
Abstract
We developed and optimized the dissolution and separation chemistry of iridium (Ir) from irradiated Ir pellets and prepared Ir counting samples for analysis by a silicon drift detector. Previous Ir chemistry techniques used on the irradiated Ir pellets are tedious, lengthy, and require extra safety precautions and planning due to the use of perchloric acid in the dissolution step. The dissolution and separations method described in this paper are safer, eliminating the use of perchloric acid, more efficient, and can successfully replace the previous procedure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Iridium is an important radiochemical detector that has been used to measure the neutron spectrum in past nuclear device tests at Los Alamos National Laboratory (LANL) [1, 2]. Isotopic ratios of Ir can be used to monitor three neutron spectral groups at once. These spectral groups include thermal/epitherthermal neutrons, 14 MeV neutrons, and neutrons at the fission spectrum (> 2 MeV) [3]. Ir is currently used as a radiochemical detector at reactor facilities to provide a spectral index for energy integral cross section measurements [4]. The irradiation uses a pellet of Ir (37.3% 191Ir and 62.7% 193Ir), composed of a mixture of potassium chloride (KCl) and potassium hexachloroiridate (K2IrCl6), inside an aluminum casing. Once irradiated, the pellet is chemically digested and the Ir is separated and purified through a lengthy complex process and prepared for radiometric analysis.
The previous chemical process used at LANL for Ir pellets is derived from a procedure developed by Gilmore to process Ir from underground nuclear test debris samples [5]. The Gilmore procedure has been modified and applied to the Ir pellets currently being used [6]. The modifications of the Gilmore procedure were never published. The modified Gilmore procedure will be referred to as the “previous procedure” throughout this paper. The previous procedure chemically digests the Ir pellet using perchloric acid fuming. The Ir is purified from K and impurities through two cation exchange columns. After the columns, the Ir sample solution is divided into two equal portions, precipitated as Cs2IrCl6, filtered and prepared for gamma and x-ray measurement. We describe this procedure in greater detail in this paper.
The useful neutron induced reactions of Ir are 191Ir(n,γ)192Ir, 191Ir(n,2n)190Ir, 191Ir(n,3n)189Ir, 193Ir(n,n’)193mIr, and 193Ir(n,2n)192Ir. The activity of the isotopes 189Ir, 190Ir, 192Ir and 193mIr are measured after irradiation. The half-lives are listed in Table 1. Ratios of the produced isotopes are used to determine the hardness of the neutron spectrum [1]. Since the results are determined based on the Ir isotopes relative to one another and the half-lives of some of the Ir isotopes are short, a rapid separations procedure is more important than a 100% chemical yield.
Traditionally at LANL, atoms of 193mIr are calculated through a deconvolution method. The combined L x-rays of 189,190,192,193mIr are measured. The 189,190,192Ir isotopes are also measured by gamma spectroscopy and subtracted from combined L x-ray measurements to determine the activity of 193mIr. Since L x-rays of all Ir isotopes are around 9–11 keV, the mass of the Cs2IrCl6 precipitate must be thin and uniform to minimize x-ray self absorption. The typical mass for the previous method of Cs2IrCl6 precipitate for counting is between 10 and 30 mg with a diameter of 11 mm. Ir must be thoroughly purified from impurities in order to prevent additional mass in the Cs2IrCl6 precipitate and to remove elements, such as osmium and platinum, which emit x-rays in the same energy range as Ir. Purification also removes potassium (K) which interferes with the formation of the Cs2IrCl6 precipitate.
