
Abstract
A pilot study to quantify 55Fe in steel from a reactor vessel of a nuclear power plant by accelerator mass spectrometry (AMS) without any chemical sample preparation was validated by liquid scintillation counting (LSC) and AMS after radiochemical separation. AMS reaches an uncertainty < 10% at the 1 kBq gFe−1 level within less than 10 min measuring time. The background was < 3 Bq gFe−1, presently limited by the short measurement time. The new instrumental AMS method for analysing 55Fe from neutron capture production is reasonable and fast compared to other analytical methods.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As iron is a very important element in the biosphere and in material sciences, labelling and tracing methods offer a wide field of applications for long-lived radioactive nuclides like 55Fe (t1/2 = 2.7563 ± 0.0004 years [1]). Among them are biomedical and biological research (e.g., iron absorption and blood disorder) and plant-soil relationship [2]. Iron-55 is also of high astrophysical interest considering that iron is the seed element for nucleosynthesis into the higher-mass region via slow neutron-capture reaction [3, 4]. Primary and secondary particles from solar and galactic cosmic radiation also produce 55Fe in the lunar surface and meteoroids. The radionuclide can be detected in freshly fallen meteorites [5, 6], hence it can be used to determine short terrestrial ages of meteorite finds (≤ 15 years) or check specific pieces for their potential relation to a recent meteorite fall.
Anthropogenic activities like running nuclear power plants and accelerator installations are the major sources for 55Fe, which is formed mainly by the nuclear reactions 54Fe(n,γ)55Fe and 56Fe(n,2n)55Fe [7]. A major component in different construction materials like steel of reactor vessels, tubes and bearing structures in buildings, steel in heavy concrete or in the concrete itself is iron. Because of the broad utilization of those materials, 55Fe can be produced in high specific and absolute activities [8]. Unintended releases to the Earth’s environment from nuclear reprocessing and by fallout from nuclear weapons are also reported [9, 10].
Besides 55Fe other radionuclides with high importance for decommissioning are produced either by activation or fission processes like 3H, 14C, 36Cl, 41Ca, 59,63Ni, 60Co, 90Sr, 93Zr, 93Mo, 94Nb, 137Cs, 152Eu and the transuranics [11]. These radionuclides are divided into:
-
(a)
Easy-to-measure ones analysed fast and easily by gamma-spectrometry often without any separation and
-
(b)
Difficult-to-measure ones analysed by alpha or beta spectrometry (liquid scintillation counting (LSC), proportional counter, ionizing chamber etc.) after necessary radiochemical separation.
Both groups can be linked by so-called scaling factors, which are typical for each power plant, but gained only by precise and often elaborate radiochemical analyses. For example, 55Fe, 63Ni and the transuranics can often be linked to 60Co, whereas 90Sr can be linked to 137Cs, and 41Ca can be linked to 152Eu [12,13,14,15].
The importance of 55Fe is based on its rather short half-life of 2.7563 a compared to the other radionuclides. Thus, 55Fe is of high relevance above all for the first years after shut-down of nuclear power plants or other nuclear installations for decommissioning. Last but not least, 55Fe is also of high relevance during the running phases of nuclear installations with respect to maintenance and repairs. For a high level of safety during decommissioning as well as during processing, temporary storage and final repository of nuclear waste generated during running and dismantling chemical and radiological properties have to be thoroughly investigated [16, 17].
Because 55Fe decays solely by electron capture to the ground state of 55Mn, gamma-rays are not emitted. Thus, routine detection of 55Fe via its characteristic 5.9 keV X-rays is performed on very thin samples to overcome self-absorption [18] or after time-consuming elaborate radiochemical separation via LSC [8, 16, 17]. The great potential of accelerator mass spectrometry (AMS) for measuring 55Fe has been firstly reported by Korschinek et al. about thirty years ago [2]. They could estimate a sensitivity for 55Fe/Fe of about 8 × 10–13 using the 14-MV tandem accelerator at Munich. More recent papers of Wallner et al. [19, 20] demonstrated first measurements of 55Fe using a smaller tandem accelerator (3 MV) while improving the detection limit by more than two orders of magnitude (< 10–15). However, AMS has never been considered as a true competitive analytical method for 55Fe because its general reputation is to be extraordinarily expensive, and comparatively slow resulting in low sample throughputs. Within this study, we explore AMS for 55Fe detection—in steel samples from nuclear decommissioning—without the need of any radiochemical separation to qualify as a routine analysing method.
