
Abstract
In 90Sr analysis, determining its daughter 90Y improves the sensitivity of the radiometric methods. We found that to imprint a cavity made of [Y(6-(4-Vinylphenylcarbamoyl)pyridine-2-carboxylate)3] into a polystyrene skeleton yields a solid phase extraction resin with high selectivity for Y and Ln(III) over transition metals, alkaline, and alkaline-earth cations. We used this resin in an automated chromatography system to extract 90Y from milk, grass, vegetables, soil, sediments, water, human bones, and milk teeth samples. We found that the ion-imprinted resin could be used to separate light Ln(III) using a pH gradient, favoring the targeting of molecules used in nuclear medicine.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
90Sr (T1/2=28.2 y) is produced by the nuclear fission of heavy atoms such as 235U and 239Pu. It emits a β-particle of 546 keV, giving rise to 90Y, a hard β-emitter (T1/2= 64.2 h, 2.28 MeV), with which it reaches secular activity equilibrium in 12 days. As a consequence of its release into the environment, for instance during nuclear accidents, nuclear bomb testing, or authorized discharges, 90Sr/90Y are found in every significant compartment of the environment and the food chain. As an alkaline-earth cation, 90Sr has the same chemistry as calcium and targets bones in humans. It is easily transmitted from soil to plants with a TF of about 1.2, and from plant to milk with a TF of 0.02 kg L−1. In Switzerland, where dairy products are a large part of the diet, TF from milk (Bq g Ca−1) to human vertebrae (Bq g Ca−1) was estimated to be 0.3 [1, 2]. This means that 90Sr determination is an important part of the annual radioactivity survey plan of the Swiss Federal Office of Public Health in samples such as soil, sediments, grass, milk, wheat, foodstuff (especially vegetables), human vertebrae and deciduous teeth. However, to determine 90Sr in such matrices is not an easy task, because the radiometric determination of a β-particle necessitates a complete isolation of the radionuclide before measurement because the β-particle is strongly absorbed into the matrix. Recently, several examples of 90Sr determination by mass spectrometry were described [3,4,5], but none with a similar detection limit as the radiometric methods [6]. For samples with high Sr content such as teeth and bones, mass spectrometry cannot resolve the 88Sr signal [7]. Currently, isolating Sr from complicated samples such as soil and milk can be done using the Eichrom® Sr.spec resin, which will specifically extract Sr over Ca as a crown ether complex in a highly acid nitric medium [8]. Nevertheless, the extracting molecule is not covalently bound in Eichrom® SPE resins, so the resin cannot be reused or the presence of extracting material in the eluted fraction is a drawback e.g. in radiolabeling. In addition, Sr.spec cartridges can suffer from aging [9]. Recently, Sr.spec resin has been used in an automated separation method using the 1-probe/4-column prepFast MC computer controlled system from ESI for a fast determination of 90Sr in urine samples [10]. Alternately, ion-imprinting polymer (IIP) is a synthesis method in which a complexing unit with specific interactions with the template ion is covalently bound to a polymer substrate. Since Wulff’s pioneering work on the molecular recognition in polymers prepared by imprinting with a template [11], numerous studies have demonstrated the advantage of the ion-imprinted concept to create a material with enhanced selectivity over a wide range of metal cations [12,13,14,15,16]. However, functionalizing an extracting ligand with a styrene functionality to create the covalent bond during the polymerization is a challenging task, often resulting in polymers with a theoretical utility only, due to the high cost of the synthesis. Here we used dipicolinic acid, a cheap starting product, in a one-pot synthesis, to obtain the functionalized ligand 6-(4-Vinylphenylcarbamoyl)pyridine-2-carboxylic acid (HL1), a ligand already described by our research group [13] This ligand was used in an emulsion co-polymerization synthesis with styrene and divinylbenzene and Y3+ as the template to obtain a resin with a cavity suitable for the selective extraction of Y3+. The resin, obtainable in quantities of tens of grams, was used in an automated system for the extraction and elution of 90Y from all samples taken in the Swiss National Radioactivity Survey Plan since 2014, with the aim to determine 90Sr in secular radioactivity equilibrium with 90Y. In addition, we observed that the resin was able to separate La3+, Ce3+, Nd3+ and Gd3+ with a pH gradient from 2 to 0.6 only, thus leaving the Ln3+ fraction free of ligand for targeting molecules of interest in nuclear medicine.
