
Abstract
P2X7 sites are emerging targets for molecular imaging research, notably in the context of neurodegeneration and inflammatory conditions. Therefore, we prepared a precursor for (radio)iodination of the P2X7 ligand VPGIS191. We then developed a radioiodination method with 123I for SPECT with a radiochemical yield of 71 ± 13% and 125I for autoradiography with a radiochemical yield of 85 ± 6% Autoradiography of [125I]VPGIS191 in mouse brain cryostat sections demonstrated approximately 36 nM binding affinity and Bmax of approximately 400 pmol/gram tissue for P2X7 binding sites. VPGIS191 (cis) had a two-fold lower affinity compared to its geometric trans-isomer TZ6019.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
P2X7 (Fig. 1) is a transmembrane ligand-gated cation channel that is activated by extracellular adenosine triphosphate (ATP) and certain other adenosine nucleotides, thereby mediating cytokine release from cells of hematopoietic lineage such as microglia and macrophages in the brain, retina, and peripheral nervous system [1]. Activation of P2X7 receptors mediates host immune responses participating in the regulation of apoptosis and inflammation [2] in a variety of pathologies including Alzheimer’s and Parkinson’s diseases [3], cardiovascular disorders [4], and cancers [5]. Due to their potential as markers or therapeutic targets in diverse disease conditions, P2X7 receptors are an emerging topic for molecular imaging by positron emission tomography (PET) and single photon computer tomography (SPECT). However, clinical molecular imaging of P2X7 receptors is in its infancy, in part due to the inadequate sensitivity of available radioligands [6].

Cartoon depicting the cellular locations of P2X7 receptors and their predicted involvement in various pathologies with an inflammatory component. Created with BioRender.com
The few P2X7 ligands developed to date for molecular imaging have called for straightforward syntheses of precursors for simple [11C]methylation or [18F]fluorination for PET, and [123I]iodination for SPECT. The main classes of these P2X7 ligands are pyroglutamic acid-based (e.g. [11C]GSK-1,482,160 [7], [123I]TZ6019 [8]), triazole-based (e.g. [18F]JNJ-64,413,739 [9], [3H]JNJ-54,232,334 [10]) and adamantane-based compounds (e.g., [11C]SMW139 [11]). The pyroglutamic acid-based radioligands identified thus far (Fig. 2) include [11C]- or [18F]GSK1482160 [7, 12], the [11C]halo-GSK-1,482,160 analogs [9], [18F]IUR-1601 and [18F]IUR-1602 [13, 14], and [123I]TZ6019 [8]. The characterization and preliminary results of their binding properties (Ki, IC50, Kd, Bmax) in HEK293 hP2X7R (HEK) cells are shown in Table 1. Apart from [11C]F-GSK1482160, [123I]TZ6019 and [18F]IUR-1602, the pyroglutamic acid-based ligands have high affinity for P2X7. Notably, [11C]GSK1482160 had 1.2 ± 0.1 nM affinity (Kd) [15] versus 19.3 ± 2.8 nM for [123I]TZ6019 [8]. These lead compounds for PET and SPECT imaging present unexplored opportunities for structural modification alternative radiosynthetic routes.

Pyroglutamic acid-based P2X7 ligands
The only published report of a SPECT ligand for P2X7 receptors indicated moderate affinity and specific binding of [123I]TZ6019 in HEK cells [8]. However, the [123I]TZ6019 was prepared from commercially unavailable (E)-(3-bromoprop-1-en-1-yl)-tributylstannane, which has likely impeded the broader use of the radioligand. Our compilation of literature data led us to note that E-3-iodoallyl-derived radioligands often demonstrate slightly higher affinity for their various receptor targets as compared to Z-3-iodoallyl-derived radioligands [17,18,19,20]. Therefore, we determined to prepare the [123I]TZ6019 geometric Z-isomer from commercially available (Z)-(3-bromoprop-1-en-1-yl)-tributylstannane, and evaluate its binding to P2X7 receptors in rodent brain sections. In this study, we describe the syntheses of a VPGIS191 precursor and the VPGIS191 chromatographic standard, and procedures for precursor iodination with 123I for SPECT and 125I for autoradiography. Finally, we made a preliminary evaluation of [125I]VPGIS191 binding to P2X7 receptors in mouse brain cryostat sections, comparing our results with previously reported KD/Bmax values for [123I]TZ6019 in HEK cells.
