
Abstract
Spherical silica particles doped with iron oxide have been synthesized via base-catalyzed one-pot sol–gel process using tetraethoxysilane (TEOS) and iron(III) ethoxide (ITE) as co-precursors. In the modified Stöber process adopted, depending on the concentration of ITE in the starting composition, materials of various morphologies were observed under a scanning electron microscope and an atomic force microscope. The presence of ITE significantly affected the formation process of particulate silica; the spherical particles were formed accompanied by the co-presence of irregular-shaped finer aggregates. The fraction of the aggregates with rough surfaces increased with an increase of the ITE content in the reaction mixture. Both the spherical particles and irregular-shaped aggregates contained iron hydroxide and they exhibited paramagnetic behavior. The chemical composition and physicochemical properties of the materials were determined using various complementary spectroscopic methods.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Over the past several years, magnetic nanoparticles of iron oxides have gained increasing attention due to the wide variety of emerging applications of these easy to prepare biocompatible materials with favorable magnetic properties [1]. They have been extensively exploited as the materials of choice for magnetic resonance imaging (MRI), tissue-specific release of therapeutic agents as well as labeling and sorting of cells, and separation of biochemical products [2–4] for immobilization of enzymes or controlled and targeted drug delivery [5]. However, most of these applications require nanoparticles which are chemically stable, uniform in size, and form stable dispersions in liquid media. Unfortunately, pristine nanoparticles of iron oxides tend to aggregate into large clusters, which results in the loss of their specific properties associated with single-domain, magnetic nanostructures [6]. One of the ways to avoid these problems is a combination of magnetic nanoparticles with other carriers. Variety of the inorganic and polymeric materials has been reported as possible carriers for magnetic materials. Among them silica, an inorganic carrier, seems to be interesting supporting material, which can be easily prepared and retains stability in most chemical and biological environments [7, 8]. It has been demonstrated that the formation of a passive coating of inert materials such as silica on the surfaces of iron oxide nanoparticles could help to prevent their aggregation in liquids and improve their chemical stability [9]. Another advantage of the silica coating is that such a surface is often terminated by a silanol group, which can serve as a reacting centers allowing for covalent attachment of specific ligands to the surfaces of these magnetic nanoparticles. That creates an opportunity to design and synthesize of magnetic carriers that can be used to deliver specific ligands to target organs via the antibody–antigen recognition [6].
There are two different approaches which have been used to generate silica coatings on the surfaces of iron oxide particles. The first method uses the well-known Stöber process [10], in which silica is formed in situ by the hydrolysis and condensation of a precursor. This method can be also useful for the creation of organic molecules-silica hybrid materials. In our previous work we have described the formation of self-assembled porphyrin microstructures covered by silica obtained in the Stöber synthesis in the presence of 5, 10, 15, 20-tetrakis(4-hydroxyphenyl)porphyrin (mTHPP) [11]. The second method is based on microemulsion synthesis, in which surfactant micelles were used to confine and control the coating of silica on core nanoparticles [12]. The drawback of that method is the necessity of separation of the core–shell nanoparticles from the large amount of surfactants present in the microemulsion system, when the reaction is completed.
In a current paper we report on the formation of interesting and stable magnetic structures of micrometer size. The materials were obtained in the sol–gel synthesis carried out in the mixture of tetraethoxysilane (TEOS) and iron(III) ethoxide (ITE). This one-pot synthesis seem to be the novel and attractive way in the producing the stable magnetic structure alternatively to the synthesis of core–shell Fe3O4/SiO2 beads. Depending on the ITE concentration applied in the synthesis, the structures with different morphologies can be obtained as was shown using Atomic Force Microscopy (AFM) and Scanning Electron Microscopy (SEM). The properties of the microstructures were characterized using thermogravimetry measurements (TG), Energy-Disperse Spectroscopy (EDS), Electron Spectroscopy for Chemical Analysis (ESCA) and Solid-state 29Si Magic-Angle Spinning Nuclear Magnetic Resonance Spectroscopy (29Si MAS NMR). Magnetic Force Microscopy (AFM/MFM) application allowed us to correlate the magnetic properties with the microstructure of the materials synthesized. This technique provides the interesting combination of simultaneous examination of the morphology and magnetic features. The conducted magnetic susceptibility and magnetization measurements characterized the magnetic properties of the obtained material.