The previous LANL method uses perchloric acid (HClO4) fuming to digest the sample and convert Ir to the + IV oxidation state [7]. Perchloric acid is a hazardous chemical that requires a certified perchloric acid fume hood at LANL which limits where the work can be performed. Use of a specialized perchloric hood involves coordination with other users and operations personnel to ensure the hood is available and currently in service. Use of the hood requires coordination with appropriate personel for cleaning of the ventilation after use in addition to the worker cleaning the hood after use. Nuclear debris or an Ir pellet is first dissolved in concentrated acids before fuming twice with HClO4. The perchloric fuming procedure takes a full working day to complete (about eight hours) under constant supervision. The subsequent separation procedure is a tedious process. After fuming, the irradiated Ir is loaded onto a cation exchange resin column in a perchloric acid solution. The Ir is eluted with hot acid while constantly rotated under a heat lamp to avoid the column from melting and the column must be placed at a distance from the lamp to ensure the solution does not boil inside the column. The Ir is held very tightly by the cation exchange resin as Ir(H2O6)4+ complex [7]. The Ir is extracted with 4.5 M hot HCl, forming a chloro-anionic complex, IrCl62−, which is not retained by the cation exchange resin. We desire to develop a safer more rapid dissolution method without the use of perchloric acid and a less tedious separations procedure.
A literature review has shown that separation and purification of Ir can be achieved through chromatography methods [5, 8, 9]. Separations of Ir have been succesfully performed using cation and anion exchange resins [5, 10,11,12,13]. Separations of Ir in the literature are often carried out with the purpose to separate Ir from platinum metals and other metal impurities [10]. Ir exists in oxidation states from −III to + IX with the most common being + III and + IV in solution [8]. For column chromatography, the oxidation state of Ir is adjusted to the + IV state and forms an anionic or cationic complex that will be retained on the resin.
Anion exchange resin has been used for Ir separations. Ir(IV) in HCl is retained on anion resin as IrCl62−. Evers et al. [10] adjusted the oxidation state of Ir in 6 M HCl at 40 °C using chlorine as an oxidant. The solution is passed through a bed of anion exchange resin Amberlite IRA-400. Base metal impurities are removed by washing the column with 0.1 M HCl. The Ir is reduced to Ir(III) and eluted with a saturated solution of SO2 as a mixed sulphito-chloro complex with a large negative charge [10]. The Ir can also be eluted from an anion column by forming stable complexes with halogen ions such as bromide ions or iodine ions which have higher stability constants than Ir chloride complexes [9, 13]. Dybczynski and Maleszewska [13] determined distribution coefficients for Pt, Ir, palladium (Pd), and rhodium (Rh) in HBr solutions containing bromine or hydrazine hydrochloride with Dowex 1 × 4 and 1 × 8 resin. The authors showed that Ir, Pd and Rh have higher distribution coefficients at lower acid concentrations and the distribution coefficients decrease with increasing acid concentrations. The distribution coefficient for Pt follows the same pattern as the other metals except the distribution coefficient increases with increasing acid concentration with HBr + 2% N2H2 · HCl until 5 N HBr and then decreases. The distribution coefficient values for the platinum metals are different enough to expect successful separations and are used to develop separations of Ir from solutions containing Rh, Pd and Pt [13]. Marsh et al. determined distribution coefficients for 58 elements, including Ir, in 0.1–8.7 M HBr, 0.1–7.4 M hydroiodic acid (HI), and 0.1–12 M HCl in Dowex resins 50–100 mesh, AG 1 × 4, AG 1 × 8 and AG MP-1 [9]. This work shows that Ir is adsorbed to the listed resins in HCl. The Ir sorption decreases with increasing HBr acidity while platinum metals, such as Os and Pt, remain highly adsorbed to the anion exchange resin at all HBr acid concentrations. Bond et al. utilized this information to design a separation using AG MP-1 resin in which the Ir is loaded onto a column in 0.1 M HCl, where it is retained, the contaminants (KCl from fusion) are washed off, and the Ir is recovered by elution with 9 M HBr [14].
In this paper we discuss the improvements in Ir dissolution and column chemistry for the previous procedure which is a modified Gilmore procedure. An ideal method for the dissolution and processing of the Ir pellets would eliminate the use of perchloric acid, reduce the overall procedure time, and produce samples with enough Ir for sufficient counting statistics. An anion exchange column, adapted from Bond et al., was used in order to eliminate the use of hazardous acids such as HClO4 or HF in the dissolution and separation steps [14]. The new procedure was applied to 10 Ir pellets and qualitatively compared to the previous method which had been applied to seven Ir pellets from past analysis.