Experimental
General methods
For thin-layer chromatography (TLC), we used aluminium silica gel sheets with detection in UV light (TLC silica gel 60 F254, Merck). For TLC visualization, we applied a dilute solution of H2SO4 in MeOH and heated the plates. For column chromatography, we used 30–60 μm silica gel (ICN Biomedicals, Costa Mesa, USA). NMR spectra were recorded using Agilent-MR DDR2 (Varian, Palo Alto, USA). HRMS were measured using an LTQ ORBITRAP VELOS with HESI+/HESI- ionization (Thermo Scientific, Waltham, MA, USA). A quadrupole LC/MS-ESI with an Infinity III LC system (Agilent Technologies, Santa Clara, USA) served for LR-MS and HPLC-MS analyses (10 μm C18 column: 100 mm; UV detection). For the characterization of radioactive products, we undertook HPLC analyses (C18 columns, UV, and RAD detections). Purification procedures employed SPE columns (Bond Elut Plexa, Agilent Technologies, Santa Clara, USA, and Strata, Phenomenex, Torrance, USA). Materials for quantitative autoradiography included brains of female Balb/c mice (Palacky University, Olomouc, Czech Republic), Tissue-Tek gel (Sakura, Torrance, USA), Superfrost plus microscope slides (Fisher Scientific, Hampton, USA), a microtome cryostat (Leica Biosystems, IL, USA), phosphor storage screens (BAS), and a Cyclone Plus Phosphor Imager (PerkinElmer, Waltham, USA).
Materials
Bachem (Bubendorf, Switzerland): L-pyroglutamic acid (> 99.0%); Lach-Ner (Neratovice, Czech Republic): dichloromethane (99%); Sigma-Aldrich (Missouri, USA): acetic acid (99.7%), benzylamine (99%), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (≥ 98%), dimethylformamide (≥ 99.8%), chloroform (≥ 99%), hexane (95%), hydrochloride acid (37%) 1-hydroxybenzotriazole hydrate (≥ 97%), lithium bis(trimethylsillyl)amide (1 M solution in THF), N-iodosuccinimide (95%), methyl trifluoromethanesulfonate (≥ 98%), sodium chloride (≥ 99.0%), (+)-sodium L-ascorbate (≥ 98%), sodium sulphite (≥ 98%), tetrabromomethane (99%), tetrabutylammonium hydroxide (≥ 99.0%), tetrahydrofuran (≥ 99.0%), trifluoroacetic acid (≥ 99.0%), triphenylphosphine (99%); Synthonix (Wake Forest, USA): (Z)-3-(tributylstannyl)prop-2-en-l-ol (> 99%); VWR (Radnor, USA): 2-chlor-3-(trifluormethyl)benzylamine (≥ 97%). The solvents for column chromatography and reactions for the synthesis of VPGIS191 were purchased from PENTA (Prague, Czech Republic) and were used as delivered (all of p.a. quality). For radio-iodinations, sodium iodide I123 (37 MBq/mL) with sodium chloride, sodium hydrogen carbonate and water for injection were from THP Medical Products Vetriebs GmbH (Wien, Austria). Sodium iodide I125 (3550 MBq/mL) solution in sodium hydroxide was purchased from Izotop (Budapest, Hungary).
Syntheses of 1, and a precursor for VPGIS191
(S)-N-(2-Chloro-3-(trifluoromethyl)benzyl)-5-oxopyrrolidin-2-carboxamide (1): To a solution of 2-chloro-3-(trifluoromethyl)phenyl)methanamine (650 mg, 3.1 mmol) in 30 mL of dry CH2Cl2 L-pyroglutamic acid (426 mg, 3.3 mmol), N-hydroxybenzotriazole (HOBt, 502 mg, 3.7 mmol) and ethyl(dimethylaminopropyl)carbodiimide (EDCI, 713 mg, 3.7 mmol) were added, respectively. The mixture was put aside at room temperature for 14 h, whereupon solvents were removed under reduced pressure and the residue was purified by column chromatography in CHCl3-MeOH (20:1–10:1, v/v) to give 1 (649 mg, 2 mmol, 61%) as a colorless solid. After lyophilization, the product was stored at 4 °C as a white foam. RF = 0.25 in CH2Cl2-MeOH 20:1 (v/v). 1 H NMR (400 MHz, acetone-d6) δ ppm: 2.07–2.33 (m, 3 H), 2.40–2.52 (m, 1 H), 4.25 (dd, J = 9.0, 3.9 Hz, 1 H), 4.53–4.65 (m, 2 H), 7.29 (br s, 1 H), 7.47–7.54 (m, 1 H), 7.72 (t, J = 6.7 Hz, 2 H), 8.10 (br s, 1 H). 13 C NMR (101 MHz, acetone-d6) δ ppm: 27.01 (CH2, pyrr), 41.82 (CH2, pyrr), 57.83 (NH-CH2), 124.50 (q, J = 272.4 Hz, CF3), 127.65 (q, J = 5.6 Hz, ArCH), 128.45, 129.21 (q, J = 30.5 Hz, ArCH), 131.65 (d, J = 1.5 Hz, ArCH), 134.10 (ArCH), 134.12 (ArCH), 140.53 (ArCH), 174.21 (-NHC = O), 179.01 (C = O, pyrr). MS-ESI: for C13H12CIF3N2O2 calcd. 320.05 Da, found 338.7 [M + NH4]+.