2 Experimental section
2.1 Materials
Tetraethoxysilane (TEOS, ≥98 %, Fluka), iron(III) ethoxide (ITE, 99 %, ABCR), ethanol (99,8 %, spectroscopic grade) and ammonium hydroxide (25 %, pure p.a., Chempur) were used as received. Millipore-quality water was used during the experiments.
2.2 Preparation of microstructures
Appropriate amounts of TEOS and ITE were dissolved in 6 mL of ethanol and the mixture was shaken for 2 days. 1.5 ml of 16 % (w/w) ammonia (obtained by mixing 1 ml of 25 % (w/w) of ammonia with 0.5 ml of water) was added to the alcoholic solution of TEOS and ITE. The sample was stirred 3 days at room temperature. Then the resulted dispersion was purified by washing with ethanol. Systems with different content of ITE in relation to TEOS were prepared. For the comparison synthesis in samples containing only TEOS or ITE were also carried out. The names and the sample composition were collected in Table 1.
2.3 Scanning Electron Microscopy (SEM)
SEM observations were carried out using a cold field emission scanning electron microscope (FESEM) HITACHI S-4700 equipped with a NORAN Vantage energy dispersion spectrometer. Samples for the measurements were prepared by placing a drop of solution on a silicon plate without any temperature treatment. The solvent was allowed to evaporate at room temperature and subsequently the thin film of carbon was deposited on the sample by sputtering.
2.4 Atomic and magnetic force microscopy (AFM and MFM)
AFM and MFM images were obtained with a NanoScope IV Multimode atomic force microscope (Veeco, Santa Barbara, CA) working in the tapping mode. Standard Veeco silicon cantilevers for measurements in the air of normal spring constant of 40 N/m were used. Samples were prepared on silicon wafers using the spin-coating method. MFM images were acquired using the same microscope and Co/Cr covered cantilevers of normal spring constant of 2 N/m. Before scanning the cantilevers were magnetized with a small magnet. All the MFM images were taken in a lift mode at 1 μm lift high.
2.5 Magnetization measurements
The measurements of magnetization were performed at room temperature using Lake Shore 7300 vibrating sample magnetometer equipped with Stanford SR 830 lock-in nanovoltameter for controlling the current and with constant-flow cryostat (Janis).
2.6 Electron spectroscopy for chemical analysis (ESCA)
ESCA analysis was carried out using a multifunctional ESCA instrument equipped with additional equipment produced by PREVAC. The vacuum chamber of the instrument was equipped with an 5-axial analytical precision manipulator, hemispherical charged particle analyzer XPS, UPS and AES (VG Scient R3000), two anticathodal X-ray tube Mg/Al (power Mg/Al 400/600 W), X-ray monochromator (3 crystals) with the radiation source (a single Al anticathode. 600 W), pumping and measuring vacuum system (provided the vacuum base <1 × 10−8 mbar after 48 h annealing at 150 °C), the charge neutralizing gun (FS40-PS) on the surface of non-conducting samples (energy range 0–500 eV, electron current 1–500 μA), the ion gun to perform depth profiles (IS 40E-PS) and electron gun for the Auger spectroscopy (ES 40C-PS). For ESCA experiment the resulted dispersions were dried at 100 °C for 24 h.
2.7 Thermogravimetry measurements (TG)
Oxidation and thermal decomposition of the dried product was studied using a Mettler-Toledo TGA/SDTA851e thermogravimeter. The samples were heated at a rate of 1 °C min−1 from zero to 1,000 °C in argon and in air flow of 110 ml min−1.
2.8 Solid-state 29Si magic-angle spinning nuclear magnetic resonance spectroscopy (29Si MAS NMR)
29Si MAS NMR spectra were collected on a pulse NMR spectrometer at 59.515 MHz (magnetic field 7.05 T). A Bruker HP-WB high-speed MAS probe was used to record spectra at 7 kHz spinning speed. The ppm scale was referenced to the 29Si resonance of TMS. The sample for 29Si MAS NMR experiment was isolated from dispersion by drying at 100 °C for 24 h.