Experimental
Reagents and instrumentation
Chemicals used for separations described here are ACS reagent grade or better. The HCl and HNO3 are TraceMetal™ grade, and the HBr is ACS grade, all from Fisher Scientific. The K2IrCl6 is 99.99% trace metals basis from Sigma Aldrich. The KCl and CsCl are TraceMetal™ grade from Fisher Scientific. All deionized (DI) water (> 18 MΩ) used is from a Milli-Q water purification system. The AG MP-1 M (50–100 mesh) and AG 50 W-X8 (50–100 mesh) resins are from Bio-Rad Laboratories, Inc.
The Ir carrier used contains 3.596 g of K2IrCl6 in 500 mL of 2 M HCl and yields 10 mg of Cs2IrCl6/mL.
Gamma samples are analyzed to determine yields throughout the procedures with a GDM sodium iodide detector (NaI) with WinDAS software. Each sample is counted long enough to receive at least 10,000 counts (between 10 and 1440 min) in the 316.5 keV gamma energy peak from 192Ir. Spectra are analyzed using Peak Easy Software and corrected for decay and background. Counting uncertainty is reported at 2σ.
Ir pellet preparation
The Ir pellets, of natural isotopic abundance, are prepared by thoroughly mixing 60% (wt) KCl and 40% (wt) K2IrCl6 using a Wig-L-Bug Grinding Mill with three teflon balls for five minutes. Roughly 90 mg of the mixture is pressed into a pellet using a KBr quick press kit with a 7 mm die set. An image of an Ir pellet is shown in Fig. 1. Pellets are wrapped in teflon tape and placed inside aluminum capsules to be irradiated. Pellets are irradiated in critical assemblies at the National Criticality Experiments Research Center (NCERC).

A 90 mg Ir pellet before irradiation
Previous procedure
After irradiation, the Ir pellet is weighed and the mass of Ir in the pellet is estimated in order to determine if the Ir carrier needs to be added. The Ir carrier is added to prevent Ir loss during the dissolution process because Ir in microgram quantities is volatile under conditions of the dissolution. The Ir carrier is also used to ensure that the end sample Cs2IrCl6 precipitate is above 30 mg total so that each counting sample will be above 10 mg. The Ir pellet is placed in a 250 mL erlenmeyer flask and fumed with 2 mL of concentrated HNO3 and 5 mL of HClO4. This step is repeated with 2 mL concentrated HNO3 and 2 mL of HClO4. The solution is fumed to a low volume of HClO4. The fuming step converts Ir to a polynuclear cationic species and adjusts the Ir oxidation state to + IV [5]. The remaining HClO4 in the flask is cooled after fuming. The fuming step typically takes an entire working day (about eight hours) to set up the fume hood, perform the fuming, and clean up after the fuming operation.
The sample is prepared for the first cation exchange column. This column uses about 5 mL AG 50 W-X8, 50–100 mesh in a plastic tube with Teflon turnings used as a frit. DI water is added to the Ir fumed solution to make 1–2 M HClO4 (~ 10 mL of DI water to 1 mL HClO4 acid solution). The column is prewashed with 20 mL 4.5 M HCl, 20 mL DI water and 20 mL 1 M HClO4. The sample is loaded onto the column using gravity. The column is washed with 10 mL of 1 M HCl + 0.1 M HF, followed by 10 mL of 1 M HCl to remove ClO4− anions. Ir is eluted from the column with 40 mL of hot (70–80 °C) 4.5 M HCl while column is rotated under a heat lamp. The heat is used to speed up the conversion of the Ir catonic complex to a chloro-anionic complex, IrCl62−. The column must be continuously rotated to avoid the column from melting and the column is placed at a distance from the lamp to ensure the solution does not boil inside the column. Then 2 mL of concentrated HCl and a few drops of HNO3 are added to the Ir eluent to help keep the Ir in the + IV oxidation state. The eluent is taken to dryness slowly at low heat on a hotplate. This column takes about 2 h to complete.