(S,Z)-N-(2-Chloro-3-(trifluoromethyl)benzyl)-5-oxo-1-(3-(tributylstannyl)allyl)pyrrolidine-2-carboxamide (2): Step 1. To a solution of (Z)-3-(tributylstannyl)prop-2-en-l-ol (1 g, 2.9 mmol) in 40 mL of THF triphenylphosphine (982 mg, 3.7 mmol) and CBr4 (1.23 g, 3.7 mmol) were added. The mixture was stirred at 0 °C (on ice) for 3 h, and then at room temperature for 14 h. The solvents were removed under reduced pressure and the residue was purified by column chromatography with hexane to give (Z)-(3-bromprop-l-en-l-yl)tributylstannane [21] (800 mg, 1.95 mmol, 68%) as a colorless liquid. The product thus obtained was used directly in the next reaction. 1 H NMR (400 MHz, CDCl3) δ ppm: 0.80–0.87 (m, 15 H), 1.18–1.30 (m, 6 H), 1.38–1.46 (m, 6 H), 3.89 (dd, J = 6.7, 0.9 Hz, 2 H), 6.05 (dt, J = 18.5, 6.6 Hz, 1 H), 6.22 (d, J = 18.7 Hz, 1 H). 13 C NMR (101 MHz, CDCl3) δ ppm: 9.6, 13.7, 27.2, 29.0, 35.8, 135.1, 143.0.
Step 2. To a solution of 1 (300 mg, 0.93 mmol) in 5 mL of DMF, stirred under an atmosphere of argon at 0 °C (on ice), 2 mL of lithium bis(trimethylsilyl)amide solution in THF (1 M) was added. Thereafter, (Z)-(3-bromprop-l-en-lyl)tributylstannane (800 mg, 1.95 mmol) was added dropwise. The mixture was stirred at 0 °C (on ice) for 10 min, and then at room temperature for 3 h, whereupon the reaction was stopped by adding 25 mL of aqueous HCl (1 M). The resultant mixture was extracted with EtOAc (3 × 50 mL). The combined organic layer was washed with a saturated solution of NaCl (2 × 50 mL) and dried over Na2SO4. The solvent was evaporated under reduced pressure and the residue was purified by column chromatography (hexane-EtOAc 4:1–1:2, v/v) to give 2 (268 mg, 3.7 mmol) as a pale-yellow viscous substance. RF = 0.3 in hexane-EtOAc 1:1. The product was visualized on TLC by gaseous iodine (brown spot). The product 2 was injected onto an analytical HPLC (YMC-Triart C18 column, 150 × 4.6 mm, 5 μm; gradient 40/60–90/10, v/v AcCN/0.1% TFA, 1 mL/min, tR = 28 min). 1 H NMR (400 MHz, CDCl3) δ ppm: 0.75–0.98 (m, 15 H, overlap 3×CH2 a 3×CH3), 1.18–1.37 (m, 6 H, 3×CH2), 1.37–1.51 (m, 6 H, 3×CH2), 1.96–2.10 (m, 1 H, CH), 2.16–2.53 (m, 3 H, overlap, 3×CH, 1×CH2), 3.51 (br dd, J = 14.9, 7.4 Hz, 1 H, CH), 4.07 (br dd, J = 9.2, 2.5 Hz, 1 H, CH), 4.21 (br dd, J = 14.9, 5.5 Hz, 1 H, CH), 4.58 (d, J = 5.9 Hz, 1 H, CH), 6.04 (br d, J = 12.5 Hz, 1 H, CH), 6.28 (ddd, J = 12.8, 7.3, 5.7 Hz, 1 H, CH), 7.07 (br t, J = 5.7 Hz, NH), 7.29–7.36 (m, 1 H, CH), 7.56 (br d, J = 7.4 Hz, 1 H, CH), 7.63 (br d, J = 7.8 Hz, 1 H, CH). 13 C NMR (101 MHz, CDCl3) δ ppm: 10.23 (d, JCSn = 343.3 Hz, 3×CH2), 13.66 (3×CH3), 24.21 (CH2, pyrr), 27.23 (d, JC-Sn = 58 Hz, 3×CH2), 28.08 (d, JC-Sn = 21.4 Hz, 3×CH2), 29.52 (1×CH2, pyrr), 41.48 (NH-CH2), 47.24 (d, JC-Sn = 38.9 Hz, N-CH2), 60.90 (CH, pyrr), 122.75 (q, J = 273.6 Hz, CF3), 126.94 (q, JC-F = 5.9 Hz, ArCH), 129.06 (q, JC-F = 31.3 Hz, ArCH), 131.60 (d, JC-F = 1.5 Hz, ArCH), 133.47 (ArCH), 134.60 (d, JC-Sn = 339.52 Hz, = CH) 134.61 (ArCH), 137.78 (ArCH), 141.61 (= CH) 171.64 (-NHC = O), 175.51 (C = O, pyrr). MS-ESI: for C28H42ClF3N2O2Sn calcd. 650.2 Da, found m/z 651.2 [M + H]+.