3 Result and discussion
3.1 Synthesis of microstructures
The main aim of our studies was to obtain magnetic microstructures composed of silica doped with iron atoms. For this reason the sol–gel synthesis was carried out in a mixture of TEOS and its structural analog containing Fe atoms, namely ITE. TEOS is a silicon alkoxide used for the preparation of silica particles in the Stöber synthesis [10]. The synthesis results in formation of the spherical monodisperse silica particles in the size range from 50 nm to 2 μm. The reaction occurs in silicon alkoxide–alcohol–water–ammonia system and it is based on hydrolysis and condensation of a precursor catalyzed by ammonia. Generally, it is also quite well established that the size and distribution of silica particles formed in Stöber synthesis are strongly dependent on the type of silicon alkoxide and alcohol used. Therefore, it was interesting to see whether ITE can participate in the sol–gel synthesis and to determine its effect on the structure of the nanoparticles formed. ITE is a highly reactive late transition metal alkoxide. This brown powder is hard to oxidize and easy to handle material. It has been previously used as a precursor in the synthesis of colloidal FePt nanoparticles (NPs) [13–15]. ITE has also been used for making Fe2O3 nanostructured thin films obtained by dipping silica slides in ethanolic solutions of Fe(OEt)3 [16].
The reaction mixtures contained various amounts of ITE (see Table 1). The content of ITE strongly affected the color of the reaction mixture, which was milky-orange for the 1 % ITE sample, brick-red for 10 % ITE sample, and dark-brown for 100 % ITE sample (Fig. 1). For all samples the sedimentation was observed. Using variety of physicochemical methods it was demonstrated that the addition of ITE to TEOS has significant, concentration dependent, effect on morphology of the product.

Photograph of the dispersions obtained in the sol–gel synthesis in samples: a 1 % ITE, b 10 % ITE, and c 100 % ITE
3.2 Morphology of microstructures
3.2.1 SEM analysis
The morphologies of synthesized materials were examined using SEM. Figure 2 presents the SEM micrographs for the objects obtained in the mixtures of various compositions. The individual silica particles synthesized from TEOS (in the absence of ITE) are of 445–667 nm (Fig. 2a). They are of regular shapes and are quite smooth. Figure 2 b–g show the images for particles obtained in the presence of ITE. One can observe that ITE significantly affects the size, shape and the morphology of the obtained materials. At low content of ITE, the formation of spherical particles is accompanied by the appearance of irregular aggregate-type of materials. The fraction of the later is increasing with increase of the ITE content in reaction mixture and it is the largest for the product of Stöber synthesis carried out for ITE (without addition of TEOS) (Fig. 2h).

SEM micrographs of the materials obtained in Stöber synthesis in the TEOS-ITE systems: a 0 % ITE, b 0.1 % ITE, c 0.5 % ITE, d 1 % ITE, e 2 % ITE, f 5 % ITE, g 10 % ITE, h 100 % ITE
Based on the SEM micrographs one could be concerned that it is very probable that during the Stöber synthesis TEOS and ITE created two different types of materials. To gain information whether the spherical objects contain the iron atoms, the energy-disperse spectroscopy (EDS) analysis were carried out for samples c–g. The results were collected in the Table 2. In all samples, the presence of iron in spherical objects, as well as in aggregates was confirmed. The content of Fe in the product is dependent on the content of ITE used in synthesis.
3.2.2 AFM analysis
Atomic force microscope (AFM) measurements were done for the 100 % ITE sample and they were helpful in determination of sizes of investigated objects. Figure 3 shows the AFM image of the objects formed in that sample. The results are in agreement with the described above findings from the SEM analyses. It can be seen some kind of aggregates, which are built from smaller components. The average size of the composite particles was about 8–12 nm in height and 30–40 nm in width, respectively.

AFM micrographs material obtained as a product of Stöber synthesis in the 100 % ITE sample. a 3D image of the surface, b cross-sections
3.3 ESCA analysis
Because of the interesting morphology obtained in the case of the system 1 % ITE we have decided to study properties of that material more carefully. The TG, ESCA analysis and some magnetic measurements have been done to characterize that structure.
