous organic–inorganic networks [11–13]. Furthermore, the sol–gel process allows tailoring the interface between organic and inorganic phases through the adequate selection of both the silane precursors of the inorganic phase and the sol–gel reaction conditions [14, 15].
In details, the use of coupling agents allows preventing phase separation phenomena of the epoxy and inorganic components, hence giving rise to the formation of silica/epoxy hybrid networks; therefore, the organic and/or inorganic precursors have to be modified in order to achieve an optimal compatibility between the organic and inorganic components of the resulting hybrid material [16, 17]. However, the preparation of epoxy hybrid materials is largely empirical and requires an efficient control of the key parameters for the assembly of the organic and inorganic domains into an optimized morphology. Hybrids containing particulate inorganic oxides are produced under conditions that ensure fast condensation rates: as a consequence, phase separation takes place by a nucleation and growth mechanism [14].
Mascia et al. [18] thoroughly studied a lot of coupling agents, including (3-aminopropyltriethoxysilane (APTS), γ-glycidoxypropyltrimethoxysilane (GOTMS), and mercaptosilanes. In particular, APTS has been frequently used, as extensively studied by Bakhshandeh et al. and Seraja et al. [19–21]. It possesses three hydrolysable ethoxy groups that, thanks to silanols condensation, can be grafted onto the surface of sol–gel silica. Conversely, the aminopropyl group is not hydrolysable and may react with the epoxy rings similarly to amine hardeners; this way, it may promote a very good compatibilization effect. It turns out that the formation of one network can exert a control on the growth of the other, thereby creating conditions that lead to a wide range of morphologies at the nanometer length scale, including the co-continuous morphology [14, 21].
Despite the mechanical properties of these hybrid systems (particularly referring to the epoxy/silica systems that exploit APTS as coupling agent) have been deeply investigated, few studies report on the thermal as well as fire behavior of these materials, that often, because of very severe regulations (i.e., in aerospace engineering), prevents their applicability [22]. It is known that the inclusion of silica into epoxy networks favors the char formation and increases its yield, hence improving the thermal stability: in fact, the carbonaceous residue layer acts as a thermal insulator and a barrier to oxygen diffusion [23, 24]. Matějka et al. [7] investigated the thermal degradation of epoxy/silica nanocomposites owing to their widespread use as high-performance materials. Thermal analysis results, coming from several experiments [23, 24], confirm that silica, being an inherently stable inorganic phase, lowers the degradation rates by favoring the formation of more voluminous and mechanically stronger char; furthermore, it also affects the pyrolytic degradation of amine-cured epoxy networks that proceeds through several overlapping mechanisms, e.g., dehydration, intramolecular cyclization, isomerization, or chain-transfer reactions [25–28].
In this paper, an “in situ” sol–gel synthesis procedure was applied to a commercial two-component epoxy resin system. The samples were characterized by means of infrared spectroscopy (FTIR), small-angle X-ray scattering (SAXS), transmission electron microscopy (TEM), dynamic mechanical analysis (DMA), nuclear magnetic resonance (NMR). The experimental results support that a very fine and even distribution of silica nanoparticles (at nanometer level) within a hybrid network structure was obtained in all cases. It is shown that a non-isothermal post curing treatment beyond the Tg of the neat epoxy affects the thermal and mechanical features of the obtained hybrid materials. In vertical flame spread tests, the presence of silica domains, even at very low loadings (2 wt.%), prevented the dripping of incandescent drops. Furthermore, a remarkable reduction (about 40%) of the heat release rate (HRR) was observed in the presence of just 2 wt.% silica in the samples not subjected to non-isothermal post curing treatments (instead of 16% for the fully cured sample).
All these findings were mainly ascribed to the nanoparticles clustering derived from the post curing treatments, the hybrids were subjected to.
2 Materials and methods
2.1 Materials
Tetraethyl orthosilicate (TEOS,>99%), (3-aminopropyl)-triethoxysilane (APTES, >98%) and ethanol (ACS reagent, anhydrous) were purchased from Sigma-Aldrich (Switzerland). A two-component epoxy resin system (SX10 by MATES S.r.l., Milan, Italy), consisting of a modified bisphenol A resin and modified cycloaliphatic polyamines, was used for fabricating composite laminates.
2.2 Preparation of the epoxy/silica hybrid nanocomposites
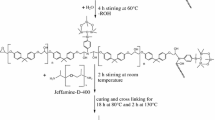
APTS and TEOS were used as silica precursors and added to the commercial two-component epoxy resin system, hence promoting an “in situ” sol–gel synthesis prior to the addition of the epoxy hardener. Samples from batches containing different TEOS loadings were prepared. The TEOS/epoxy weight ratio was changed in the range 0–0.15 at constant TEOS/APTS weight ratio (equal to 3). The synthesis route is inspired to one already reported in the literature [29]. However, in the present work, the TEOS/epoxy weight ratio was remarkably increased (0.12 instead of 0.04) and acetone was avoided; this required substantial changes of the synthesis procedure. So as indicated in the second step reported below, silica formation required a higher temperature (80 °C instead of room temperature) and reflux conditions.