(S,Z)-N-(2-Chloro-3-(trifluoromethyl)benzyl)-1-(3-iodoallyl)-5-oxopyrrolidine-2-carboxamide (VPGIS191):
Method I. To a solution of 2 (6.5 µg, 10 nmol) in 20 µL 1% acetic acid in MeOH was added 4 µL N-iodosuccinimide (10 mg/mL 1% acetic acid in MeOH). The mixture was stirred at RT for 15 min, whereupon the reaction was stopped with sodium thiosulfate (5 mg, 1.6 nmol). Subsequently, the mixture was diluted with 1 mL of distilled water and passed through a preconditioned SPE cartridge (2 mL, 20% EtOH), and eluted with 2 mL of 80% EtOH. The product 3 was injected onto an analytical HPLC (YMC-Triart C18 column, 150 × 4.6 mm, 5 μm; gradient 40/60–90/10, v/v AcCN/0.1% TFA, 1 mL/min, tR = 12 min).
Method II. To a solution of 2 (32.5 mg, 0.05 mmol) in 3 mL of CHCl3 was added 0.5 mL solution of iodine in CHCl3 (0.1 M) dropwise. The mixture was stirred at RT for 20 h, and then diluted with 4 mL of CHCl3, and washed with saturated solution of first Na2S2O3 (6 × 15 mL), and then with a saturated solution of NaCl (15 mL). The combined organic layer was dried over Na2SO4. The solvents were removed under reduced pressure and the residue was purified by column chromatography CHCl3-MeOH (20:1–10:1, v/v) to give VPGIS191 (12.7 mg, 52%) as a brown solid. The product was injected onto an analytical HPLC (YMC-Triart C18 column, 150 × 4.6 mm, 5 μm; gradient 40/60–90/10, v/v AcCN/0.1% TFA, 1 mL/min, tR = 12 min). 1 H NMR (400 MHz, CDCl3) δ ppm: 2.02–2.12 (m, 1 H), 2.27–2.41 (m, 2 H), 2.50–2.61 (m, 1 H), 3.75 (br dd, J = 15.5, 7.6 Hz, 1 H), 4.00 (br dd, J = 8.6, 2.7 Hz, 1 H), 4.25 (br dd, J = 15.3, 5.1 Hz, 1 H), 4.63 (d, J = 5.9 Hz, 2 H), 6.11 (br q, J = 7.4 Hz, 1 H), 6.38 (br d, J = 7.8 Hz, 1 H), 7.37 (t, J = 7.4 Hz, 1 H), 7.66 (br dd, J = 7.6, 2.5 Hz, 2 H).13 C NMR (101 MHz, CDCl3) δ ppm: 23.76, 29.40, 41.83, 46.17, 61.02, 86.62, 121.36, 124.08, 127.01, 127.31 (q, JC-F = 5.3 Hz), 129.34, 134.34, 134.89, 137.44, 171.31, 175.76. MS-ESI: for C16H15ClF3IN2O2 calcd. 486.7 Da, found m/z 504.7 [M + NH4]+.
Manual syntheses of iodine-123 or iodine-125 labelled VPGIS191
(S,Z)-N-(2-Chloro-3-(trifluoromethyl)benzyl)-1-(3-[125I]iodoallyl)-5-oxopyrrolidine-2-carboxamide (1 [125I]VPGIS191): To a solution of 2 (6.5Sch µg, 10 nmol) in 20 µL 1% acetic acid in MeOH were added 23.5 MBq [125I]NaI and chloramine-T (2 µL, 10 mg/mL). The mixture was stirred at RT for 10 min, and the reaction was stopped with sodium ascorbate (5 µL, 50 mg/mL). Subsequently, the mixture was diluted with 2 mL of distilled water and 1 mL of 1% acetic acid in MeOH and passed through a preconditioned SPE cartridge (2 mL, 20% EtOH, Bond Elut Plexa, Agilent) and eluted with 1.5 mL of 60% EtOH. The product [125I]VPGIS191 was isolated 20 MBq with 85% radiochemical yield (RCY, Tab. S1). The product was injected onto an analytical HPLC (YMC-Triart C18 column, 150 × 4.6 mm, 5 μm; gradient 40/60–90/10, v/v AcCN/0.1% TFA, 1 mL/min, tR = 12 min). (S,Z)-N-(2-Chloro-3-(trifluoromethyl)benzyl)-1-(3-[123I]iodoallyl)-5-oxopyrrolidine-2-carboxamide ([123I]VPGIS191) ([123I]VPGIS191): To a solution of 2 (6.5 µg, 10 nmol) in 20 µL 1% acetic acid in MeOH were added 12.3 MBq [123I]NaI and chloramineT (2 µL, 10 mg/mL). The mixture was stirred at RT for 20 min, and the reaction was then stopped with sodium ascorbate (5 µL, 50 mg/mL). Subsequently, the mixture was diluted with 2 mL of distilled water and 1 mL of 1% acetic acid in MeOH, passed through a preconditioned SPE cartridge (2 mL, 20% EtOH, Strata, Phenomenex), and then eluted with 1.5 mL of 70% EtOH. The product [123I]VPGIS191 was isolated at 10.4 MBq, corresponding to 85% RCY (Tab. S2).