Experimental
Ligand synthesis (HL1)
10 g (60 mmol) of dipicolinic acid were refluxed overnight in 100 ml of SOCl2. Excess SOCl2 was distilled (and reused in further synthesis) under vacuum, leaving a pale brown solid. The solid was dissolved in 500 mL of dry CH2Cl2 and 12.7 g (60 mmol) of K3PO4 was added as a solid. 7.14 g of vinylphenylamine (60 mmol) dissolved in 200 ml of dry CH2Cl2 was added dropwise for 2 h. The mixture was stirred overnight, then CH2Cl2 was evaporated under vacuum and the residue dissolved in 500 ml of 0.1 M NaOH. Insoluble di-substituted pyridine-2,6-dicarboxylic acid bis[(4-vinylphenyl)amide] formed as a minor by-product of the reaction was filtered off and 6-(4-Vinylphenylcarbamoyl)pyridine-2-carboxylic acid (HL1) was precipitated upon acidification. The product was filtered and dried in a desiccator under vacuum to yield 12 g (75 %) of HL1 as a white powder (Fig. 1). 1 H NMR (CDCl3): δ = 9.91 (s, 1 H, HNCO), 8.37 (d, 1 H, Ha), 8.13 (dd, 1 H, Hb), 8.55 (d, 1 H, Hc), 7.70 (d, 2 H, Hd), 7.38 (d, 2 H, He), 6.66 (dd, 1 H, CH = CHfH), 5.70 (d, 1 H, CH = CHHg), 5.22 (d, 1 H, CHh = CH2). Elemental analysis (%), calcd for HL1·H2O (C15H12N2O3·H2O): C 62.93, H 4.93, N 9.78; found: C 63.55, H 5.0, N 9.74.
Emulsion polymerization synthesis of the SPE resin IIP-Y
Divinylbenzene (DVB) and styrene were purified from stabilizer on Al2O3. A mixture of 40 mL of DVB, 6 mL of styrene, 1 g of polyvinylpirrolidone, 3.2 g of sodium dodecyl sulfate and 24 mL of dibutylphtalate was emulsified in 300 mL of water for 15 min in an ultrasonic bath. 5 g (18.6 mmol) of HL1 was dissolved in 50 mL of water by adding 19 mL of 1 M NaOH and added to the mixture. The mixture was poured in a 500 mL three-neck flask and degassed with N2 for 30 min under mechanical stirring (300 rnd per minute). 2.5 g of azobisisobutyronitrile (AIBN) dissolved in 20 ml of toluene was added as polymerization initiator and the temperature was increased to 70 °C. After 15 min, 1.9 g (6.2 mmoles) of YCl3·6H2O dissolved in 20 ml of water was added. After three hours, the mechanic stirrer rate was lowered to 150 rnd per minute and the mixture was left at 70 °C overnight to terminate the polymerization. The polymer particles were filtered off the mixture and washed with water and ethanol. The polymer particles were refluxed in THF overnight to remove all unreacted material, then filtered off and dried in an oven at 60 °C. The particles were sieved at 75 μm using a Fritsch 3 SPARTAN pulverisette apparatus. The polymer particles were wetted in a mixture (1:10) of ethanol:water and the Y3+ template cation was removed in 5 M HCl. After 2 h, the polymer particles were filtered off, washed with water until pH reached 4, ethanol and acetone, then dried in an oven at 70 °C to yield 35 g (75 %, based on DVB, styrene and HL1) of the resin IIP-Y. Based on batch extraction experiments, the resin has a loading capacity of 4 mg g−1 IIP-Y.