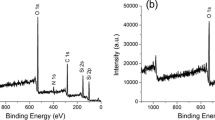
ESCA was used to reveal the changes in chemical composition of the ITE, which occurred during Stöber synthesis. ESCA spectrum for 1 % ITE sample was difficult to interpret because of the low iron content. Thus, the 10 % ITE sample was used for these studies. Full-scanning ESCA spectrum of the 10 % ITE sample is shown in Fig. 4a. Signals from four detectable elements (silicon, carbon, oxygen, and iron) were recorded at 103.4 (Si 2p), 285.4 (C 1s), 532.8 (O 1s) and about 708–730 eV (Fe 2p). The Fe 2p peak is the least detectable compared to rest of the peaks as shown in Fig. 4b (high resolution ESCA spectra of Fe 2p). Some of the iron oxide structures can be identified on the basis of their core line binding energies. The small differences in the binding energy values of the core-electrons of the three oxides FeO, Fe3O4 and Fe2O3 have been observed since they contain FeII and FeIII ions in different amounts. FeO contains exclusively FeII ions, which are octahedrally coordinated in a cubic lattice. Fe3O4 contains both FeII and FeIII ions in an inverse cubic spinel structure, with the FeII ions in octahedral sites and FeIII ions half in octahedral and half in tetrahedral sites, whereas the α-Fe2O3 contains only FeIII ions in a lattice having the rhombahedral A12O3 structure. That is the reason why the binding energies of the photoelectron peaks can be used to provide an identification of FeII and FeIII species and of iron oxides themselves. The use of the more intense Fe 2p3/2 and Fe 2p1/2 peaks plus associated satellite peaks are particularly useful for that purpose [17].

ESCA spectra of: a full scan, b high-resolution of Fe 2p of the 10 % ITE sample
In the FeO spectra the Fe 2p3/2 and Fe 2p1/2 binding energies are observed at 710.7 and 724.4 eV respectively. In a case of the Fe3O4 oxide the Fe 2p3/2 and Fe 2p1/2 binding energy values are 710.8 and 724.7 eV, respectively. For Fe2O3 the Fe 2p3/2 peak has a binding energy of 711.6 eV and the Fe 2p1/2 peak is 725.1 eV. The satellite peaks occurring at approximately 8.0 eV above the Fe 2p3/2 peak is characteristic of FeIII species, while the occurrence of a satellite peak at approximately 6.0 eV above the Fe 2p3/2 peak is characteristic of an FeII species [18]. In fact, the oxide species can be distinguished by the about 1.0 eV difference in their core line binding energies. It has been noticed that for the examined material the Si 2p, C 1s and O 1s are predominant peaks. The Fe 2p spectrum is detected in spite of low intensity, as it is shown in the Fig. 4b. We have observed the considerable broadening of the Fe 2p peaks: the Fe 2p3/2 is in the range of the 708–716 eV and Fe 2p1/2 are in about 721–730 eV. The maximum value for Fe 2p3/2 peak is about 711.9 and 725.6 eV for Fe 2p1/2 peak which might be assigned to the presence of the Fe2O3 oxide. Nevertheless, taking into account the very small changes in binding energy of the above mentioned iron oxide structures on the basis of the presented ESCA spectra it is difficult to identify precisely the kind of Fe species, which are formed during synthesis.
3.4 29Si MAS NMR analysis
To gain more insight into the chemical structure of the obtained material 29Si MAS NMR technique was applied. Spectra obtained for 0, 2, and 5 % ITE samples are shown in Fig. 5. Because of the strong magnetism it was not possible to analyze sample with higher content of iron oxide. For all these samples the observed signals are located at the same chemical shift but their intensities differ from each other. The integrated peak area of the 29Si spectrum for 2 and 5 % ITE samples represent 70 and 55 %, respectively, of that characteristic for peak area of 0 % ITE sample. Because the amount of TEOS used in synthesis was the same for all samples the number of silicon atoms can be assumed to be the same in each product. The lowering of the 29S integrated peak area in samples containing the iron species reflect their effect on silicon atoms.