In vitro autoradiography
We followed an autoradiographic procedure much as described by Kuhar et al. [22]. Saline-perfused mouse brains were frozen by immersion to isopentane at − 40 °C, and stored at − 80 °C until use. A cerebral hemisphere was mounted in the sagittal orientation in a cryostat for cutting at − 20 °C into 20 μm-thick sections, which were thaw-mounted onto Superfrost glass slides. After air-drying and storage overnight at − 80 °C, the slides were preincubated in buffer (50 mM buffer: Tris-HCl, pH 7.4, 1 mM EDTA, 0.1% BSA) for 15 min. After removal of the excess buffer, the slides were incubated with the same buffer (1 mL) modified by addition of radiotracer [125I]VPGIS191 (Am = 4.6 MBq/nmol) at final concentrations of 2, 10, 50 and 100 nM. Nonspecific binding was evaluated in consecutive brain sections by the addition of GSK1482160 to a final concentration of 10 µM. After one-hour incubation, the sections were washed by immersion in ice cold buffer (3 × 1 min) and finally dipped in distilled water (30 s, 5 °C) to remove buffer salts, followed by rapid drying under an air stream. The dried slides were then exposed to a phosphor storage screen for 1 − 6 min together with slides bearing dried drops of buffer of known concentrations of The imaging screens were read using a Cyclone phosphor imager (Perkin-Elmer, USA), and the brain binding results (total, non-specific, and specific) converted into units of pmol/gram tissue (wet weight).
Data analysis
Data from autoradiography experiments were analyzed using Microsoft Excel (Microsoft, Redmond, WA, USA). The saturation binding parameters (Bmax and Kd) were determined using linear regression of the Scatchard plot.
Results and discussion
Chemical synthesis and radiochemistry
The preparation of amide 1 was carried out according to the procedure previously described using the reagent 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) with the addition of N-hydroxybenzotriazole (HOBt) as a catalyst [7]. The reaction and isolation of the product were unproblematic, although the 61% yield fell short of the 76% reported in the literature [7]. We produced (Z)-(3-bromoprop-1-en-1-yl)tributylstannate from the commercially available (Z)-3-(tributylstannyl)prop-2-en-1-ol using the Appel reaction, following a literature method [21] (Scheme 1).

Synthesis of the P2X7 receptor ligand VPGIS191 and its radio-iodination. Reagents and conditions: (a) PPh3, CBr4, THF, 0 °C → 20 °C, 17 h; (b) EDCI, HOBt, CH2Cl2, RT, 12 h; (c) (Z)-(3-bromoprop-1-en-1-yl)-tributylstannane, LHMDS in THF, DMF, 0 °C → RT, 3 h; (d) Method (I) NaI, chloramine-T, MeOH, AcOH, sodium ascorbate, RT, 5 min; Method (II) I2, CHCl3, RT, 20 h; e) Na125I, chloramine-T, MeOH, AcOH, sodium ascorbate, RT, 5 min; f) Na123I, chloramine-T, MeOH, AcOH, sodium ascorbate, RT, 5–20 min
The preparation of the (E)-stereoisomer of substituted lactam 2 was as previously described [8], through N-alkylation in DMF with tributyl-(3-chloropropenyl)stannane in the presence of lithium bis(trimethylsilyl)amide (LHMDS) as a base. In our case, we used a bromine-substituted analog as the alkylation reagent (Scheme 1). Reaction and isolation of the product were unproblematic, with 68% yield, compared to 88% as reported for the (E)-stereoisomer [8]. We suppose that our replacement of chlorine for bromine accounts for the lower yield.