Our earlier studies have proven AMS being reasonable, fast and easily accessible for 7Be [21] and 41Ca [22] analysis, which was mainly based on largely reducing efforts for sophisticated radiochemical separation. To our knowledge there are only a few cases in AMS where wet chemical separation could be completely omitted such as 10Be/9Be in a Be containing mineral (phenakite, Be2SiO4) [23] and 14C/12C by laser-ablation AMS of stalagmites and corals [24].
Usually, AMS sample preparation is needed to isolate the radionuclide of interest either together with its naturally occurring stable isotope(s), such as 26Al/27Al from e.g., stony meteorites and sediments, or for e.g., quartz after the addition of a stable isotope carrier such as 9Be to allow 10Be/9Be measurements. The separation results also in depletion of matrix-elements (e.g., Si from quartz) and—importantly—isobars, finally yielding to a thermally and electrically conductive powder. For 55Fe we are in the fortunate situation that the isobar, 55Mn, does not form negative ions [2]. Additionally, in the case of steel, from e.g., projects related to nuclear decommissioning, stable iron nuclides (54,56,57,58Fe) constitute already to a large fraction of the original sample. Steel is thermally and electrically conductive and the target element of interest—with the nuclides 54Fe and 56Fe dominating 55Fe production—is homogeneously distributed relative to 55Fe, hence smaller subsamples (milligram instead of typical gram(s)) will not produce any bias. To conclude, there is no reason why 55Fe determination in steel samples without any chemical separation should fail. Though, the approach is validated in this pilot study by comparing AMS measurements of unprocessed steel samples to LSC and AMS measurements of radiochemically processed samples.
Experimental
Sample preparation, LSC and ICP-MS measurements
Two fine and two coarse grained steel chips originating from the reactor vessel of a nuclear power plant were obtained either by drilling or rasping. There was no further pretreatment before preparation for AMS and LSC measurement, respectively.
Pristine small solid aliquots from these chips weighing about 2.5–13 mg (Table 1; without adding of and mixing with any metal binder, without any backing), have been hammered—with the help of a punch—from the backside into Cu sample holders (∅: 1.3 mm; length: 3.8 mm; volume: 5 mm3—maximum dimensions for sample material) temporarily closed by a ball for the hammering process.
For comparison of this non-chemical (instrumental) approach with the conventional chemical approach, radiochemical separation has been performed using bigger solid aliquots from the chips (1.52–1.60 g). The chemicals used throughout the procedure are of analytical grade and water is deionized to 18 MΩ cm. The steel chips were covered with 6 M HCl and carefully heated to generate a steady hydrogen development. For complete dissolution 12 M HCl is added dropwise. The resulting solution is filled-up to a defined volume of 50 ml. From these solutions representative aliquots are taken for the analysis of 55Fe, which corresponds to a total mass of about 40–60 mg of iron. The radiochemical separation procedure for LSC measurements of 55Fe (and 63Ni) comprises an enrichment of Fe, Co and Ni via hydroxide precipitation with NaOH, an anion exchange chromatography for separation of Fe, Co and Ni, and several further purification steps as repeated hydroxide precipitation, extraction chromatography or liquid–liquid extraction. For LSC measurements 55Fe is prepared by Fe(OH)3 precipitation (with ammonia) from its corresponding fraction, redissolving the dried precipitate in 200–300 µl HF (40%) and 5–10 ml H2O. The fluoride ions form a colourless complex with Fe(III) resulting in a strong colour quench suppression [25, 26].
The chemical recovery is obtained by measuring the stable Fe concentration by inductively coupled-plasma mass spectrometry (ICP-MS) in a 10% aliquot from the analysed sample amount before and after radiochemical separation. The ICP-MS measurements are carried out using an Element 2 of Thermo Fisher Scientific, which is a high-resolution sector field mass spectrometer with inductively coupled plasma (HR-ICP-MS). The measurements were done according to DIN EN 17294–2 (2005) after calibration of a suitable working range with a certified single-element, i.e., iron, solution or a multi-element standard solution using 103Rh as an internal standard.