The synthesis was performed in one pot involving the following three steps:
-
1.
Mixtures of epoxy (DGEBA) and APTS with weight ratios epoxy/APTS changing from 100/3 to 100/5 were stirred vigorously at 80 °C for 2 h to get a silanized epoxy
-
2.
Tetraethoxysilane (TEOS), distilled water and ethanol (EtOH) were added to the silanized epoxy and stirred vigorously at 80 °C under reflux for 90 min. The reaction vessel was, then, opened and kept at 80 °C for 30 min in order to remove ethanol and water
-
3.
The amount of hardener needed for the curing was then added to the mixture at room temperature and mixed for 5 min. The resulting mixtures were degassed under vacuum and poured into a Teflon® mold. The curing process was carried out at 30 °C for 24 h; then, the curing was completed by treating the samples at 80 °C for 4 h.
The silica contents estimated from the stoichiometry were 2, 4, 6 wt.%. The typical reaction batches are reported in Table 1 together with their acronyms that will be used throughout the paper.
A post cure treatment was also performed by thermally treating the samples in a tube furnace, at 3 °C/min from 25 to 100 °C, i.e., 15 °C higher than the neat epoxy glass transition temperature. These samples will be coded by adding “t” to the acronyms: as an example, EPO (EPO is the acronym for the commercial two-component epoxy resin system containing 0 wt.% of silica.)_2%Si_t is the post cured hybrid system containing 2 wt.% of silica.
2.3 Characterization
FTIR transmittance spectra were recorded with a Nicolet 5700 FTIR spectrometer (Thermo Fisher, Waltham, MA, USA), using a single reflection Attenuated total reflectance (ATR) accessory with a resolution of 4 cm−1 and 32 scans and Thermo Scientific™ OMNIC™ Software Suite (v7.2, Thermo Fisher, Waltham, MA, USA, 2005). All the obtained spectra were normalized to the strong absorption bands at 1607 and 1509 cm−1, related to the C=C bonds of the benzene rings present in the epoxy resin structure, that are not expected to change after the curing reaction.
Simultaneous small-angle and wide angle X-ray scattering analyses (SAXS-WAXD), for both fully cured and uncured hybrid epoxy samples, were carried out using an Anton Paar SAXSpace camera equipped with a 2D imaging plate detector. CuKα X-Rays with 1.5418 Å wavelength were generated by a sealed tube source (40 kV, 50 mA) and slit collimated. All scattering data were corrected for background and normalized for the primary beam intensity. In order to remove the inelastic scattering from the data, SAXS profiles were corrected for both Porod constant and desmearing effect.
Bright field TEM analyses were performed using a FEI TECNAI G12 Spirit-Twin (LaB6 source) equipped with a FEI Eagle-4k CCD camera, operating with an acceleration voltage of 120 kV.
Dynamic mechanical tests were carried out on a DMA3300 (TA Instruments). The tests were run in a three-point bending mode with a span of 40 mm and a frequency of 1 Hz; the width of samples was about 10 mm and the temperature was ramped from 25 to 100 °C at a heating rate of 3 °C/min.
NMR spectra were acquired with a 300 MHz (7.0 Tesla) Bruker Avance magnet (Bruker Biospin, Rheinstetten, Germany), composed by a wide-bore system and equipped with a CPMAS (Cross-Polarization Magic-Angle-Spinning) probe, working at 29Si and 13C frequencies of 59.62 and 75.47 MHz, respectively. The samples (two replicates per treatment) of hybrid polymer before (EPO_2%Si) and after the thermal treatment (EPO_2%Si_t) were loaded into 4 mm zirconia rotors, closed with KelF caps and spun at a rate of 10,000 ± 1 Hz. 13C NMR spectra were acquired by applying a Cross-Polarization technique and consisted in 1814 time domain points, a spectral width of 300 ppm (22727.3 Hz), a recycle delay of 2 s, 4000 scans and 1 ms of contact time. 13C CPMAS pulse sequence was conducted by using a 1H Ramp pulse to account for non-homogeneity of the Hartmann–Hahn condition. 29Si NMR spectra were acquired by using a direct polarization and consisted in 2048 time domain points, a spectral width of 500 ppm (29762 Hz), 40 s of recycle delay and 5600 scans.