We carried out the syntheses of the non-radioactive chromatographic VPGIS191 standard by two different methods, using either 82.2 µmol (Method I) or 10 nmol of precursor (Method II) (Scheme 1). Method I followed the procedure of Jin et al. [8], but replacing CHCl3-MeOH (20:1–10:1, v/v) for hexane-EtOAc (4:1, v/v) as solvent in the silica-gel column purification. We obtained the product in 54% yield, as compared to the 90% reported by Jin et al. [8]. In the slightly modified Method II, we stopped the reaction with sodium ascorbate instead of sodium thiosulfate, with reaction time of 5 min instead of 15 min. We developed a new procedure for the SPE purification of the radiotracer, using 10% ethanol in water for SPE washing and 60% ethanol to elute the product, and had optimized the SPE purification for Bond Elute Plexa and alternately for Strata Phenomenex cartridges. Despite these measures, the product contained up to 0.01% tin-containing precursor according to HPLC-UV (Suppl. Mat. Fig. S1). We estimate a 100-fold margin of safety for the maximum possible carryover of tin from the synthesis, were this method eventually applied for human SPECT studies. We also tested the Sep pack C18 cartridge for SPE purification, which proved to retain the product even in 95% ethanol. Therefore, we used the optimized SPE procedure above for the radiosyntheses with 125I and 123I.
In vitro autoradiography of [125I]VPGIS191 in mouse brain tissue
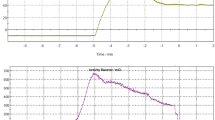
We performed in vitro autoradiography of [125I]VPGIS191 in mouse brain sagittal cryostat sections (Fig. 3), following an established procedure with optimized washing conditions by Kuhar et al. [22] as modified in [8]. The specific binding was invariably less than 50% of total binding, which disfavored quantitation by Scatchard analysis. Nonetheless, we were able in one trial to obtain estimates of Kd = 36 nM and Bmax = 402 pmol/g, which closely matched corresponding affinity for [123I]TZ6019 in HEK cells (Kd = 19 nM) [8]. In what seems to be the first quantitation of P2X7 sites in rodent brain cryostat sections, we find a rather high Bmax, comparable to that seen for [3H]PK11195 binding at microglial TSPO sites [23]. Despite this high Bmax, the low affinity and high non-specific binding of [125I]VPGIS191 disfavored its use in quantitative autoradiography, and may likewise predict low signal-to-background in SPECT studies with this ligand (Fig. 4).

In vitro autoradiography of [125I]VPGIS191 in sagittal mouse brain cryostat Sect. (20 μm-thick). Non-specific binding was determined in the presence of excess of unlabeled GSK1482160 (10 µM). Exposure time 1 min (A) and 6 min (B). TB – total binding, NSB – nonspecific binding

Scatchard analysis of the saturable binding of [125I]VPGIS191 in mouse brain sagittal cryostat sections. Each point indicates the mean of four determinations at 2, 10, 50, and 100 nM [125I]VPGIS191.
Conclusions
Several structural classes of radioligands for the P2X7 receptor intended for PET or SPECT imaging have affinities in the range 1–20 nM (Table 1); among these, only the pyroglutamic acid derivative [123I]TZ6019 is intended for SPECT imaging had 20 nM affinity in HEK cells [8], which may not suffice for visualization on P2X7 sites in living brain. As an alternative to [123I]TZ6019, we prepared its geometric isomer VPGIS191 as reference standard, and also labelled with iodine-123 for SPECT, or iodine-125 for autoradiography in vitro. Unlike the literature synthesis for TZ6019, our synthesis of VPGIS191 employs a commercially available intermediate, namely (Z)-3-(tributylstannyl)prop-2-en-1-ol. We obtained the precursor in 68% yield, the reference standard VPGIS191 VPGIS191 in 52% yield, 123I labelled radiotracer in 71 ± 13% radiochemical yield, and 125I labelled radiotracer in 85 ± 6% radiochemical yield. Despite its high non-specific binding in vitro, we obtained adequate estimates of the saturation binding parameters Kd (36 nM) and Bmax (402 pmol/g) in mouse brain sections. The two-fold lower affinity of [125I]VPGIS191 compared to the literature value for its geometric isomer TZ6019 seems in agreement with previously published comparisons of Z- and E-3-iodoallylated PET and SPECT tracers [17,18,19,20].