The remaining solution (81%) is filled into a 20-ml vial (polyethylene) and mixed with 10 ml of the liquid scintillator Instagel (PerkinElmer®). The LSC vial is stored cool and dark for at least 0.5 h before measuring in a 1220 Quantulus Low-Level scintillation counter (PerkinElmer®) for 150 min. The count rate in the channel range 25–200 of the low-energy window is used for activity calculation. The counting efficiency is determined using a quench curve and the SQP(E) value that shifts around 0.3 for a SQP(E) value of 710–720.
It has to be noted that the four LSC samples have been treated to the “standard operation procedure” for (commercial) samples of totally-unknown radioactivity, i.e., the time for the LSC measurement is predefined to meet the demanded decision thresholds.
About 4% of the digestion solution has been used to produce AMS targets by the following steps [27]: (1) addition of NH3aq (25%); (2) rest overnight and centrifuging; (3) two times washing with ~ 10 ml very dilute ammonia solution (pH 8-9); (4) parts of washed hydroxides have been transferred to quartz crucibles, dried at 110–125 °C; (5) ignite for 15 min at 400 °C and 2 h at 600 °C in a muffle furnace; (6) mixed with Ag powder (Aldrich, −325 mesh, 99.99 + %); and (7) pressed into Cu sample holders with Cu pins as backing (Table 1).
For blank corrections (machine blank, Table 1), iron oxide has been produced from pre-nuclear, i.e., from the beginning of the last century iron metal, kindly provided by Peter Steier (U Vienna, Austria) [28] by dissolution in HCl/HNO3 and evaporation, followed by further treatment as mentioned above. AMS standard-type material “A0” and “A2” [20] provided as Fe2O3 has been mixed with Ag and pressed into Cu sample holders with Cu backing (Table 1). “A2” has been used for quality assurance.
AMS measurements
For the AMS measurement a thermally and electrically conductive material is required. When the material is non-conductive it is mixed with additional (metal) powder and pressed (or hammered) into sample holders. It has to be noted that one of the major advantages of AMS is the small amount of sample material required for a measurement, typically a few mg are sufficient. Details of AMS measurements at the DREsden AMS (DREAMS) facility have been described earlier [29, 30]. We took advantage of the works of Wallner et al. [19, 20] describing high-sensitivity isobar-free 55Fe AMS measurements and applied the knowledge to DREAMS.
The 16 AMS sample holders together with those containing blanks, standards “A0” and quality assurance samples “A2” [20] are placed repeatedly one-by-one in a Cs-sputter ion source. Unknown samples were inserted once or twice into the ion source. By selecting Fe− ions isobaric interference from 55Mn is completely eliminated. Single-charged negative ions of 54Fe, 55Fe, and 56Fe are quasi-simultaneously (100 times per second each) injected into the accelerator by the fast-bouncing system that switches between the beams via changing the potential of the vacuum chamber of the beam injection magnet. The negative ions are accelerated towards the positively charged terminal of 3.89 MV. In the terminal, the particles pass the gas stripper (a small argon gas volume) resulting in a charge exchange and a distribution of positive ions. A 90° analysing magnet selects ions of charge state 3+ with an energy of 15.6 MeV. Sayer [31] predicts that this charge state is the most-populated one when stripping with argon gas at this energy and we found a transmission of 22%. It is also the most populated charge state in an oxygen gas stripper at a slightly lower beam energy of 3.18 MeV (23% transmission) [19]. Stable iron isotopes 54 and 56 are measured quasi-simultaneously in Faraday cups after the analysing magnet on the high-energy side. The radioactive nuclide 55Fe is counted, after passing additional background filters (35° electrostatic analyser, vertical 30° magnet), in a gas ionisation chamber. The measured ratios of our in-house standard material “A0” and quality assurance material “A2” were as high as 85% of their nominal true value, which was caused by transport losses of the ion beam between Faraday cup and gas ionisation chamber in this case.
Hence, results are normalised to the in-house standard material “A0” [20], which is decay-corrected to the date of the AMS measurement with the quoted overall uncertainty of 1.6%. For the conversion of 55Fe atoms into activity units, the half-life of (2.7563 ± 0.0004) years [1] is used.
Results and discussion
Sample preparation
The small inner diameter (∅: 1.3 mm) of our standard AMS sample holders and the small inner diameter of the corresponding funnel to fill the sample into the holder is currently the bottleneck for easy and fast sample preparation of steel samples, which are not radiochemically processed at all. The more coarse grain the chips are, the more tedious the work gets. An optimised set-up will reduce AMS sample preparation to 5–10 min per sample. It will also allow more material to be in one sample holder omitting the need for measurements of multiple aliquots of the same sample material.