Free induction decays (FIDs) were processed by Bruker Tospin (v2.1) and MestreNOVA (v. 9.0, Mestrelab Research) software. Prior to be phase and baseline corrected, 29Si and 13C spectra were Fourier Transformed by applying a twofold zero-filling and adopting an exponential filter function with a line broadening of 500 and 250 Hz, respectively.
Cone calorimeter tests (Fire Testing Technology, East Grinstead, London, UK) were performed according to the ISO 5660 standard, by using squared samples (5.0 × 5.0 × 0.3 cm3), with a heat flux of 35 kW/m2, in horizontal configuration. Time to ignition (TTI, s), total heat release (THR, MJ/m2), peak of the heat release rate (pkHRR, kW/m2) were measured. Total smoke release (TSR, m2/m2), carbon monoxide yield (CO yield, kg/kg), carbon dioxide yield (CO2 yield, kg/kg) and specific extinction area (SEA, m2/kg) were evaluated, as well. For each sample, the experiments were repeated at least three times in order to ensure reproducible and significant data.
UL94 tests were performed according to the D3801−10 standard; the size of the specimens was 120 × 10 × 2 mm3.
3 Results and discussion
3.1 Characterization of the epoxy/silica hybrid nanocomposites
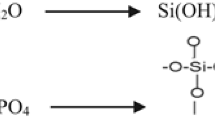
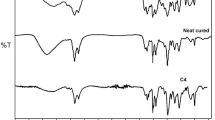
Figure 1 shows the FTIR spectra of the different samples. In the presence of silica, a remarkable evolution of the IR absorption bands in the frequency range between 1050 and 1150 cm−1 is observed. The absorption in this frequency range keeps on increasing in the hybrid samples (see the FTIR spectra of EPO_2%Si, EPO_4%Si and EPO_6%Si): this finding can be ascribed to the presence of a progressively higher silica phase amount, thus proving its formation from the APTS and TEOS added to the batch through the following well-known reactions:

FTIR spectra of the uncured resin (a) and of all the hybrid systems (b–e), either non-thermally treated (red) or subjected to a non-isothermal post curing (dark gray) (color figure online)
It is known, in fact, that SiO4 stretching vibration modes of fused silica give rise to a sharp band at 1100 cm−1 [30–32]. When alkaline or earth-alkaline oxides are added, this band gradually shifts towards lower wavenumbers and broadens. This finding is due to the build-up of SiO4 tetrahedral units bearing a progressively higher number of non-bridging oxygens and therefore to a lack of network connectivity [30–32]. In the case of silica gel produced through the sol–gel process, this lack of connectivity may be due to incomplete condensation of silanol groups. Accordingly, the bands appearing at 1070 and 1100 cm−1 may be ascribed to partially uncondensed and condensed silica phases, respectively [14, 33, 34]. The band at 1070 cm−1 may be interpreted also on the basis of the formation of bi-continuous nanocomposites consisting of particles in a hybrid silica-epoxy network, thus associated with the existence of a “diffused” silica network [35]. It is worth noticing that the characteristic bands of the epoxy group, located at 970, 913, and 870 cm−1 are intense and well defined in the uncured system and disappear in all the other samples. Taking into account that the cure involves the reaction of the oxirane ring with the amine present in the hardener, the disappearance of these bands confirms the completeness of the polymer curing reactions [29, 33].

3.2 Morphology of the obtained hybrid systems
The hybrid systems, before and after the thermal treatment, are transparent, as shown in Fig. 2 for EPO_2%Si. TEM micrographs, displayed in Fig. 3, show a very fine and even distribution of particles. In all samples, some particles tend to aggregate into clusters or bigger particles.

Photograph of an epoxy/silica hybrid material containing 2 wt.% silica

TEM micrographs of EPO_2%Si (a), EPO_6%Si (b), EPO_4%Si (c), and EPO_4%Si at high magnification (d)
This clustering effect is very limited for the hybrids containing 2 wt.% silica but increases with increasing the filler loading, as observed when comparing TEM micrographs reported in Fig. 3a, b.
The higher magnification micrograph (Fig. 3d) further supports the presence of a hybrid co-continuous structure. This may be attributed [16, 17, 29] to the first step of the synthesis procedure, when epoxy resin and APTS are left to react to form the silanized epoxy through the reaction of the APTS amino group with the epoxy oxirane ring:
3.3 Small angle X-ray diffraction analysis of the epoxy/silica hybrid nanocomposites
The WAXD and SAXS results are displayed in Fig. 4. In particular, Fig. 4a shows for the composite with 6 wt.% of silica the typical epoxy features at q vector equal to 4 and 12 nm−1, which are assigned to local fluctuations of the epoxy network. This confirms that the epoxy network of hybrids is not affected by the functionalization reaction of epoxy resins macromolecules with APTS and silica adducts produced by the sol–gel method. Different results were obtained by some of the authors when 3-glycidyloxypropyl)trimethoxysilane (GOTMS) was used as coupling agent. In this case, the presence of 3:1 GOTMS:TEOS brought about a significant structural modification of the epoxy network with the disappearance of the peak at q vector equal to 4 nm−1 [15].