29Si MAS NMR spectra of the 0 % ITE, 2 % ITE and 5 % ITE sample
Bruni et al. [19] have investigated the interactions of iron oxide with the silica matrix in a Fe2O3–SiO2 nanocomposite prepared by a sol–gel method and subsequently annealed at high temperature (up to 900 °C). Using Near-, Mid-, and Far-IR, 29Si MAS- and 1H NMR techniques they have shown that the Fe2O3 nanoparticles interacted with the silica or silanol groups at the surface of the cavities in which they were form. These authors have noticed that for the nanocomposite treated at higher temperature the integrated peak area of the 29Si spectrum decreased and in the Mid-IR spectra they have observed the signal at 590 cm−1 indicating formation of Fe–O–Si bonds at the nanoparticle-matrix interface. Additionally, they have also shown that some kind of interactions between iron oxide nanoparticles and silica matrix operate even at low temperature. However, due to the presence of iron (III) paramagnetic ions in close proximity to the Si atoms, the 29Si signal broadens while its intensity decreases and the signal becomes beyond detection. This broadening takes place only if the distance between iron and silicon atoms is not large. That might suggest that at low temperature, even in the absence of Fe–O–Si bonds, the interactions between Si and Fe can occur through SiOH groups present at the matrix surface. We have also carried out the FTIR studies (the supplementary figure) but have not observed the signal which could be ascribed to Fe–O–Si bond. On the basis of the above mentioned findings we have postulated that our synthesis resulted in formation of the materials in which the silica particles with –OH groups at the surface interact with iron oxide species. The distance between them are small enough to cause the changes which we have observed in 29Si MAS NMR spectra.
3.5 Calcination
To determine whether the properties of obtained materials have changed after heat treatment for sample 1 % ITE the calcination was performed. The sample was calcinated on the silica plates in argon and in air atmosphere, respectively. The calcination was performed in 600 °C, with the 10 °C min−1 rate. SEM analyses for the calcination products were conducted. Figure 6 presents the SEM images before and after calcination. Further condensation of the silica matrix had occurred on heating. After calcination the siloxane network is more highly condensed and more highly ordered, but there are no observable changes in the morphology. The energy-disperse spectroscopy (EDS) analysis was carried out and the results did not show any noticeable differences.

SEM micrographs of the 1 % ITE sample after calcination in: a air atmosphere, b argon atmosphere
3.6 TG measurements
It is well established that there are two distinct mass loss steps in the thermograms of silicas. The first step is mainly connected with the removal of the physically adsorbed water from the silica surface, whereas the second step is due to slow condensation of silanols. The temperature where physically adsorbed water was considered to be completely released was found to be in the range from 100 to 130 °C and was dependent on the type of silica [20]. However, according to Zhuravlew [21] the temperature corresponding to the ending of dehydratation and the beginning of dehydroxylation is in the range of 190 ± 10 °C, which means that small amount of physically adsorbed water remains at the sample surface up to 200 °C. Heating of the silica samples at 1,100 °C results in considerable decrease in the amount of isolated hydroxyls, but for the complete removal of the rest of silanols, the temperatures has to be higher than 1,200 °C [20].
The thermograms for the 1 % ITE sample are depicted in Fig. 7. TG profiles have been obtained in oxidative and inert atmosphere. In both cases three steps of the mass losses have been observed. TG profile in oxidative atmosphere shows onset of oxidation at about 60 °C. First rapid oxidation step accounts for 2.1 % of total 10.2 % mass loss, the second step is at about 250 °C and accounts for 4.0 % mass loss and the last one occurs at about 500 °C with mass loss of 4.1 %. TG profiles obtained in inert atmosphere show very similar decomposition and desorption. We observed three steps of the mass loss: at 60 °C (1.8 %), 250 °C (4.2 %) and about 525 °C (4.8 %). Total mass loss in an inert atmosphere accounts for about 10.8 %.