For comparison, radiochemical processing of steel samples for 55Fe AMS measurements of Fe2O3 will need less than an hour working time per sample distributed over 2–3 days for best chemical yield. As low chemical yields are not influencing the result and only less than 10 mg Fe2O3 is needed for AMS analysis, there is much room for cutting down to less than 24 h. Total costs for consumables are in the order of 15 € compared to 5 € for instrumental, i.e., without chemistry, AMS sample preparation. The costs of the consumables (chemicals, vials, pipette tips, scintillation cocktail) for LSC are also in the range of 15 € per sample. However, all these values do not comprise manpower, operating, maintenance and repairs of laboratories and measurement equipment/devices, overhead expenses etc. These calculations neither include potential Fe measurements in liquid aliquots by ICP-MS or other techniques.
The pure working time for the complete radiochemical procedure starting with the digestion and ending with the LSC and ICP-MS measurement comprises about 20–25 h distributed over 4–5 working days. Normally 4–8 samples are worked on in parallel, i.e., as one batch, in this time period.
Hence, the determination of 55Fe by AMS is definitely a competitive alternative to LSC determination when balancing costs for consumables, efforts in working time and manpower as well as expenses for operation, repair and maintenance of the different instruments.
Stable isotope measurements
Stable isotopes have been measured by AMS as Fe3+ in Faraday cups with currents of 4–17 nA for 54Fe and 60–260 nA for 56Fe, respectively, which is comparable to experiments elsewhere reporting several hundreds of nA for 56Fe [20]. Measured 54Fe/56Fe ratios of samples are normalised to ratios of the standard material “A0” set assuming to have a natural ratio of 0.0637 [32]. The scatter of this ratio in the whole set of unknown sample materials is only 0.3% (at 1-sigma).
The difference in fractionation from steel vs. Fe2O3 is less than 5% monitored by the difference in 54Fe/56Fe. Hence, there is no need for matrix-matched standards and blanks in AMS, as it is absolutely required for other analytical methods like secondary ion mass spectrometry (SIMS) or laser ablation ICP-MS.
Results of ICP-MS measurements of iron of liquid aliquots of chemically-processed samples are presented in Table 2. They confirm within 1-sigma uncertainties the steel samples to be “100% Fe”. The uncertainty of 10–11% for the ICP-MS measurement of natural iron is a combined uncertainty consisting of following parts: wet digestion (8%), calibration (5–7%), pipetting/diluting (1%) and the measurement uncertainty (1.5–2.5%).
55Fe measurements by AMS
Results of 55Fe measurements by AMS are presented in Table 2. AMS measurements of blank material (Fe2O3) resulted in the determination of zero counts within 10 min corresponding to a 1-sigma upper limit of 3 × 10–14 55Fe/56Fe, which is actually between blank values of earlier experiments at Vienna and Munich [2, 19, 20]. Longer measurements of the blank material would decrease the background level but were not anticipated in this work as 55Fe/56Fe ratios of samples are 2–3 orders of magnitude above the present blank level, resulting in negligible blank corrections. These results clearly indicate the strength of AMS over other mass spectrometry methods as isotopic suppression from e.g., 54FeH− is highly effective and does not contribute to any background.
Sample values of 55Fe/56Fe in Table 2 are presented as blank-corrected values, so are the specific activities using an Fe concentration of the steel material to be 100%. The present 55Fe blank value is an upper limit of 3 Bq gFe−1. The estimate is based on the analysis of a single sample with limited measurement time. A reduction of the blank value by factor of 3–6 is expected when increasing the measurement time from a few minutes to the maximum sputter time of the sample. The final limitation as for most other AMS measurements [30] is defined by long-term memory and cross-contamination in the ion source, which is in the order of 10–4 for 55Fe. Therefore, by using standard material of 10–11 55Fe/56Fe, the background is expected to be in the order of 10–15, resulting in a theoretical limit of 0.08 Bq gFe−1. However, as expressed by the normalisation to Fe, the natFe content is influencing this limit for the specific activity. Hence, for samples containing less natFe than 100%, the limits are in the range of LSC or slightly higher, e.g., for graphite (0.091 Bg g−1) or concrete (0.018 Bq g−1) samples elsewhere [16], or 0.01 Bq g−1 at the VKTA lab (using 0.5–1 g concrete, soil or graphite).