a WAXD scattering profiles of epoxy/silica nanocomposites containing 6 wt.% of silica before and after post cure thermal treatment (3 °C/min till 100 °C) and b SAXS profile of epoxy/silica nanocomposites before and after post cure thermal treatment
In the SAXS region, at q value lower than 2 nm−1, the spectra of the different hybrids depend on the silica content and the thermal treatment the samples underwent. In details, as can be seen from Fig. 4b, the EPO_4%Si and EPO_6%Si SAXS spectra show, before the thermal treatment, a typical Guinier knee feature, which is ascribed to the presence of discrete silica particles homogeneously dispersed in epoxy matrix. After the post cure non-isothermal treatment, this diffraction feature disappears and the diffracted intensity becomes linear with the scattering vector for all samples. This evidence may be assigned to the formation of an inorganic fractal structure: its density, which is measured as slope of the diffracted intensity in the log I(q)-log q graph, does not depend on the silica content and seems to be approximately similar for all hybrids. The EPO_2%Si shows a SAXS spectrum characterized by a linear diffracted intensity, also before the thermal treatment. This is likely due to the presence of smaller silica particles homogeneously dispersed throughout the sample, giving rise to a homogeneous co-continuous hybrid morphology. This latter does not change upon the thermal treatment from 25 to 100 °C. From the Guinier feature it is possible to estimate the size of silica particles and the results confirm what is seen from the TEM micrographs: the samples consist of very small particles. In particular, the silica size increases by increasing the silica content from 2% to 6 wt.%, being the geometrical radius equal to 1.15 nm for EPO_4%Si and 1.25 nm for EPO_6%Si. Referring to EPO_2%Si sample, the SiO2 size is too small to be measured.
The SAXS data indicate that the final morphology of the hybrids depends on the silica content and on the effect of post curing thermal treatment. Before this latter, all samples consist of nanostructured particles, whose size increases with TEOS content, which are homogeneously dispersed in an organic–inorganic network made by epoxy resin and diffused epoxy APTS functionalized/siloxane moieties (i.e., not fully condensed). The morphology of the samples with higher silica loadings significantly shifts towards a fractal structure. The silica particles aggregate during the post cure thermal treatment forming a co-continuous hybrid morphology, which appears as a fractal structure in the SAXS analysis.
3.4 Dynamic mechanical analysis
All the prepared samples were subjected to dynamic mechanical analyses (DMA); each test was repeated twice on the same sample. Tanδ vs. temperature curves referring to the first and second DMA runs are shown in Fig. 5. Furthermore, the glass transition temperature (Tg) values, calculated as the maximum of Tanδ curve, are plotted as a function of the composition in Fig. 6. It is worthy to note that, as the DMA analyses were carried out from room temperature to 100 °C at a heating rate of 3 °C/min, the first repetition of DMA analyses can mimic the non-isothermal “post cure” treatment defined in “Preparation of the epoxy/silica hybrid nanocomposites” section. This is the reason, for which in the legend of Fig. 6, some curves are coded with “t”, according to the acronyms defined in “Preparation of the epoxy/silica hybrid nanocomposites” section.

Tanδ vs. temperature curves of cured epoxy a and epoxy/silica nanocomposites b–d at different silica content derived from the first (red) and second (dark gray) DMA run

Glass transition temperatures as a function of silica content, for cured epoxy and epoxy/silica hybrid materials: first (squares) and second DMA (triangles) run
As can be seen, only the cured epoxy curve shows a sharp peak that does not change in the second DMA run, hence indicating that the polymer network is fully cured in the adopted experimental conditions. Conversely, the Tanδ peaks for the hybrid samples are broader, possibly showing shoulders; the modifications observed in the successive heating cycles are an indication of the occurrence of structural changes during the second DMA run. This finding can be ascribed to the presence of two co-continuous phases, one consisting of more flexible and the other of more rigid segments [14, 29, 35, 36], as supported by TEM images of the hybrid system (see Fig. 3), where very fine silica nanoparticles are embedded in a hybrid co-continuous network. According to the FTIR results reported in Fig. 1, in all samples the oxirane ring absorption bands almost disappear. This means that the changes should not be related to polymer cure reactions completion. However, two other explanations can be given for the Tanδ peaks changes observed in the case of “treated” samples. First, it must be taken into account that the final temperature of the thermal treatment at 3 °C/min (first DMA run) is, for all samples, above the glass transition temperature of the neat polymer network. The mobility that, in the glass transformation range, all the hybrid segments are expected to acquire may well allow silanols (≡Si–OH) or silanols and unhydrolysed ≡Si–O–C2H5 groups to get closer and condensate according to reactions (2) or (3). This could well explain the changes of Tanδ peaks. SAXS results concerning the greater size of silica in the post cured samples may find a similar explanation: taking into account the very low particles diameter (maximum 1.25 nm), when the hybrid segments acquire enough mobility, nanoparticles clustering may easily occur. However, these structural rearrangements may, by themselves, explain the Tanδ peaks changes. A third explanation is, by this way, found.