The dry Ir sample is rehydrated in 2 mL of 0.05 M HCl and loaded onto a 15 mL glass column (20 cm in length, with a diameter of 9 mm) containing 5 mL of AG 50 W-X8 (50–100 mesh) resin. The purpose of this column is to remove any cations present in the solution. The column had been prewashed with five column volumes of 4.5 M HCl and 20 mL of 0.05 M HCl. The Ir solution is loaded onto the column as an anionic complex which does not interact with the resin. The column is washed in 2 mL increments with 0.05 M HCl until the eluent changes color from brown to clear, about 10 mL, and collected in a graduated glass centrifuge tube.
Concentrated HCl is added to the sample to bring up the concentration to 6 M and the solution is roughly divided into two fractions. A few drops of concentrated HNO3 is added to each fraction to ensure Ir is in the + IV oxidation state. The Ir is precipitated as Cs2IrCl6 by adding 1 mL of a CsCl solution (1 g of CsCl per 15 mL of 6 M HCl) [5]. The fractions are placed on a hot plate at 90 °C for several hours to overnight to allow the Cs2IrCl6 precipitate to form. The Cs2IrCl6 precipitate varies in color from bright red to a very dark almost black color. The darker color is preferred because it is more crystalline, granular and easy to centrifuge and filter. The black Cs2IrCl6 precipitate will settle to the bottom of the centrigue tube and the supernatant will be mostly clear with a brownish tinge. The bright red Cs2IrCl6 precipitate is harder to filter because the Cs2IrCl6 precipitate is much finer. The red Cs2IrCl6 precipitate will remain somewhat suspended in the solution and the supernate will have a reddish tinge. Once the Cs2IrCl6 precipitate is formed, the fractions are centrifuged for 10 min. The supernatant is decanted. The sample is washed with 5 mL of 6 M HCl, centrifuged for 10 min and decanted. The sample is washed with 5–10 mL of ethanol twice, centrifuged and decanted.
The Cs2IrCl6 precipitate is filtered onto a preweighed Whatman grade 50 filter paper for one of the sample fractions [15]. The filtration aparatus is shown in Fig. 2. It consists of a filter flask and a glass frit. The filter paper is set on the frit, a rubber O-ring with ID 7/16″ is centered on top of the filter paper, and a glass chimney with an ID of 7/16″ is placed on top of the O-ring and filter. The chimney is held in place with a horseshoe clamp. Approximately 2 mL of ethanol is pipetted into the glass chimney. The Cs2IrCl6 precipitate is suspended in 2–5 mL ethanol and the resulting slurry is transferred to the filtration apparatus using a transfer pipette. Additional ethanol is added to the chimney and the Cs2IrCl6 precipitate is resuspended in the solution while applying vacuum until the deposit is distributed uniformly onto the filter paper.

Filter aparatus
Once dry, the Cs2IrCl6 precipitate is weighed and one drop of a dilute rubber cement solution is applied at 120° increments, for a total of three drops, around the Cs2IrCl6 precipitate onto the filter to adhere the sample to the filter paper. It is very important that the glue is not directly applied to the Cs2IrCl6 precipitate because it will create a coating attenuating the Ir x-rays and can physically disturb the Cs2IrCl6 precipitate. The filter is dried and mounted onto a 3″ × 5″ aluminum plate for counting as shown in Fig. 3. A piece of double sided tape is placed in the middle of the aluminum plate and the filter paper with the Cs2IrCl6 precipitate is placed on top of the tape. The entire sample is covered with Mylar film and the edges of the Mylar are taped in place.

Cs2IrCl6 precipitate on filter, mounted to an aluminum plate
The second fraction of the Cs2IrCl6 precipitate is prepared for analysis by a γ-ray well detector. This is done by transferring the Cs2IrCl6 precipitate with ethanol into a small Teflon counting container (2″ long, inner diameter = 1/4″, with a fitted Teflon cap). The Cs2IrCl6 precipitate is dried and a few drops of the glue solution is added to the sample to keep the sample in place.