Internal consistency of AMS measurements were tested by measuring the “A2” standard-type sample as an unknown. The measured 55Fe/56Fe ratio of (1.46 ± 0.11) × 10–11 overlaps within 1-sigma with the nominal value (19th February 2020; (1.344 ± 0.023) × 10–11) after a short measurement time of 7 min.
The comparison of AMS data of four different samples of chemically untreated, pristine chips (weighted mean of three individual targets, each 7 min measurement time) with AMS data of processed Fe2O3 (one target, 11–17 min measurement time) of the corresponding samples is shown in Fig. 1. Individual uncertainties are between 4–13% and 6–12% for oxide and metal samples, respectively.

Comparison of isotopic ratios by AMS for radiochemically treated and unprocessed chip material
We treat remeasurements of steel from the same material as measurements that are expected to give the same result. The standard deviation of the mean is thus the standard deviation divided by the square-root of the number of measurements (N = 3 samples). The mean is estimated using the absolute uncertainty as the inverse weight. The uncertainty given for the mean in Table 2 is the higher from (1) the combination of uncertainties of the single measurements or (2) the weighted standard deviation of the mean. Uncertainties of the single results are not uniform and thus the weighted standard deviation of the mean was estimated by taking into account the uncertainty of the single data point via a weighted standard deviation of the mean. Again, we used the absolute uncertainty as the weight.
Data for two out of four samples overlap within 1-sigma, one sample within 2-sigma uncertainties. Metal samples have systematically lower ratios than chemically-treated oxide samples. Hence, our pilot study might point to a possible (systematic or random) additional uncertainty in the order of 5–10%, which is not a function of 55Fe concentration. Further experiments are needed to evaluate these effects.
55Fe measurements by LSC and comparison with AMS
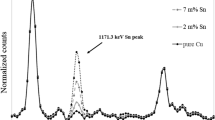
Measured specific activities of 55Fe by LSC and associated overall uncertainties in the range of 12–13% are included in Table 2 and Fig. 2. The uncertainty for the determination of 55Fe by LSC after radiochemical separation is also a combined uncertainty, with following components: chemical handling (weighing, wet digestion, pipetting, chemical separation, ICP-MS) with 10–12%, quench curve calibration with 5–8%, pure measurement uncertainty with 0.10–0.65% for these four samples (but could be higher with lower count numbers). In the case of the four samples used for this comparison, the resulting decision thresholds range from 0.35–0.42 Bq g−1 due to the composition (almost only iron) and high total activity of the sample material, which restricts the analytically reasonable amount to 40–60 mg. The four LSC spectra are presented in Fig. 3. They show no interferences from other radionuclides or luminescences, thus, the radiochemical procedure is characterised by a high separation performance for disturbing radionuclides as well as chemically interfering elements. In the 1220 Quantulus Low-Level scintillation counter (PerkinElmer®) the count rates of the chemical blanks range from 1.5–2.0 cpm, the luminescences range from 0.1–0.75 cpm and the measured count rates for the four investigated samples range from 168–6385 cpm in the evaluated channel region. With SQP(E) values about 705–715 the measurement efficiency lies in the region of 0.29–0.31.

Comparison of specific activities resulting from AMS data for radiochemically treated and unprocessed chip material with LSC data

LSC spectra of 55Fe samples after radiochemical separation of iron. Measurement times are 150 min, each
For comparison of the LSC data with AMS data, we did not use individual Fe data measured by ICP-MS but set Fe to 100% to calculate 55Fe in units of Bq per gram sample material (see above). The resulting data is presented in Table 2 and Fig. 2.
Most importantly, the complete AMS data set of the chemically-treated samples is comparable with the LSC data within 1-sigma uncertainty. Three out of four AMS data of the metal chips are comparable within 1-sigma with the LSC data; one sample (VKTA-5) within 2-sigma. As mentioned above, the uncertainties of the AMS data seem to be partially underestimated and are in this pilot study not (yet) a function of the Fe isotopic ratio. They can reach levels as high/low as LSC data uncertainties. The combined uncertainties of the LSC data are in the range of 12–13% for this study, but can also lie between 10–35% and comprises not only the counting statistics from LSC but also uncertainties from sample pretreatment, chemical handling and recovery determination by ICP-MS. Further systematic AMS work would be necessary to reduce the AMS uncertainties to a “routine” 5%-level, however, for some scientific projects and commercial analyses a reasonable and fast detection method with a 15%-uncertainty might be of more interest.