Finally, the trends of Tg curves reported in Fig. 6 can be justified as well: in fact, both completion of condensation reaction and/or particles clustering may well justify the Tg increases of the post cure treated samples.
Figure 7 plots the room temperature storage modulus of the samples before and after the post cure non-isothermal treatment. A remarkable increase (+30%) of the modulus is observed, particularly for EPO_6%Si after post curing.

Room temperature storage modulus of untreated and thermal treated samples
This finding is likely to be ascribed to the nanoparticles clustering occurring in the samples, after the thermal treatment: the clustering may act as a reinforcement for the resin, increasing the mechanical properties with respect to the untreated sample.
3.5 Nuclear magnetic resonance
The above discussed hypothesis about the occurrence of silicate condensation reactions during the post curing thermal treatment was checked in the case EPO_2%Si by means of solid state NMR spectroscopy. The 29Si NMR and 13C CPMAS NMR spectra are shown in Figs. 8 and 9.

29Si NMR spectra of hybrid polymer before (a, blue) and after (b, red) the thermal treatment acquired at a spin rate 10,000 Hz (color figure online)

13C CPMAS NMR spectra of hybrid polymer before (a, blue) and after (b, red) the thermal treatment acquired at a spin rate 10,000 Hz (color figure online)
The 29Si NMR spectrum of studied hybrid material (Fig. 8a) shows a relatively broad and intense signal ranging within −95 and −125 ppm and attributable to the Q4 silicon nuclei. The high-field resonance of this peak is due to the strong shielding-effect resulting for the involvement of observed silicon nuclei in four –Si–O–Si– linkages. Remarkably, the fact any peak was detected in the spectral regions of Q2 (−85 ppm) and Q3 (−95 ppm) forms suggests that most of silicon nuclei composing the added TEOS were converted into Q4 forms during the gel formation. Additionally, the lack of Si peaks in forms different than Q4 also suggests to exclude a relevant presence of residual APTES (3-aminopropyltriethoxysilane), which, depending on the extent of siloxane formation, may polymerize to produce Q3 forms. However, the comparison with the 29Si NMR spectrum of thermally treated material (EPO_2%Si_t) did not reveal any significant change attributable to the thermal treatment (Fig. 8b). The same result was observed when a replicate was analyzed for both sample types. This confirms that the origin of the peculiar properties of studied hybrid material, revealed by other analytical techniques, cannot be explained by a different arrangement of the siloxane network, since most of silicon atoms appear to have been fully involved in the condensation reactions (2) and (3).
Most signals detected in the 13C CPMAS NMR spectrum of EPO_2%Si (Fig. 9a) are attributable to the DGEBA (bisphenol A diglycidyl ether) structure. In detail, the alkyl region appears dominated by two peaks corresponding to both the two magnetically-equivalent methyls (at 32 ppm) and the quaternary carbon, which the methyls are bound to (at 42 ppm). The four peaks in the range 110–165 ppm correspond to the aromatic carbons of phenol ring. Specifically, the signals at 158 and 143 ppm are due to the two aromatic quaternary carbons which are bound to the oxygen involved in an ether bond and an alkyl quaternary carbon, respectively. The signals at 128 and 143 ppm correspond to protonated aromatic carbons positioned in ortho and meta, in respect of the quaternary aromatic C–O at 158 ppm. The peak at 70 ppm corresponds to the methylene carbon bound to the phenolic oxygen. Finally, the signals at 58, and 44 ppm rise from the methylene and methine carbons composing the DGEBA epoxy group, respectively. The peaks dominating the spectral region 40–60 ppm appear relatively broadened since the methine and methylene signals of unreacted DGEBA resonate at very similar frequencies as compared to the corresponding carbons, which resulting from the opening of the epoxy ring and coupling with APTES or curing agent amino group (Fig. 9a). The comparison with the 13C NMR spectrum of EPO_2%Si_t did not reveal any significant difference depending on thermal treatment (Fig. 9). Remarkably, the fact that intense ethoxy peaks were not detected in the most shielded alkyl-C region, suggests an approximately complete hydrolysis of ethoxy groups in both APTES and TEOS reagents. This is in agreement with the results achieved by 29Si spectra about the fully involvement of silicon atoms in the condensation reactions (Fig. 9).