Chromatographic studies
The overall purpose for the chromatographic studies is to replace the perchloric fuming dissolution and first column of the previous procedure with an anion exchange column. An 192Ir tracer, used for the chromotographic studies, was produced by irradiating K2IrCl6 at the research reactor at University of Massachusetts Lowell. The K2IrCl6 was dissolved in 6 M HCl after the irradiation.
Initial Ir study (192Ir tracer + Ir carrier)
The goal of the intial Ir study is to identify losses in procedure and to ensure a sufficient Cs2IrCl6 precipitate is produced for analysis. A different first column is used in our procedure in comparison with the previous procedure. The load is prepared by adding 100 µL (2 × 105 dpm) aliquot of the tracer and enough Ir carrier to produce 80 mg of the Cs2IrCl6 precipitate end product to a 15 mL plastic centrifuge tube containing 10 mL of 3 M HCl. The load is transferred to a polypropylene gravity column containing 20 mL of AG MP-1 M (50–100 mesh) resin. The column is prewashed with five column volumes of DI water, 9 M HBr, and 6 M HCl. The column is 18 cm in height, a diameter of 16.5 mm, with a 3 mm frit. The interaction of the IrCl62− with the column is visible as a dark ring. The column is rinsed with 100 mL of 6 M HCl after the sample is loaded. This step removes K and any impurities from the K2IrCl6 such as platinum and osmium. The Ir is eluted as a bromo complex, IrBr62− [9, 14] with 100 mL of 9 M HBr, in 10 mL fractions. The 10 mL fractions are collected and measured with the GDM NaI detector to determine the volume at which the Ir elutes completely from the column. The HBr eluents are combined and taken to dryness on a hotplate, reconstituted in 10 mL concentrated HCl, and taken to dryness a total of three times in order to convert the Ir to the chloride form, IrCl62−.
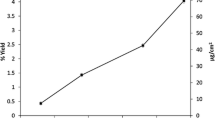
The sample is prepared for column 2 and the remaining steps from the previous method are repeated to produce the filtered Cs2IrCl6 precipitated sample. Aliquots are taken throughout the procedure and their activity is measured with the GDM NaI detector to monitor for losses. The Cs2IrCl6 precipitate is also counted with the NaI detector to determine method yield (Fig. 4).

Separation schematic for Ir
Column 1 tracer volume studies (192Ir tracer only)
The purpose of this study is to compare Ir yields when using smaller volumes of resin in the first column to determine which resin volume to use for purification of Ir from irradiated Ir pellets. As mentioned previously it is more important to produce a radiochemically pure Ir fraction for counting than having a 100% yield, because the results are determined based on ratios of the Ir isotopes present. However, increasing the yield will allow us to reduce the column volume which will decrease the amount of reagents used, decrease the waste produced, and decrease the procedure time. Column 1 tracer volume studies containing 2, 10 and 20 mL of AG MP-1 M (50–100 mesh) resin are performed using 50 µL of 192Ir tracer containing 0.03 ng of 192Ir. The Ir carrier was not used in the column volume studies. The 10 and 20 mL columns used are of the same dimensions as the previous section. A 12.7 cm polypropelene column with a 9 mm diameter is used for the 2 mL resin. The first column procedure in the previous section is repeated for each column. Columns are loaded with the tracer in 10 mL 3 M HCl. The wash and eluting acid volumes are scaled down for each column (30 mL for the 10 mL column and 6 mL for the 2 mL column). Aliquots of column feeds and eluents are measured with the GDM NaI detector to determine yields.
New procedure
The purpose of this research is to apply the new procedure which replaces the fuming and the first column of the previous procedure with an HCl digestion and an anion exchange column (resin volume of 10 or 20 mL), and apply the procedure to irradiated Ir pellets.