References
Leeson HC, Chan-Ling T, Lovelace MD, Brownlie JC, Gu B, Weible MW (2019) P2X7 receptor signaling during adult hippocampal neurogenesis. Neural Regen Res 14:1684–1694. https://doi.org/10.4103/1673-5374.257510
Surprenant A, Rassendren F, Kawashima E, North RA, Buell G (1996) The cytolytic P2Z receptor for extracellular ATP identified as a P2x receptor (P2X7). Sci (Washington D C) 272:735. https://doi.org/10.1126/science.272.5262.735
Takenouchi T, Sekiyama K, Sekigawa A, Fujita M, Waragai M, Sugama S, Iwamaru Y, Kitani H, Hashimoto M (2010) P2X7 receptor signaling pathway as a therapeutic target for neurodegenerative diseases. Arch Immunol Ther Exp 58:91–96. https://doi.org/10.1007/s00005-010-0069-y
Chen Z, He L, Li LF, Chen LX (2018) The P2X7 purinergic receptor: An emerging therapeutic target in cardiovascular diseases. Clin Chim Acta 479:196–207. https://doi.org/10.1016/j.cca.2018.01.032
Adinolfi E, Raffaghello L, Giuliani AL, Cavazzini L, Capece M, Chiozzi P, Bianchi G, Kroemer G, Pistoia V, Di Virgilio F (2012) Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res 72:2957–2969. https://doi.org/10.1158/0008-5472.can-11-1947
Zheng Q-H (2020) Radioligands targeting purinergic P2X7 receptor. Bioorg Med Chem Lett 30:127169. https://doi.org/10.1016/j.bmcl.2020.127169
Gao MZ, Wang M, Green MA, Hutchins GD, Zheng QH (2015) Synthesis of C-11 GSK1482160 as a new PET agent for targeting P2X(7) receptor. Bioorg Med Chem Lett 25:1965–1970. https://doi.org/10.1016/j.bmcl.2015.03.021
Jin H, Han JB, Resing D, Liu H, Yue XY, Miller RL, Schoch KM, Miller TM, Perlmutter JS, Egan TM, Tu ZD (2018) Synthesis and in vitro characterization of a P2X7 radioligand I-123 TZ6019 and its response to neuroinflammation in a mouse model of Alzheimer disease. Eur J Pharmacol 820:8–17. https://doi.org/10.1016/j.ejphar.2017.12.006
Gao M, Wang M, Meyer JA, Territo PR, Hutchins GD, Zarrinmayeh H, Zheng Q-H (2019) Synthesis and in vitro biological evaluation of new P2X7R radioligands [11 C]halo-GSK1482160 analogs. Bioorg Med Chem Lett 29:1476–1480. https://doi.org/10.1016/j.bmcl.2019.04.018
Lord B, Ameriks MK, Wang Q, Fourgeaud L, Vliegen M, Verluyten W, Haspeslagh P, Carruthers NI, Lovenberg TW, Bonaventure P, Letavic MA, Bhattacharya A (2015) A novel radioligand for the ATP-gated ion channel P2X7: [3H]JNJ-54232334. Eur J Pharmacol 765:551–559. https://doi.org/10.1016/j.ejphar.2015.09.026
Janssen B, Vugts DJ, Wilkinson SM, Ory D, Chalon S, Hoozemans JJM, Schuit RC, Beaino W, Kooijman EJM, van den Hoek J, Chishty M, Domene A, Van der Perren A, Villa A, Maggi A, Molenaar GT, Funke U, Shevchenko RV, Baekelandt V, Bormans G, Lammertsma AA, Kassiou M, Windhorst AD (2018) Identification of the allosteric P2X7 receptor antagonist [11 C]SMW139 as a PET tracer of microglial activation. Sci Rep 8:1–10. https://doi.org/10.1038/s41598-018-24814-0
Huang G, Lu X, Qiu Y, Bi L, Ye P, Yang M, Shen Y, Jin H, Han J (2022) Hetero-aryl bromide precursor fluorine-18 radiosynthesis and preclinical evaluation of a novel positron emission tomography (PET) tracer [18F]GSK1482160. Bioorg Med Chem 73:116996. https://doi.org/10.1016/j.bmc.2022.116996
Gao M, Wang M, Glick-Wilson BE, Meyer JA, Peters JS, Territo PR, Green MA, Hutchins GD, Zarrinmayeh H, Zheng Q-H (2018) Synthesis and preliminary biological evaluation of a novel P2X7R radioligand [18F]IUR-1601. Bioorg Med Chem Lett 28:1603–1609. https://doi.org/10.1016/j.bmcl.2018.03.044
Gao M, Wang M, Glick-Wilson BE, Meyer JA, Peters JS, Territo PR, Green MA, Hutchins GD, Zarrinmayeh H, Zheng Q-H (2019) Synthesis and initial in vitro characterization of a new P2X7R radioligand [18F]IUR-1602. Appl Radiat Isot 144:10–18. https://doi.org/10.1016/j.apradiso.2018.11.006
Territo PR, Meyer JA, Peters JS, Riley AA, McCarthy BP, Gao M, Wang M, Green MA, Zheng Q-H, Hutchins GD (2017) Characterization of 11 C-GSK1482160 for targeting the P2X7 receptor as a biomarker for neuroinflammation. J Nucl Med 58:458. https://doi.org/10.2967/jnumed.116.181354