TG profiles obtained in oxidative and inert atmosphere for 1 % ITE sample
Based on the literature, we can assume that the first step of decomposition is associated with the removal of physically adsorbed water and solvents used in the synthesis. Although the sample before TG experiments was dried at 100 °C, it is possible that the some amounts of the mixture of water, ethanol and ammonium were still trapped in the material before the TG experiment. The continuation of the thermal treatment causes the condensation of silanol groups of the silica surface resulting in the release of water and formation of siloxane group. The dehydratation and dehydroxylations reactions overlap to some extent so it is difficult to define the remarkable border between each of them. In fact, the TG profile for our sample differs from that observed in typical experiment for the silica. That reflects the presence of iron compound produced during Stöber synthesis.
Barick et al. [22], in his work on fabrication of composite materials in situ generation of γ- and ε-Fe2O3 nanoparticles in a SiO2 matrix through sol–gel process, has found that TG profiles are strongly dependent on the concentration of Fe3+. He has observed two main steps of thermal decomposition for samples with lower Fe3+ concentration (x = 0.05 and 0.1) and three steps for sample with higher Fe3+ concentration (x = 0.2). The alcogel samples exhibit significant weight loss in the low temperature range (25–100 °C) mainly assigned to the removal of solvent (water and ethanol absorbed into the matrix). The thermal decomposition in the temperature range of 100–1,000 °C was explained by the evolution of organic species and structurally bonded –OH groups. For samples with the highest content of Fe3+ these authors observed the thermal decomposition in the temperature range of 100–235 °C which was accompanied with an exothermic effect at 224 °C assigned to the formation of γ-Fe2O3 from the thermal decomposition of γ-FeOOH originally formed during sol–gel synthesis [23].
Taking into account the above information we can assume that the concentration of ITE in analyzed sample was high enough to affect the TG profile of the silica matrix. The thermal-treatment removes the volatile substances and simultaneously causes the densification of silica matrix along with the formation of some iron oxide nanoparticles.
3.7 Magnetic measurements
3.7.1 MFM analyses
Magnetic force microscopy technique (MFM) was used to confirm magnetic properties of the samples. Microscope worked in interleave mode scanning all points on sample twice—in the tapping mode to obtain AFM topography and phase images and in the lift mode (1 μm) to obtain pictures of MFM phase [24]. The images obtained for 1 % ITE sample, 1 % ITE after calcination in argon and air are shown in Fig. 8. It was confirmed that all of them are magnetic materials.

Images of AFM: a topography, b AFM phase and c corresponding MFM phase of Stöber synthesis products for 1 % ITE sample (1), the same sample after calcination in argon (2) and sample after calcination in air (3)
There is very good correlations between AFM and MFM images (Fig. 8a–c) for all samples. The aggregates shown on AFM topography and phase images reveal contrast also in magnetic mode (MFM), which occurs only for magnetic sample which interacts with tip. That suggests the material shows homogeneous distribution of the iron oxide within the sample.
3.7.2 Measurements of magnetic susceptibility
One of the value which characterizes the magnetic features of the material is the magnetic susceptibility (χ). In the case of the paramagnetic materials the value of magnetic susceptibility χ > 0, whereas for diamagnetic substances χ < 0 (χ = 0 in vacuum) [25]. For example, the magnetic susceptibility for paramagnetic materials is in the range: +0.65 × 10−6 emu/g for aluminium, +106.37 × 10−6 emu/g for oxygen. For diamagnetic materials the value for copper is equal –0.09 × 10−6 emu/g and for silicon –0.13 × 10−6 emu/g [26].
Figure 9 shows the magnetic susceptibility for our sample obtained in system containing 1 % ITE. The magnetic susceptibility value for that sample was found to be equal to 5.01 × 10−6 emu/g-sample, which is in the range characteristic for paramagnetic material. That small value of magnetic susceptibility is due to the low content of iron in the sample. Nevertheless the magnetic susceptibility calculated for 1 g of iron present in the sample is equal 1.73 × 10−3 emu/g-Fe.