After irradiation, the Ir pellets are removed from the capsule and Teflon tape and weighed in order to determine the amount of Ir and to estimate how much Cs2IrCl6 precipitate will potentially form. If less than 30 mg of Cs2IrCl6 precipitate is estimated, then the Ir carrier is added during the dissolution. The pellet is placed in a 125 mL erlenmeyer flask with 10 mL 3 M HCl and heated on a hotplate at 40 °C for 1–1.5 h until fully dissolved, yielding a brown solution indicating the formation of IrCl62−.
The solution is loaded onto a plastic gravity column containing 10 or 20 mL of AG MP-1 M (50–100 mesh). The complete procedure previously described is applied to the Ir pellets. After the second column, the eluent is visually divided into two fractions of equal volume and precipitated, centrifuged and filtered as described previously.
Results and discussion
Chromatographic studies
Initial Ir study (192Ir tracer + Ir carrier)
The motivation for doing this work is to remove the perchloric fuming step which will result in a faster separation as well as removing the use of a hazardous chemical. We decided to adapt the anion exchange separation from Bond et al. to test if it could be used to achieve these goals. Modifications of Bond’s method include increasing the column volume from 5 to 20 mL, increasing the concentration of HCl from 0.1 to 3 M for the load and 6 M for the wash, and reducing the wash volume from 100 to 50 mL. Results are reported in Table 2. The first column, 20 mL of AG-MP 1 M (50–100 mesh), has an eluent total yield of 76 ± 3% which is slightly higher than the 72% and 70% yields reported by Bond [14]. The first 30 mL of the eluting acid contains 75 ± 2% of the Ir wtih 1 ± 2% in the following 20 mL. The remaining 50 mL of the eluting acid was not counted. No Ir was detected in the column load eluent. There is 22 ± 2% of Ir in the wash fraction. The Ir in the wash could be associated with Ir(III) not fully oxidizing to the extractable complex of IrCl6− in HCl and instead forming mixed aquo-chloro complexes [10]. After the first column is completed, the second column separation is conducted as described in the experimental section. The total column yield for the second column (AG 5 W-X8) is 103 ± 1% and is higher than 100% possibly due to errors in volume measurements. The overall method yield is 75 ± 1%. The method yield was determined by the amount of Ir counted with NaI detector in the Cs2IrCl6 precipitate.
Our yields are consistent with previous work and will be discussed below. This study proved that an adequate Cs2IrCl6 precipitate mass can succesfully be prepared using this procedure and can be applied to irradiated Ir pellets.
Modifications to increase the method yield should be focused on the first column. Increasing the yield will allow for a smaller column which will reduce the volume of reagents used, waste produced, and procedure time which is important for the short lived isotopes of Ir. Improvements in the retention of Ir on the first column during the wash will be further investigated in the future by adding an oxidation step to ensure the Ir is in the + IV state before running through the column. Other concentrations of acid will be investigated for the wash to see the effect on Ir retention. Mass spectrometry studies will also be performed to monitor K throughout the procedure.
Column 1 tracer volume studies (192Ir tracer only)
The column 1 tracer volume studies compares the separation of the Ir tracer using three different volumes of resin. The Ir carrier is not used in the tracer volume study because the Ir in the tracer will only undergo the first column procedure to monitor the effectiveness of the colume and not the entire procedure to produce the Cs2IrCl6 precipitate. The average Ir in the wash and the eluent for each column volume are listed in Table 3. The Ir in the eluent increases with the increase in column volume. The Ir in the wash is consistent throughout the different size columns. The 2 mL column may be applied to experiments where having a faster column is more important than yield. The 60% Ir in the eluent from the 2 mL column is still greater than the 50% yield reported by Gilmore et al., applied to nuclear debris samples, and can be used for the analysis of irradiated Ir pellets. The amount of Ir found in the eluent in the 20 mL column that contained only 192Ir tracer shown in Table 3 is consistent with the amount of Ir eluted from the 20 mL column discussed in Table 2, which had additional Ir mass from the Ir carrier. These results indicate that added mass from the Ir carrier does not effect recovery. As shown in Table 3, the Ir observed in the eluent for the 10 and 20 mL columns was greater than the 2 mL column. Therefore, we chose to apply the 10 and 20 mL columns to the irradiated Ir pellet analyisis, which is discussed in the next section. As mentioned previously, future studies will be done to adjust the Ir oxidation state to ensure the Ir is in the + IV state, reducing the amount lost in the wash and overall increasing the column yield so that a smaller volume column can be applied.