Loading solution preparation
Every sample (except water and urine, see below) was ashed at 550 °C for at least 48 h in an oven. 50 g of ashes (soil, sediments) or 10 g of ashes (grass, wheat, human vertebrae) or at least 5 g of ashes (vegetables, teeth) were traced with 10 mg of stable Y3+ and 100 mL of 8 M HNO3 and 6 mL of 30 % H2O2 were added. The mixture was introduced into a Teflon beaker for microwave digestion under pressure (50 bars) for 30 min at 180 °C using an UltraClave IV apparatus (MLS, Milestone, Germany). The mixture was centrifuged to remove solids (3000 rnd per minute for 10 min) and the supernatant was diluted to 500 mL with ultrapure water and filtered if necessary. 15 g of oxalic acid were added and alkaline-earth oxalates were precipitated at pH 1.5 by adding NH4OH 25 %. After decantation and removal of the supernatant the oxalate precipitate was dissolved in 20 ml (or more) of hot concentrated HNO3 and the solution diluted to 500 mL. A new fraction of 15 g of oxalic acid was added and the alkaline-earth oxalates were precipitated once again to completely remove the phosphates. After decantation and centrifugation, the oxalates were wet-ashed in 20 ml of concentrated HNO3 and 6 mL H2O2 in a Teflon beaker at 200 °C for 15 min under a pressure of 50 bars using the UltraClave IV apparatus. The resulting solution was diluted to 300 mL and the pH adjusted to 3 with 25 % NH4OH.
Urine samples
500 mL of urine were acidified with 100 mL of concentrated HNO3 and 10 mg of Y3+ as a carrier was added, afterward the mixture was heated at 80 °C for 2 h. After cooling, 2 mL of 85 % H3PO4 85 % and 1 mL of a solution of Ca (100 mg/mL) were added and the calcium phosphate precipitated at pH 8 upon the addition of 25 % NH4OH. After decantation and centrifugation, the precipitate was wet-ashed in 15 ml of concentrated HNO3 for 15 min at 200 °C and a pressure of 50 bars in the UltraClave IV. The solution was diluted to 300 mL with water and the pH adjusted to 3 with 25 % NH4OH.
Water samples
To a 1 l water sample, acidified with HNO3 and filtered at 0.45 μm 10 mg of Y3+ carrier was added and the pH adjusted to 3 with NH4OH.
SPE procedure
The solution (at pH 3) was loaded at a rate of 1 mL minute-1 on a column made of 3.5 g of IIP-Y in a 12 mL column (∅ 1 cm) using the automaton (see below) to extract Y3+. The column was washed with 100 mL of water at pH 3, afterward Y3+ is eluted with 20 mL of 1 M HCl and 50 mL of water. Because 210Bi3+ and 228Ac3+ could interfere in some cases, a further purification was carried out as followed: 2 mg of Ba2+ and 2 mg of Fe3+ were added to the elution solution along with 2 drops of a solution of methyl orange pH indicator (pH 3.1–4.4) and 5 mL of 0.25 M ammonium acetate. Upon the addition of 5 drops of concentrated H2SO4, a precipitate of BaSO4 formed. NH4OH 25 % was added until the solution turned from pink to orange (avoid yellow coloration). The precipitate was filtered off and Y3+ oxalate was precipitated from the filtered solution upon adding 10 mL of an ethanol solution of oxalic acid (12 g in 60 mL). An Y-oxalate source was obtained through micro-filtration on Millipore filter (GSWOP 02400).
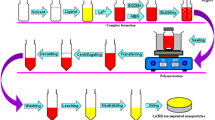
Automating of the SPE procedure
The extraction and elution flow chart was fully automated. We constructed an apparatus which can load the initial solution, select the washing solutions and elute Y3+ in a medium appropriate for preparing a source for radiometric counting. The regeneration of the column was also included in the flow chart. The extraction/elution/regeneration procedure can be executed overnight so that the purified 90Y solution is available for the operator the next morning. Details of the apparatus are given in SI.
90 Y counting
90Y oxalate sources were counted in a low-level gas proportional counter (Tennelec LB 4100w) with 4 drawers and 16 counters in steps of 4 h, 30 times. The counting efficiency of this counter was 0.48 for the β-particle of 90Y. Background was 0.4 dpm. Chemical recovery was determined by ashing the oxalate source and dissolving the residue in 1 % HNO3, followed by quantification of Y with ICP-OES (ThermoFisher iCAP 7000). The radionuclide purity was checked by determining the 90Y half-life based on the 30 counting steps and must be between 60 and 69 h for the source to be considered pure enough. When necessary (e.g. bone and teeth), a 1 mL aliquot of the loading solution was collected to determine Ca by ICP-OES to normalize the 90Sr activity to the Ca weight.