The results of magnetic susceptibility measured for the 1 % ITE sample using a magnetic balance
3.7.3 Magnetization measurements
In order to compare the magnetic properties of our material with these determined for iron oxides-silica composites reported in literature [22, 27] we have performed the additional magnetization measurements. Figure 10 presents magnetization curves for the 1 % ITE sample. The curve has a symmetrical sigmoid shape—when the magnetic induction decreased, the magnetization decreased and eventually reached zero, no hysteresis is observed. Nevertheless the material does not exhibit saturation behavior up to a maximum magnetic induction used in the experiment. There was only possible to determine the maximum magnetization which is equal 0.028 emu/g-sample, what translates to 10.01 emu/g-Fe. It has been demonstrated previously that the magnetization of core–shell superparamagnetic Fe3O4/silica nanoparticles have reached the saturation [27]. The value of the saturation magnetization for pristine Fe3O4 nanoparticles is about 60.5 emu/g–Fe3O4 (which can be expressed as a 83.6 emu/g-Fe) whereas for core–shell magnetite/silica composite particles with 17.3 wt. % of magnetite that value decreased to 7.5 emu/g-sample (59.6 emu/g-Fe) [27]. It has been shown also that coating of magnetic particles with non-magnetic materials such as silica influences the value of magnetization due to the quenching of surface moments [28]. The thicker silica shell—the lower the saturation magnetization [8].

Magnetization curves in a function of magnetic induction for the 1 % ITE sample
Barick et al. [22] reported on the preparation of dispersed iron oxide Fe2O3 nanoparticles in silica matrix using sol–gel process. The resulting samples contained various amounts of iron oxide. The synthesized samples were thermally annealed. Magnetization measurements have shown that all samples exhibit non-saturation behavior up to a maximum field of 20 kOe. The observed maximum magnetization for samples treated at 800 °C was equal: 0.27, 0.38 and 1.23 emu/g-sample depending on the Fe3+ concentration. The authors have explained the low values of maximum magnetization by taking into account the presence of large amount of non-magnetic SiO2 matrix. The lack of the saturation has been interpreted as resulting from a coexistence of fraction of superparamagnetic particles with ferri/ferromagnetic coupled particles.
Taking into account the above mentioned studies we can concluded that maximum magnetization value of the 1 % ITE (0.028 emu/g-sample) is about ten times lower than that presented in Barick studies [22] for the smallest iron oxide content (0.27 emu/g-sample). That reflects the high content of silica in our material. To make a comparison of our results with these presented in literature we have expressed magnetization in units emu per gram of iron present in the samples. The value obtained for 1 % ITE sample was found to be 10.01 emu/g-Fe and it is of the same order of magnitude as that presented in the literature—59.6 emu/g-Fe [27].
Our material is attractive as it can be obtained using a simple one-pot synthetic strategy. Additionally, even if only 1 % wt. of ITE was used in synthesis the process resulted in preparation of material with magnetic properties. Furthermore our synthetic strategy can be used for preparation of other types of magnetic composite microstructures.
4 Conclusions
The silica materials containing various amounts of iron oxide incorporated were obtained from TEOS and ITE mixtures in the Stöber synthesis. The presence of iron in all materials synthesized was confirmed by EDS analyses. The morphology of these materials is strongly dependent on the amount of Fe present. The SEM micrographs and AFM measurements indicated that at low Fe content the formation of regular, spherical particles is accompanied by the appearance of irregular aggregate-type of materials. The fraction of the later is increasing with increase of the Fe content in reaction mixture. TG analyses indicated that even small amount of iron present in the particles can affect their thermal behavior. ESCA analysis has not provided an unambiguous answer concerning the creation of defined phase of iron oxide. 29Si MAS NMR spectra have suggested that obtained materials consist of silica nanoparticles which interacted with iron oxide species. MFM, magnetic susceptibility and magnetization measurements have demonstrated that our materials have magnetic properties. Values of magnetic susceptibility indicated that the material is paramagnetic. The magnetization measurements have revealed that sample obtained in a presence of 1 % ITE did not exhibit saturation behavior up to a maximum magnetic induction used. The high content of silica in our material has affected the maximum magnetization value which was equal to 0.028 emu/g-sample, what translates to 10.01 emu/g-Fe. MFM experiment suggests that the homogenous distribution of iron oxide in the silica matrix obtained. Overall, it can be concluded that even the small content of ITE (1 %wt) in the reaction mixture resulted in formation of the paramagnetic materials. It is, however, expected that the material with more homogeneous morphology would show better properties. The studies aimed at obtaining such materials are currently in progress.