Conclusions
Our first AMS pilot study of pristine steel samples for 55Fe determination can be described as very successful. Additional improvements can be expected in the range of 50% by further optimising ion source output and ion transport efficiency through the AMS system. The current background can be lowered by longer measurement times. Fine-grained material is favourable over coarse chips not only for easier filling of AMS sample holders, but it will also improve reproducibility by avoiding different sample geometries that influence fractionation while sputtering. The latter is also a prime suspect for the difference between metal and oxide samples of the same material.
AMS without chemical sample treatment has impressive advantages over LSC measurements after radiochemical separation of iron and will surely work for all thermally and electrically conducting materials. In the future, there will be a clear correlation of 55Fe specific activity level and the anticipated uncertainty of the 55Fe result with the overall analysing costs as the major contribution will not be from sample preparation but from accelerator time. To overcome even these costs, there are possibilities for external research projects at several accelerator facilities via a proposal system, e.g., funded by the Helmholtz Association (large-scale-facility [33]) or Trans-National-Access funded by the EU (RADIATE [34]).
As this technique is omitting any chemical preparation, it is not only more reasonable and faster, but we can also exclude any systematic errors due to incomplete sample digestion and there is no need to monitor the chemical yield. However, the method only works for pure neutron capture nuclides within the sample—such as 55Fe in steel—as we cannot exclude sample inhomogeneities for other production ways: This could be either neutron activation “elsewhere” followed by contamination (by liquid or dust transport) of the taken sample or any other e.g., nuclear spallation reaction on non-homogeneously distributed heavier elements. For sure, any errors due to non-representative sample-taking will be as erroneous for our new method as for all other analytical methods.
Last but not least, it is obvious that for non-pure iron materials an Fe determination by other analytical techniques will be essential to calculate activity levels of 55Fe from the Fe ratios determined by AMS. This could be either classical ICP techniques after sample digestion—ruining partially the above-mentioned advantages—or non-destructive methods like X-ray fluorescence (XRF) or particle-induced X-ray emission (PIXE) using a micro-beam focussed on material pressed in AMS sample holders before AMS measurement.
References
Pommé S, Stroh H, Van Ammel R (2019) The 55Fe half-life measured with a pressurised proportional counter. Appl Radiat Isot 148:27–34. https://doi.org/10.1016/j.apradiso.2019.01.008
Korschinek G, Müller D, Faestermann T, Gillitzer A, Nolte E, Paul M (1990) Trace analysis of 55Fe in biosphere and technology by means of AMS. Nucl Instrum Methods Phys Res Sect B 52:498–501. https://doi.org/10.1016/0168-583X(90)90465-7
Wallner A, Buczak K, Belgya T, Bichler M, Coquard L, Dillmann I, Golser R, Käppeler F, Karakas A, Kutschera W, Lederer C, Mengoni A, Pignatari M, Priller A, Reifarth R, Steier P, Szentmiklosi L (2017) Precise measurement of the thermal and stellar 54Fe(n,γ)55Fe cross sections via accelerator mass spectrometry. Phys Rev C 96:025808. https://doi.org/10.1103/PhysRevC.96.025808
Slavkovská Z, Wallner A, Reifarth R, Pavetich S, Bott L, Brückner B, Göbel K, Al-Khasawneh K, Koll D, Merchel S, Reich M, Volknandt M, Weigand M (2020) Investigation of 54Fe(n,γ)55Fe and 35Cl(n,γ)36Cl reaction cross sections at keV energies by accelerator mass spectrometry. EPJ Web Conf 232:02005. https://doi.org/10.1051/epjconf/202023202005