Therefore, taking into account the NMR results, the structural changes evidenced by the DMA curves and the Tg changes during the post cure thermal treatment must be, mainly, attributed to the clustering of silica nano-particles evidenced by SAXS analyses.
3.6 Fire behavior
The fire behavior of a flame retarded system is usually investigated by performing either flammability (according to UL94 standard, in vertical or horizontal configuration) or forced-combustion (i.e., cone calorimetry) tests: in fact, their combination allows assessing the overall fire performance of the considered system, hence evaluating the reaction of the material towards the application of a flame or a heat flux. In this context, vertical flame spread and cone calorimetry tests were exploited. As far as vertical flame spread tests are concerned, all the samples investigated cannot be classified, as they do not achieve self-extinction before the flame reaches the top of the sample. However, the presence of the silica domains in the hybrid structures allows preventing melt dripping phenomena, which, at variance, occur in the neat epoxy network. Undoubtedly, melt flow and dripping of the pyrolyzing polymer can be detrimental during a fire, as they often provide an additional ignition source, an additional flame spread process and even favor the starting of a pool fire independent from the original burning item. As clearly reported in the scientific literature [37], the addition of micro- to nano-particles to thermoplastic or thermosetting polymer matrices can prevent these undesired phenomena, because of the increase of the melt viscosity of the burning system: unlike the systems described in the literature, the high homogeneity of distribution of the silica nanoparticles in the epoxy network allows limiting the nanofiller loading to a very small extent that is capable of inhibiting the formation of incandescent drops. Furthermore, it is noteworthy that the flammability behavior of all the samples investigated is not affected by the non-isothermal treatment they possibly underwent. The pictures of the residues after vertical flame spread tests are shown in Fig. 10: once again, they demonstrate the high char-forming character of the in situ synthesized silica nanoparticles, which allow achieving a high residue after flammability test.

Residues of the different hybrid systems after vertical flame spread tests, before a and after b the post cure non-isothermal treatment
As far as forced-combustion tests are considered, Table 2 collects the cone parameters in terms of TTI, HRR, pkHRR, THR and final residue. First of all, it is noteworthy that all the hybrid systems, irrespective of the possible non-isothermal post curing treatment, anticipate the ignition as compared to the neat cured EPO. Conversely, the presence of the silica domains well distributed in the polymer matrix promotes a remarkable decrease of HRR and THR as well: this finding can be explained in terms of formation of a ceramic silica layer, which acts as a thermal shield, hence protecting the underlying materials, slowing down both the diffusion of the volatile flammable degradation products towards the gas phase and the oxygen diffusion towards the degrading material [38].
The higher reduction of HRR and THR observed for the samples that did not undergo the non-isothermal post curing could be the result of two different phenomena taking place during the thermal treatment: more specifically, this latter can favor further condensation reactions of partially condensed hybrid structures, hence consuming the condensation by-products, which provide an endothermic effect that lowers the heat-related cone parameters.
In addition, the non-isothermal post curing treatment can promote some rearrangements in the silica domains (i.e., silica clustering phenomena, as revealed by SAXS analyses), which, upon the exposure to the heat flux of the cone, give rise to the formation of a less protective ceramic layer that should slow down the heat transfer from the material to the surroundings and vice-versa. Therefore, as a consequence of the silica clustering, the efficiency of the ceramic layer decreases, hence increasing THR and HRR values of the samples subjected to the non-isothermal post curing treatment. It’s worth underlining that this effect appears to be particularly strong in the case of EPO_2%Si sample. In fact, for the not post cured sample, the HRR reduction is 40%, whereas it approaches 16% only for the post cured counterpart. NMR results suggest that the second effect is predominant. In addition, the formation of an inorganic phase within the hybrid material is responsible for the increase of the final residue at the end of the tests.
Finally, as far as the smoke parameters (see Table 3) are concerned, the hybrid structure, regardless of the possible non-isothermal post curing treatments, is responsible for an overall slight decrease of the TSR, SEA, CO, and CO2 yields.