Comparison of previous and the new procedure applied to Ir pellets
In order to compare our new procedure yields with the previous procedure, we obtained information of the analysis of Ir pellets by reviewing data from past lab notebooks processed between 2018 and 2019. The data for the previous procedure and the new procedure are presented in Table 4.
The desired weights for x-ray counting samples are between 10 and 30 mg due to the strong x-ray attenuation for thick samples. The Cs2IrCl6 precipitates from the previous and new procedures fall within the desired range. The average Cs2IrCl6 precipitate masses listed in Table 4 for the previous procedure are 19.7 ± 5 mg compared to 17.8 ± 4 mg for the new procedure. Therefore, the Cs2IrCl6 precipitate weights from the new procedure are equivalent to the weights from the previous procedure.
The filter recovery listed in Table 4 shows the percentage of mass on the filter from half of the sample in relation to the total Cs2IrCl6 potentially formed based on the starting pellet mass. As discussed in the methods section, the eluent from the second column is roughly divided into two portions before placing onto the hotplate for precipitation. These two portions are not measured in anyway, therefore the resulting recovery data is somewhat qualitative. However, the filter recovery is higher in the new procedure than the previous procedure.
Pellets 1–5 listed in Table 4 under the “New Procedure” are processed using a 20 mL column volume for column 1 while pellets 6–10 were processed using the 10 mL column volume for column 1. The mass of Cs2IrCl6 precipitate on the filter sample and filter recovery are similar for both column volumes. Around 3–6% of the Ir is lost in the wash fraction in the 20 mL column while 20–30% is lost in the wash fraction in the 10 mL column. The Ir yield is between 30 and 75% in the 10 mL column volume for column 1. The overall Ir yield for column 2 (AG 5 W-X8) of the procedure is 80–98%. These results prove the new procedure can effectively replace the previous procedure for the analysis of Ir pellets and either column volume (10 or 20 mL) can be used for column 1 (AG MP-1 M).
In addition to the efforts to modify and improve the separations chemistry as discussed here, our team has also updated the detectors used for measurement of the Ir by using a silicon drift detector (SDD) rather than a HPGe and gas proportional detectors. The details of the SDD measurements will be discussed in a separate paper. The SDD has a higher resolution than a gas proportional counter formerly used to count Ir x-rays. The 9.2 keV x-ray peak of 193mIr and 9.4 keV x-ray peak of 192Ir are used to calculate the atom ratio of 193mIr/192Ir using the SDD. The previous procedure used at LANL requires two samples to be produced for each Ir pellet. The SDD replaces the deconvoluted counting method that couples a gas proportional detector and an HPGe. Since analysis using an SDD only requires half of the sample to be processed for analysis, this leaves the other half of the Ir pellet sample in reserve which is beneficial in case there are issues during the filtration and mounting process and an additional sample is needed.
Time comparison
The new procedure decreases the time for the dissolution step and eliminates the hazards associated with the use of perchloric acid. The perchloric fuming requires the constant attention of the worker while the dissolution in the new method can be unattended. The dissolution portion of the procedure is accomplished with 3 M HCl on a hotplate. The dissolution with HCl takes 1–1.5 h to complete and does not require the constant supervision as perchloric fuming which takes about eight hours. The first column of the current method takes two hours from preconditioning to eluting the Ir when using the 20 mL column. The 10 mL column further reduces this time to one hour. Incorporating the new digestion and column procedure reduces the total method length by two days for the processing of two samples. The two samples can be digested simultaneously, and the first column separation can be performed in one working day.