Han J, Liu H, Liu C, Jin H, Perlmutter JS, Egan TM, Tu Z (2017) Pharmacologic characterizations of a P2X7 receptor-specific radioligand, [11 C]GSK1482160 for neuroinflammatory response. Nucl Med Commun 38:372–382. https://doi.org/10.1097/mnm.0000000000000660
Elmaleh DR, Fischman AJ, Shoup TM, Byon C, Hanson RN, Liang AY, Meltzer PC, Madras BK (1996) Preparation and biological evaluation of iodine-125-IACFT: a selective SPECT agent for imaging dopamine transporter sites. J Nucl Med 37:1197–1202
Xu R, Lord SA, Peterson RM, Fergason-Cantrell EA, Lever JR, Lever SZ (2015) Ether modifications to 1-[2-(3,4-dimethoxyphenyl)ethyl]-4-(3-phenylpropyl)piperazine (SA4503): Effects on binding affinity and selectivity for sigma receptors and monoamine transporters. Bioorg Med Chem 23:222–230. https://doi.org/10.1016/j.bmc.2014.11.007
Lever SZ, Xu R, Fan K-H, Fergason-Cantrell EA, Carmack TL, Watkinson LD, Lever JR (2012) Synthesis, radioiodination and in vitro and in vivo sigma receptor studies of N-1-allyl-N´-4-phenethylpiperazine analogs. Nucl Med Biol 39:401–414. https://doi.org/10.1016/j.nucmedbio.2011.10.001
Lever JR, Scheffel UA, Stathis M, Musachio JL, Wagner HN Jr (1990) In vitro and in vivo binding of (E)- and (Z)-N-(iodoallyl)spiperone to dopamine D2 and serotonin 5-HT2 neuroreceptors. Life Sci 46:1967–1976. https://doi.org/10.1016/0024-3205(90)90513-q
Thompson AM, Sutherland HS, Palmer BD, Kmentova I, Blaser A, Franzblau SG, Wan BJ, Wang YH, Ma ZK, Denny WA (2011) Synthesis and structure-activity relationships of varied ether linker analogues of the antitubercular drug (6S)-2-nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824). J Med Chem 54:6563–6585. https://doi.org/10.1021/jm200377r
Kuhar MJ (2001) In vitro autoradiography. Curr Protoc Pharmacol Chap 8. https://doi.org/10.1002/0471141755.ph0801s00.
Pedersen MD, Minuzzi L, Wirenfeldt M, Meldgaard M, Slidsborg C, Cumming P, Finsen B (2006) Up-regulation of PK11195 binding in areas of axonal degeneration coincides with early microglial activation in mouse brain. Eur J Neurosci. https://doi.org/10.1111/j.1460-9568.2006.04975.x
Acknowledgements
The authors appreciate valuable discussions with nuclear chemist prof. Ing. Jan John, CSc.; toxicologist prof. RNDr. Jiří Patočka, DrSc., PD Francisco Rafael López Picón, Ph.D.; Mag. Iris Maria Aichhorn, and nuclear chemist prof. Emma Hilda Kristina Aneheim, Ph.D. during the course of this research.
Funding
Open access publishing supported by the National Technical Library in Prague. This research was supported by a grant from the UCT Prague: A2_FPBT_2022_067 and the Ministry of Education, Youth and Sports of the Czech Republic (project EATRIS-CZ LM2023053, project CZ-OPENSCREEN LM2023052) and by project no. LX22NPO5107 (MEYS): Financed by EU – Next Generation EU.
Author information
Authors and Affiliations
Contributions
Conceptualization: R.P., M.J., A.P. Synthesis and characterization of the compounds: A.M., M.J., T.Z. Experiments on animal samples: A.M., M.P., P.D. Supervision: M.J., P.B.D., S.L., P.C., R.P. and A.P. Original draft preparation: A.M., M.J., P.C., and A.P. Revision of the paper: A.M., M.J., P.C., M.P. and A.P.
Corresponding author
Ethics declarations
Animal experimentation approval
Animal experiments were conducted in accordance with regulations and guidelines of the Czech Animal Protection Act (No. 246/1992), and with the approval of the Czech Ministry of Education, Youth, and Sports (MSMT-41830/2018-7) and the institutional Animal Welfare Committee of the Faculty of Medicine and Dentistry of Palacky University in Olomouc.
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplementary Information
Additional supporting information may be found online in the Supplementary material.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Marešová, A., Jurášek, M., Zimmermann, T. et al. Improved syntheses of P2X7 ligands based on substituted benzyl amide of pyroglutamic acid motif labelled with iodine-123 or iodine-125. J Radioanal Nucl Chem 332, 4191–4199 (2023). https://doi.org/10.1007/s10967-023-09081-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-023-09081-2