4 Conclusions
In the present work, new hybrid silica/epoxy composites were prepared, using a tailored sol–gel process. The samples were thoroughly investigated by means of infrared spectroscopy (FTIR), SAXS, TEM, DMA, NMR. The experimental results suggest that the new hybrid structures consist of very fine silica nanoparticles, homogeneously dispersed in an epoxy/silica hybrid network; their size increases with increasing TEOS content (approaching a maximum size of about 1.25 nm). A significant increase of the Tg values was observed for the hybrids, subjected to a non-isothermal post curing treatment up to end temperatures (100 °C) higher than the Tg range temperature of the neat epoxy (85 °C). Taking into account that the oxyrane ring FTIR band has already disappeared during the curing process and that no significant changes of 29Si NMR spectra chemical shifts were observed during the post curing, the Tg changes are to be ascribed mainly to the structural changes evidenced by SAXS: when entering the glass transformation range, the mobility that all the hybrid segments are expected to acquire may well allow clustering of the very small silica particles, as evidenced by SAXS; otherwise the clustering may well stiffen the network, hence promoting the Tg increase.
The presence of the silica domains in the hybrid organic–inorganic networks was found to prevent melt dripping phenomena in vertical flame spread tests. In addition, as assessed by forced-combustion tests, the inorganic domains acted as a thermal protective layer, hence improving the heat and smoke-related parameters. Finally, the efficiency of the protective ceramic layer was found to decrease after the non-isothermal post curing treatment carried out on the hybrid systems. This finding was ascribed to two different phenomena taking place during the thermal treatment, namely: (i) the consumption of condensation by-products, which cannot promote endothermic phenomena during their evaporation under the exposure to the heat flux of the cone and (ii) the clustering phenomena of the silica phases, which, in the post cured samples, may reduce the effectiveness of the ceramic layer in the protection of the underlying material. Also in this case, NMR results suggest that the second effect is predominant.
References
Stöver HDH (1996) Polymeric materials encyclopedia. CRC Press, Boca Raton
Potter WG (1970) Epoxide resins. Springer-Verlag Inc, New York, NY
Masayuki G, Kazuo T, Yoshiki C (2017) Creative synthesis of organic inorganic molecular hybrid materials. Bull Chem Soc Jpn 90:463–474. https://doi.org/10.1246/bcsj.20170005
Shimizu T, Kanamori K, Nakanishi K (2017) Silicone-based organic–inorganic hybrid aerogels and xerogels. Chem-A Eur J 23:5176–5187. https://doi.org/10.1002/chem.201603680
Wang Y, Mayorga-Martinez CC, Pumera M (2017) Polyaniline/MoS X supercapacitor by electrodeposition. Bull Chem Soc Jpn 90:847–853. https://doi.org/10.1246/bcsj.20170076
Zhao Y, Zhu K (2016) Organic–inorganic hybrid lead halide perovskites for optoelectronic and electronic applications. Chem Soc Rev 45:655–689. https://doi.org/10.1039/C4CS00458B
Matějka L, Dušek K, Pleštil J et al. (1998) Formation and structure of the epoxy-silica hybrids. Polym (Guildf) 40:171–181
Dasari A, Yu ZZ, Cai GP, Mai YW (2013) Recent developments in the fire retardancy of polymeric materials. Prog Polym Sci 38:1357–1387. https://doi.org/10.1016/j.progpolymsci.2013.06.006
Laoutid F, Bonnaud L, Alexandre M et al. (2009) New prospects in flame retardant polymer materials: From fundamentals to nanocomposites. Mater Sci Eng R Rep 63:100–125. https://doi.org/10.1016/j.mser.2008.09.002
Afzal A, Siddiqi HM, Iqbal N, Ahmad Z (2013) The effect of SiO2 filler content and its organic compatibility on thermal stability of epoxy resin. J Therm Anal Calorim 111:247–252. https://doi.org/10.1007/s10973-012-2267-9
Innocenzi P, Kidchob T, Yoko T (2005) J Sol-Gel Sci Technol 35:225. https://doi.org/10.1007/s10971-005-2290-4
Piscitelli F, Lavorgna M, Buonocore GG et al. (2013) Plasticizing and reinforcing features of siloxane domains in amine-cured epoxy/silica hybrids. Macromol Mater Eng 298:896–909
Alonso B, Massiot D, Valentini M et al. (2008) Design of hybrid organic--inorganic materials through their structure control: the case of epoxy bearing alkoxides. J Non Cryst Solids 354:1615–1626
Mascia L, Prezzi L, Lavorgna M (2005) Peculiarities in the solvent absorption characteristics of epoxy-siloxane hybrids. Polym Eng Sci 45:1039–1048. https://doi.org/10.1002/pen.20372