Light Ln(III) separation
Ln3+ (50 µg each) were extracted at pH 3 on the IIP-Y resin loaded in a 25 cm long (4 mm internal diameter) JR-68185NF peek column for intermediate pressure chromatography (VCIJour, Switzerland). Ln3+ elution by pH gradient was obtained by mixing ultrapure water and 0.5 M HCl with a Varian Pro Star gradient pump. Fractions of 10 ml were sampled automatically and Ln3+ determined in each fraction with ICP-OES spectrometry.
Quality control
The quality control was carried out by participating to several inter-laboratory comparison exercises and by measuring several reference materials (see "Results and Discussion" section.)
Results and discussion
IIP-Y synthesis
The ion-imprinted polymer synthesis presented in this work is an ameliorated synthesis procedure of a similar IIP published previously by our research group [13]. In our previous work, the HL1 synthesis followed a three-step synthesis-isolation procedure with BuLi as activator of the amide synthesis step. The procedure involved isolating the ethyl ester, the amide substitute and the final product HL1, after hydrolysis. Here, we successfully produced HL1 in a one-pot synthesis by activating the dipicolinic acid with SOCl2, following previous work by Devi et al. [17]. Hydrolysis of the acyl chloride took place during the isolation step of the final product. One major drawback of the polymer synthesized by Chauvin et al. [13] was the polymerization step in which the Y(6-(4-Vinylphenylcarbamoyl)pyridine-2-carboxylate)3 complex was introduced as an insoluble species in DVB and styrene mixture diluted in pyridine and DMSO. As a result, to reach an acceptable loading capacity necessitated a high doping percentage (30 % HL1). Moreover, the synthesis in organic solvent (DMSO and pyridine) resulted in a large particle size distribution, including many very small particles, hindering the use of the IIP in solid phase extraction (SPE). Here we used an emulsion polymerization procedure to obtain IIP-Y, yielding larger particles allowing SPE in columns containing 3.5 g of polymer. Gravity flow could be obtained because of the absence of very small particles. Moreover, we formed the Y(L1)3 complex in-situ during the polymerization step, after L1 had already reacted with forming polymer particles. Doing this enabled the formation of large particles with the extracting cavity of Y(L1)3 situated preferentially on the surface of the PS particles. Thus our IIP-Y has a larger extraction capacity, compared to the IIP-Y synthesized in Chauvin et al., for a lower doping percentage. In this work, for a doping percentage of 5 g of HL1 for 45 mL of DVB/Sty, or 9.0 %, the loading capacity of IIP-Y was 4.4 mg g−1 of resin. In comparison, the IIP-Y polymer synthesized by us previously [13] had a loading capacity of 8.6 mg Y3+ g resin−1, but with a much higher doping percentage of 33 %. In conclusion, we considerably simplified the synthesis of HL1 and the emulsion polymerization procedure yielded an IIP with a larger extraction capacity for Y3+ and larger particles making it usable in automated SPE.

One-pot synthesis of ligand HL1. (1) SOCl2, reflux, formation of the acyl chloride (2) vinylphenylamine, r.t., formation of the mono-amide (3) H2O, hydrolysis of the remaining acyl chloride to yield HL1
90 Y SPE in environmental and human bone and teeth samples
In most environmental and human samples of a radioactivity survey plan, 90Y is in secular equilibrium with 90Sr because of the time elapsed from the sampling to the beginning of the analysis. In addition, γ-analysis is usually carried out on most samples prior to 90Sr analysis. Most often, the time for the transfer of the sample to the lab, for drying, milling, freeze-drying, ashing, etc., usually exceeds the 12 days necessary to obtain a secular equilibrium between 90Sr activity and 90Y activity in a sample. Thus, the 90Sr activity can be obtained by determining the 90Y activity. Results are summarized in Table 1 for the samples of the Swiss radioactivity survey plan, from the years 2015 to 2019, after extraction and measurement of 90Y using IIP-Y and proportional counting.