Conclusions
We have successfully proven that the new procedure can effectively replace the previous procedure for the processing of irradiated Ir pellets. The new procedure replaces the dissolution method and first ion exchange column while producing Cs2IrCl6 precipitate samples of similar mass. The digestion and first column were replaced with methods that eliminate the use of perchloric acid which makes the overall new procedure safer and does not require special protocols associated with perchloric acid. The new procedure takes two working days less to complete than the previous procedure which is important for the analysis of the Ir isotopes with short half-lives.
References
Lee AS (2016) Determination of the spectral index in the fission spectrum energy regime. Los Alamos, NM. https://doi.org/10.2172/1253539
Nelson RO, Fotiades N, Devlin MJ, Talou P, Chadwick MB, MacFarlane RE, Trellue HR, Hayes AC, Jungman G, White MC, Frankle SC, Garrett PE, Younes W, Becker JA (2005) New 193Ir(n,n'y)193mIr evaluated nuclear cross sections for radchem, UCRL-CONF-209032. Livermore, CA
Cowell S, Talou P, Chadwick MB (2008) Evaluation of iridium (n,xn) reactions. Paper presented at the international conference on nuclear data for science and technology 2007, Nice, France, https://doi.org/10.1051/ndata:07571
Keith C, Selby H, Lee A (2018) Nuclear data uncertainty propagation for spectral reaction ratios. Ann Nucl Energy. https://doi.org/10.1016/j.anucene.2018.08.009
Gilmore J (1990) Iridium. In: Kleinberg J (ed) Collected radiochemical and geochemical procedures. 5th edn. Los Alamos, NM, pp I107–I109. https://doi.org/10.2172/6013858
Sanchez RG, Attrep MJ, Bounds JA, Bredeweg TA, Favorite JA, Goda JM, Hayes DK, Jackman KR, Myers WL, Oldham WJ, Rundberg RS, Schake AR (2012) Radiochemistry Results from IER-163 Comet Irradiation. United States. https://doi.org/10.2172/1054681
Fine DA (1970) Studies of the iridium(III) and (IV)—chloride system in acid solution. J Inorg Nucl Chem. https://doi.org/10.1016/0022-1902(70)80323-2
Leddicotte GW (1961) The radiochemistry of iridium. United States. https://doi.org/10.2172/4834231
Marsh SF, Alarid JE, Hammond CF, McLeod MJ, Roensch FR, Rein JE (1978) Anion exchange of 58 elements in hydrobromic acid and in hydriodic acid. Los Alamos, NM https://doi.org/10.2172/5149421
Evers AP, Edwards RI, Fieberg MM (1978) Recovery and purification of iridium. United States Patent
Despotopulos JD, Kmak KN, Shaughnessy DA (2018) Production and isolation of 197m, gHg. J Radioannal Nucl Chem. https://doi.org/10.1007/s10967-018-5927-9
Berman SS, McBryde WAE (1958) The separation of rhodium and iridium by ion exchange. Can J Chem. https://doi.org/10.1139/v58-123
Dybczyński R, Maleszewska H (1974) Separation of four platinum metals by anion-exchange in hydrobromic acid medium. J Radioanal Nucl Chem. https://doi.org/10.1007/BF02520865
Bond EM, Moody WA, Arnold C, Bredeweg TA, Jandel M, Rusev GY (2016) Preparation of iridium targets by electrodeposition for neutron capture cross section measurements. J Radioanal Nucl Chem. https://doi.org/10.1007/s10967-015-4607-2
Attrep MJ, Bowen SM, Smith JE (2001) Preparing Samples for Beta Counting Los Alamos, NM
Acknowledgements
This work supported by the Office of Experimental Sciences Campaign 4, National Nuclear Security Administration (NNSA), US Department of Energy (DOE) through LANL. LANL is operated by Triad National Security, LLC, for NNSA of U.S. DOE (Contract No. 89233218CNA000001).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests to declare that are relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Marenco, A.M., Bond, E.M., Rusev, G. et al. Improvements in iridium target chemistry. J Radioanal Nucl Chem 332, 377–385 (2023). https://doi.org/10.1007/s10967-022-08717-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-022-08717-z