Honda M, Arnold JR (1964) Effects of cosmic rays on meteorites. Science 143:203–212. https://doi.org/10.1126/science.143.3603.203
Mabuchi H, Tobailem J, Leger C, Bibron R, Blettmann D (1968) Radioactivite induite par le rayonnement cosmique dans la meteorite Granès. Geochim Cosmochim Acta 32:949–963. https://doi.org/10.1016/0016-7037(68)90060-4
Wallner A, Buczak K, Faestermann T, Klix A, Korschinek G, Lederer C, Plompen A, Poutivstev M, Rugel G, Seidel K, Semkova V, Vonach H (2011) Production of long-lived radionuclides 10Be, 14C, 53Mn, 55Fe, 59Ni and 202gPb in a fusion environment. J Korean Phys Soc 59:1378–1381. https://doi.org/10.3938/jkps.59.1378
Schumann D, Neuhausen J, Eikenberg J, Rüthi M, Wohlmuther M, Kubik PW, Synal HA, Alfimov V, Korschinek G, Rugel G, Faestermann Th (2009) Radiochemical analysis of a copper beam dump irradiated with high-energetic protons. Radiochim Acta 97:123–131. https://doi.org/10.1524/ract.2009.1585
Palmer HE, Beasley TM (1965) Iron-55 in humans and their foods. Science 149:431–432. https://doi.org/10.1126/science.149.3682.431
United Nations Scientific Committee on the Effects of Atomic Radiation (2000) Report to the General Assembly, Volume I: Sources, Annex C: Exposures from man-made sources of radiation, New York. https://www.unscear.org/docs/publications/2000/UNSCEAR_2000_Annex-C-CORR.pdf. Accessed 29 Dec 2020
Radiological characterization of shut down nuclear reactors for decommissioning purposes (1998) Technical reports series, No. 389, International Atomic Energy Agency, Vienna
INTERNATIONAL ATOMIC ENERGY AGENCY (2007) Strategy and methodology for radioactive waste characterization. IAEA-TECDOC-1537, IAEA, Vienna
INTERNATIONAL ATOMIC ENERGY AGENCY (2009) Determination and use of scaling factors for waste characterization in nuclear power plants. IAEA Nuclear Energy Series No. NW-T-1.18, IAEA, Vienna
Hou X (2007) Radiochemical analysis of radionuclides difficult to measure for waste characterization in decommissioning of nuclear facilities. J Radioanal Nucl Chem 273:43–48. https://doi.org/10.1007/s10967-007-0708-x
Zaffora B, Magistris M, Chevalier J-P, Luccioni C, Saport G, Ulrici L (2017) A new approach to characterize very-low-level radioactive waste produced at hadron accelerators. Appl Radiat Isot 122:141–147. https://doi.org/10.1016/j.apradiso.2017.01.019
Hou X, Frøsig Østergaard L, Nielse SP (2005) Determination of 63Ni and 55Fe in nuclear waste samples using radiochemical separation and liquid scintillation counting. Anal Chim Acta 535:297–307. https://doi.org/10.1016/j.aca.2004.12.022
Leskinen A, Salminen-Paatero S, Räty A, Tanhua-Tyrkkö M, Iso-Markku T, Puukko E (2020) Determination of 14C, 55Fe, 63Ni and gamma emitters in activated RPV steel samples: a comparison between calculations and experimental analysis. J Radioanal Nucl Chem 323:399–413. https://doi.org/10.1007/s10967-019-06937-4
Curioni A, Dinar N, La Torre FP, Leidner J, Murtas F, Puddu S, Silari M (2017) Measurements of 55Fe activity in activated steel samples with GEMPix. Nucl Instrum Methods Phys Res Sect A 849:60–71. https://doi.org/10.1016/j.nima.2016.12.059
Wallner A, Bichler M, Dillmann I, Golser R, Käppeler F, Kutschera W, Paul M, Priller A, Steier P, Vockenhuber C (2007) AMS measurements of 41Ca and 55Fe at VERA – two radionuclides of astrophysical interest. Nucl Instrum Methods Phys Res Sect B 259:677–682. https://doi.org/10.1016/j.nimb.2007.01.207
Wallner A, Bichler M, Buczak K, Fink D, Forstner O, Golser R, Hotchkis MAC, Klix A, Krasa A, Kutschera W, Lederer C, Plompen A, Priller A, Schumann D, Semkova V, Steier P (2013) High-sensitivity isobar-free AMS measurements and reference materials for 55Fe, 68Ge and 202gPb. Nucl Instrum Methods Phys Res Sect B 294:374–381. https://doi.org/10.1016/j.nimb.2012.03.029