References
Morales MA, Mascarenhas AJS, Gomes AMS, Leite CAP, Andrade HMC, de Castilho CMC, Galembeck F (2010) J Colloid Interface Sci 342:269–277
Babes L, Denizot B, Tanguy G, Jacques J, Jeune L, Jallet P (1999) J Colloid Interface Sci 212:474–482
Xu ZP, Niebert M, Porazik K, Walker TL, Cooper HM, Middelberg APJ, Gray PP, Bartlett PF, Lu GQ (2008) J Control Release 130:86–94
Fornara A, Johansson P, Petersson P, Gustafsson S, Quin J, Olsson E, Ilver D, Krozer A, Muhammed M, Johansson C (2008) Nano Lett 8:3423–3428
Chen F, Shi R, Xue Y, Chen L, Wan Q-H (2010) J Magn Magn Mater 322:2439–2445
Lu Y, Yin Y, Mayers BT, Xia Y (2002) Nano Lett 2:183–186
Wang Ch, Yan J, Cui X, Wang H (2011) J Colloid Interface Sci 354:94–99
Sun X, Liu F, Sun L, Wang Q, Ding Y (2012) J Inorg Organomet Polym 22:311–315
Butterworth MD, Illum L, Davis SS (2001) Colloids Surface A 179:93–102
Stober W, Fink A, Bohn E (1968) J Colloid Interface Sci 26:62–69
Staszewska M, Dzieciuch M, Lewandowska J, Kepczynski M, Zapotoczny S, Oszajca M, Łatkiewicz A, Nowakowska M (2011) J Sol-Gel Sci Technol 59:276–282
Santra S, Tapec R, Theodoropoulou N, Dobson J, Hebard A, Tan W (2001) Langmuir 17:2900–2906
Saita S, Maenosono S (2005) Chem Mater 17:3705–3710
Saita S, Maenosono S (2005) Chem Mater 17:6624–6634
Maenosono S, Suzuki T, Saita S (2008) J Magn Magn Mater 320:L79–L83
Armelao L, Granozzi G, Tondello E, Colombo P, Principi G, Lottici PP, Antonioli G (1995) J Non-Cryst Solids 192&193:435–438
Mills P, Sullivan JL (1983) J Phys D Appl Phys 16:723–732
Iijima Y, Miyoshi K, Saito S (1999) Surf Interface Anal 27:35–42
Bruni S, Cariati F, Casu M, Lai A, Musinu A, Piccaluga G, Solinas S (1999) Nanostruct Mater 11:573–586
Ek S, Root A, Prussa M, Niinisto L (2001) Thermochim Acta 379:201–212
Zhuravlew LT (1993) React Kinet Catal L 50:15–25
Barick KC, Varaprasad BSDChS, Bahadur D (2010) J Non-Cryst Solids 356:153–159
Yen FS, Chen WC, Yang JM, Hong ChT (2002) Nano Lett 2:245–252
Moskalenko AV, Yarova PL, Gordeev SN, Smirnov SV (2010) Biophys J 98:478–487
Gupta AK, Gupta M (2005) Biomaterials 26:3995–4021
Kobayashi Y, Saeki S, Yoshida M, Nagao D, Konno M (2008) J Sol-Gel Sci Technol 45:35–41
Xu H, Tong N, Cui L, Lu Y, Gu H (2007) J Magn Magn Mater 311:125–130
Acknowledgments
This work was supported by the European Union from the resources of the European Regional Development Fund under the Innovative Economy Programme (grant coordinated by JCET-UJ, No POIG.01.01.02-00-069/09).
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lewandowska, J., Staszewska, M., Kepczynski, M. et al. Sol–gel synthesis of iron oxide–silica composite microstructures. J Sol-Gel Sci Technol 64, 67–77 (2012). https://doi.org/10.1007/s10971-012-2828-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-012-2828-1