Results of Table 1 show that the overall chemical yield of the automated method was above 80 % for all samples (n = 652). The 90Y radionuclide purity, checked by determining the half-life, never surpassed 69 h or was lower than 61 h, demonstrating a very good radiochemical separation (Fig. 2). Most often, samples like soils contained an average of 30 Bq kg−1 of 238U and 232Th series, each series containing up to 20 other radioactive elements. These activities largely exceeded the average 0.8 Bq kg−1 determined in this work for 105 soils samples. Thus, our procedure was able to completely separate natural radionuclides from 90Y. Results of Table 1 also show that the SPE procedure was able to reach a very low detection limit necessary for the current determination of 90Sr in the calciferous tissues in human (bone and teeth). For instance, the current level of activity in human vertebrae in Switzerland is close to 10 mBq g Ca−1. Thus, the determination of 90Sr in this type of sample, using 10 g of bone ash, required a detection limit of a few mBq g Ca−1. This was achieved here by using SPE with a high yield (> 80 %), by using a large sample weight (10 g of bone ash) and a low level background counting of 0.4 dpm. Considering the high Ca content of bone, teeth, and milk ashes, there is comparatively no other SPE method that can separate the Ca and Sr with a reasonable time and price. In addition, determining 90Sr in water at the detection limit of 8 mBq L−1, well below the Euratom recommendation for drinking water (4.9 Bq L−1), can be carried out without any preparation, to the exception of pH adjustment to 3. The EU 2013/51 Euratom legislation imposes a method to be able to determine 90Sr at the 0.4 Bq L−1 level [18]. In this work, we show that SPE using an ion-imprinted polymer can reach this goal with a fully automated method. To the opposite of some other automated separation method for the determination of 90Sr [10], we choose to work with a large volume of loading solution (from 300 ml to 1 l) to simplify the working procedure and minimize the dead volume percentage. Because the IIP particles can be obtained easily in large quantity (tens of grams) and can be reused, larger columns can be implemented in the automated system. In addition, 90Sr determination can be carried out directly on 1 l of water, without additional work, except the addition of the stable Y carrier and the pH adjustment (at 3). Finally, the extraction and elution procedure is carried out overnight, which leaves the laboratory technician with a solution ready for preparation of the source (either for liquid scintillation of for proportional counting, depending on preferences).

90Y half-life determined on experimental counts (Tennelec LB4100w) of a 90Y oxalate source prepared after SPE starting from 50 g of soil containing 2.05 Bq kg−1 of 90Sr
Results of the quality control
Results are presented in Table 2. Unfortunately, there is no reference material for 90Sr in teeth to our knowledge. However, since teeth are chemically analogous to bone, reference material IAEA-A-12 can be considered equivalent. Results of Table 2 show that the SPE IIP-Y method is particularly well adapted to determining 90Sr in difficult samples such as soil and sediment as well as in samples with high Ca content such as bone and milk. In addition, results from four inter-laboratory comparison exercises organized by the PROCORAD association [19] demonstrate that the SPE resin IIP-Y is particularly well adapted to determining 90Sr in urine samples. The simplicity of the method makes it eligible for the rapid determination of 90Sr in urine in emergency situations. During the two BfS inter-laboratory exercises 2015 and 2017 [20], 90Sr had to be determined in tandem with 89Sr and several actinides (238U, 241Am, 239Pu). 89Sr was present at about ten times the activity level of 90Sr, mimicking the real activities found in burnt nuclear fuel. Accordingly, our SPE resin IIP-Y was able to determine accurately the 90Sr activity in these complicated conditions.
Light Ln 3+ separation
The ion-imprinted polymer synthesized in this work contained Y3+ as a template cation. Thus the imprinted complexation cavity is formed by [Y(L1)3] complex. Because Y3+ has a cationic radius similar to the Ln3+ from the middle of the Ln3+ series, the possibility of IIP-Y to have an enhanced selective for some of the Ln3+ has been demonstrated earlier by Chauvin et al. [13]. In our previous work, the selectivity of the IIP-Y resin for Y3+ over Ln3+ was established as La > Nd > Eu, Gd in batch experiments using Y/Ln pair. Here we show that the IIP-Y resin can separate light Ln3+ through elution with a pH gradient only (Fig. 3).