Tiessen C, Bemmerer D, Rugel G, Querfeld R, Scharf A, Steinhauser G, Merchel S (2019) Accelerator mass spectrometry (AMS) for beryllium-7 measurements in smallest rainwater samples. J Radioanal Nucl Chem 319:965–973. https://doi.org/10.1007/s10967-018-6371-6
Hampe D, Gleisberg B, Akhmadaliev S, Rugel G, Merchel S (2013) Determination of 41Ca with LSC and AMS: method development, modifications and applications. J Radioanal Nucl Chem 296:617–624. https://doi.org/10.1007/s10967-012-2145-8
Merchel S, Bremser W, Bourlès DL, Czeslik U, Erzinger J, Kummer N-A, Leanni L, Merkel B, Recknagel S, Schaefer U (2013) Accuracy of 9Be-data and its influence on 10Be cosmogenic nuclide data. J Radioanal Nucl Chem 298:1871–1878. https://doi.org/10.1007/s10967-013-2746-x
Welte C, Wacker L, Hattendorf B, Christl M, Fohlmeister J, Breitenbach SFM, Robinson LF, Andrews AH, Freiwald A, Farmer JR, Yeman C, Synal HA, Günther D (2016) Laser ablation - accelerator mass spectrometry: an approach for rapid radiocarbon analyses of carbonate archives at high spatial resolution. Anal Chem 88:8570–8576. https://doi.org/10.1021/acs.analchem.6b01659
Moghissi AA, Whittaker EL, McNelis DN, Lieberman R (1974) Improved liquid scintillation technique for environmental monitoring of iron-55. Anal Chem 46:1355–1356. https://doi.org/10.1021/ac60345a060
Fusban HU, Rühle H, Gans I (1997) Procedure for determining iron-55 in wastewater, H-Fe-55-AWASS-01, Federal coordinating office for drinking water, groundwater, wastewater, sludge, waste and wastewater of nuclear power plants, ISSN 1865–8725. https://www.bmu.de/fileadmin/Daten_BMU/Download_PDF/Strahlenschutz/leitstelle_h_fe_55_awass_01_v1997_05_en.pdf. Accessed 23 Sept 2021
Merchel S, Beutner S, Opel T, Rugel G, Scharf A, Tiessen C, Weiß S, Wetterich S (2019) Attempts to understand potential deficiencies in chemical procedures for AMS. Nucl Instrum Methods Phys Res Sect B 456:186–192. https://doi.org/10.1016/j.nimb.2019.05.005
Steier P, Hain K, Klötzli U, Lachner J, Priller A, Winkler S, Golser R (2019) The actinide beamline at VERA. Nucl Instrum Methods Phys Res Sect B 458:82–89. https://doi.org/10.1016/j.nimb.2019.07.031
Akhmadaliev S, Heller R, Hanf D, Rugel G, Merchel S (2013) The new 6 MV AMS-facility DREAMS at Dresden. Nucl Instrum Methods Phys Res Sect B 294:5–10. https://doi.org/10.1016/j.nimb.2012.01.053
Rugel G, Pavetich S, Akhmadaliev S, Enamorado Baez SM, Scharf A, Ziegenrücker R, Merchel S (2016) The first four years of the AMS-facility DREAMS: status and developments for more accurate radionuclide data. Nucl Instrum Methods Phys Res Sect B 370:94–100. https://doi.org/10.1016/j.nimb.2016.01.012
Sayer RO (1977) Semi-empirical formulas for heavy-ion stripping data. Rev Phys Appl 12:1543–1546. https://doi.org/10.1051/rphysap:0197700120100154300
National Nuclear Data Center, information extracted from the NuDat 2 database, https://www.nndc.bnl.gov/nudat2/. Accessed 29 June 2021
http://www.hzdr.de/ibc. Accessed 20 Apr 2021
https://www.ionbeamcenters.eu/radiate/. Accessed 20 Apr 2021
Acknowledgements
Parts of this research were carried out at the Ion Beam Centre (IBC) at the Helmholtz-Zentrum Dresden-Rossendorf e. V., a member of the Helmholtz Association. We would like to thank the DREAMS operator team for supporting the AMS measurements, Peter Steier (U Vienna) for providing pre-nuclear iron, Matthias Köhler (VKTA) for his constant interest and support for new method validation, Gerlinde Bierig (VKTA) for doing all the complex lab work and Uta Czeslik (VKTA) for ICP-MS measurements. We thank three anonymous reviewers for helpful comments and questions.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Merchel, S., Rugel, G., Lachner, J. et al. Evaluation of a sensitive, reasonable, and fast detection method for 55Fe in steel. J Radioanal Nucl Chem 330, 727–735 (2021). https://doi.org/10.1007/s10967-021-08000-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-021-08000-7