Piscitelli F, Buonocore GG, Lavorgna M et al. (2015) Peculiarities in the structure–Properties relationship of epoxy-silica hybrids with highly organic siloxane domains. Polym (Guildf) 63:222–229
Davis SR, Brough AR, Atkinson A (2003) Formation of silica/epoxy hybrid network polymers. J Non Cryst Solids 315:197–205
Matějka L, Dušek K, Pleštil J et al. (1999) Formation and structure of the epoxy-silica hybrids. Polym (Guildf) 40:171–181
Prezzi L, Mascia L (2005) Network density control in epoxy-silica hybrids by selective silane functionalization of precursors. Adv Polym Technol 24:91–102
Bakhshandeh E, Jannesari A, Ranjbar Z et al. (2014) Anti-corrosion hybrid coatings based on epoxy–silica nano-composites: toward relationship between the morphology and EIS data. Prog Org Coat 77:1169–1183
Bakhshandeh E, Sobhani S, Jannesari A et al. (2015) Structure–property relationship in epoxy-silica hybrid nanocomposites: The role of organic solvent in achieving silica domains. J Vinyl Addit Technol 21:305–313
Seraj S, Ranjbar Z, Jannesari A (2014) Synthesis and characterization of an anticratering agent based on APTES for cathodic electrocoatings. Prog Org. Coatings 77:1735–1740
Troitzsch J (1983) International plastics flammability handbook: principles-regulations-testing and approval. Hanser, Munich
Visakh PM, Arao Y (2015) Flame retardants: polymer blends, composites and nanocomposites. Cham, Switzerland, Springer
Phonthammachai N, Chia H, He C (2012) One-step synthesis of oval shaped silica/epoxy nanocomposite: process, formation mechanism and properties. In: Dr. Abbass A. Hashim. The delivery of nanoparticles. InTech 497-508. Rijeka, Croatia.
Ramirez C, Rico M, Barral L et al. (2007) Organic/inorganic hybrid materials from an epoxy resin cured by an amine silsesquioxane. J Therm Anal Calorim 87:69–72
Singh D, Narula AK (2010) Studies on the curing and thermal behaviour of diglycidyl ether of bisphenol-A (DGEBA) in the presence of aromatic diimide-diacids. J Therm Anal Calorim 100:199–205
Wu Q, Zhang C, Liang R, Wang B (2010) Combustion and thermal properties of epoxy/phenyltrisilanol polyhedral oligomeric silsesquioxane nanocomposites. J Therm Anal Calorim 100:1009–1015
Villanueva M, Martin-Iglesias JL, Rodriguez-Añón JA, Proupin-Castiñeiras J (2009) Thermal study of an epoxy system DGEBA (n=0)/mXDA modified with POSS. J Therm Anal Calorim 96:575–582
Jiao J, Liu P, Wang L, Cai Y (2013) One-step synthesis of improved silica/epoxy nanocomposites with inorganic-organic hybrid network. J Polym Res 20:202
Šimon I, McMahon HO (1953) Study of some binary silicate glasses by means of reflection in infrared. J Am Ceram Soc 36:160–164
Yu P, Kirkpatrick RJ, Poe B et al. (1999) Structure of calcium silicate hydrate (C-S-H): Near-, Mid-, and Far-infrared spectroscopy. J Am Ceram Soc 82:742–748
Simon I (1960) Modern aspects of the vitreous state. Butterworths Scientific Publications Ltd., London
Torrens F, Solar L, Puchol V et al. (2008) Incorporation of silica nanospherical particles into epoxy-amine crosslinked materials. Polym Polym Compos 16:139–151
Kang S, Hong SIl, Choe CR et al. (2001) Preparation and characterization of epoxy composites filled with functionalized nanosilica particles obtained via sol--gel process. Polym (Guildf) 42:879–887
Mascia L, Prezzi L, Haworth B (2006) Substantiating the role of phase bicontinuity and interfacial bonding in epoxy-silica nanocomposites. J Mater Sci 41:1145–1155. https://doi.org/10.1007/s10853-005-3653-5
Li Y, Mao S (1996) A study on the glass transition behavior and morphology of semi-interpenetrating polymer networks. J Polym Sci Part A Polym Chem 34:2371–2375
Crompton TR (2013) Characteristics and analysis of non-flammable polymers. Smithers Rapra Technology Ltd., Shawbury
Malucelli G (2016) Surface-engineered fire protective coatings for fabrics through sol-gel and layer-by-layer methods: An overview. Coatings 6:33
Acknowledgements
The Italian Ministry of University and Research (MIUR) and the Aerospace District of Campania Region are greatly acknowledged for funding through the research program “IMM–Interiors with Multifunctional Materials” PON03PE_00138_1 (CUP B88F12000990005) this research activity.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bifulco, A., Tescione, F., Capasso, A. et al. Effects of post cure treatment in the glass transformation range on the structure and fire behavior of in situ generated silica/epoxy hybrids. J Sol-Gel Sci Technol 87, 156–169 (2018). https://doi.org/10.1007/s10971-018-4710-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-018-4710-2