Light Ln3+ separation on IIP-Y resin using a pH gradient from 2 to 0.6
The fact that Ln3+ can be separated on the IIP-Y without the help of a co-ligand is an important result because the separation resulted in a Ln3+ fraction free of ligand. The neutralization of the slightly acidic fractions with NaOH yields a Ln3+ fraction with NaCl only, which is the desired medium for targeting molecules in nuclear medicine. In a future work, we hope to be able to separate the complete Ln3+ series by pH gradient only, using a combination of resins with a specific Ln3+ cation used as the template. The use of Ln3+ cations as templates will increase the specificity of the resin due to the imprinting of well-defined [Ln3+(L1)3] cavity size and due to the difference in Ln-L1 bond strength. For instance, it is possible that the heaviest Ln3+, Lu, will tolerate only two L1 ligands in its complexing cavity due to the small ionic radius of this cation, as already observed by others for the sterically hindered 2,6-bis(1-methylbenzimidazol-2-yl)pyridine ligand [21].
Conclusions
In this study, we present the use of a resin based on the ion-imprinted concept in the analysis of 90Sr in environmental and human samples. To our knowledge, it is the first example of a resin synthesized on a large scale for use in automated SPE as a (radio)analytical technique, using the ion-imprinted method. The resin can be produced in quantities of tens of grams using basic lab material and does not necessitate any sophisticated synthesis steps. The major advantage of using dipicolinic acid as the starting product, aside from its low price, is the versatility of the carboxylic function that can be readily activated, e.g. as acyl, to react with numerous nucleophiles. Here we chose an amide functionalization because of its high stability in the acidic condition necessary for the efficient removal of the template cation. In addition, the complexing ability and versatility of the dipicolinic acid framework has been demonstrated previously for Ln3+ and lanthanoïds [22]. Thus, HL1 ligand forms complexes such as [Ln3+(L1)3] with high stability constant (log β3 > 19) [13]. The formation of a Y(L1)3 complex in-situ during the polymerization step also increases the selectivity of the resin because it creates three covalent bonds between the imprinted cavity and the resin styrene skeleton. These three covalent bonds produce the necessary rigidity to the cavity size to adapt cations with a corresponding ionic radius. Most metal cations, especially from the transition metal series (d orbitals), form ionic bonds in a given specific geometry with ligands. For instance, Fe3+ favors an orthogonal geometry similar to a bipyramid with a square base with donor oxygen atoms. In contrast, Ni2+ favors a planar geometry or square base pyramid with donor nitrogen atom. This means it is possible to imprint complexes with a specific geometry and interaction to enhance the selectivity of the SPE resin. Thus, our lab continues to work forward to produce ion-imprinted resins with covalently bound ligands offering donor atoms creating an ionic bond with Fe3+ or Ni2+ in an orthogonal or square planar geometry. These resins will be used for the SPE of 55Fe and 63Ni, two radionuclides of relevant importance in the dismantling of nuclear power plants.
References
Froidevaux P, Bochud F, Haldimann M (2010) Retention half times in the skeleton of plutonium and 90Sr from above-ground nuclear tests: a retrospective study of the Swiss population. Chemosphere 80(5):519–524. doi:https://doi.org/10.1016/j.chemosphere.2010.04.049
Froidevaux P, Geering JJ, Valley JF (2006) Sr-90 in deciduous teeth from 1950 to 2002: the Swiss experience. Sci Total Environ 367(2–3):596–605. https://doi.org/10.1016/j.scitotenv.2006.02.011
Furukawa M, Takagi K, Matsunami H, Komatsuzaki Y, Kawakami T, Shinano T, Takagai Y (2019) Rapid quantification of radioactive strontium-90 in fresh foods via online solid-phase extraction-inductively coupled plasma-dynamic reaction cell-mass spectrometry and its comparative evaluation with conventional radiometry. ACS Omega 4(6):11276–11284. https://doi.org/10.1021/acsomega.9b01381
Ito C, Shimode R, Miyazaki T, Wakaki S, Suzuki K, Takagai Y (2020) Isotope dilution-total evaporation-thermal ionization mass spectrometric direct determination of radioactive strontium-90 in microdrop samples. Anal Chem 92(24):16058–16065. https://doi.org/10.1021/acs.analchem.0c03673
Satou Y, Sueki K, Sasa K, Matsunaka T, Takahashi T, Shibayama N, Izumi D, Kinoshita N, Matsuzaki H (2015) Technological developments for strontium-90 determination using AMS. Nucl Instrum Methods B 361:233–236. https://doi.org/10.1016/j.nimb.2015.04.032
Tomita J, Takeuchi E (2019) Rapid analytical method of Sr-90 in urine sample: rapid separation of Sr by phosphate co-precipitation and extraction chromatography, followed by determination by triple quadrupole inductively coupled plasma mass spectrometry (ICP-MS/MS). App Radiat Isotopes 150:103–109. https://doi.org/10.1016/j.apradiso.2019.05.026
Kavasi N, Sahoo SK (2019) Method for Sr-90 analysis in environmental samples using thermal ionization mass spectrometry with daly ion-counting system. Anal Chem 91(4):2964–2969. https://doi.org/10.1021/acs.analchem.8b05184
Horwitz EP, Chiarizia R, Dietz ML (1992) A novel strontium-selective extraction chromatographic resin. Solvent Extr Ion Exch 10(2):313–336. https://doi.org/10.1080/07366299208918107
Piraner O, Jones RL (2021) The effect of Sr resin cartridge age on stable Sr recovery methods used in Sr-90 analysis. J Radioanal Nucl Chem 328(1):369–375. https://doi.org/10.1007/s10967-021-07628-9
Piraner O, Jones RL (2021) Urine strontium-90 (Sr-90) manual and automated pre–analytical separation followed by liquid scintillation counting. J Radioanal Nucl Chem. https://doi.org/10.1007/s10967-021-07759-z
Wulff G (1995) Molecular inprinting in cross-linked materials with the aid of molecular templates-a way towards artificial antibodies. Angew Chem Int Ed 34(17):1812–1832. https://doi.org/10.1002/anie.199518121
Biju VM, Gladis JM, Rao TP (2003) Ion imprinted polymer particles: synthesis, characterization and dysprosium ion uptake properties suitable for analytical applications. Anal Chim Acta 478(1):43–51. https://doi.org/10.1016/s0003-2670(02)01416-2
Chauvin AS, Bunzli JCG, Bochud F, Scopelliti R, Froidevaux P (2006) Use of dipicolinate-based complexes for producing ion-imprinted polystyrene resins for the extraction of yttrium-90 and heavy lanthanide cations. Chem Eur J 12(26):6852–6864. https://doi.org/10.1002/chem.200501370
Huang K, Li BB, Zhou F, Mei SR, Zhou YK, Jing T (2016) Selective solid-phase extraction of lead ions in water samples using three-dimensional ion-imprinted polymers. Anal Chem 88(13):6820–6826. https://doi.org/10.1021/acs.analchem.6b01291
Monier M, Youssef I, El-Mekabaty (2019) A preparation of functionalized ion-imprinted phenolic polymer for efficient removal of copper ions. Polym Int 69:31–40. https://doi.org/10.1002/pi.5915
Shirazifard P, Sadjadi S, Ahmadi SJ, Faghihi F (2016) Selective solid-phase extraction of trace Pb(II) from aqueous solution using Pb(II)-ion imprinted polymer. Sep Sci Technol 51(2):248–254. https://doi.org/10.1080/01496395.2015.1085882
Devi P, Barry SM, Houlihan KM, Murphy MJ, Turner P, Jensen P, Rutledge PJ (2015) Synthesis and structural characterisation of amides from picolinic acid and pyridine-2,6-dicarboxylic acid. Sci Rep. https://doi.org/10.1038/srep09950
Council Directive 2013/51/Euratom of 22 October 2013 laying down requirements for the protection of the health of the general public with regard to radioactive substances in water intended for human consumption (2013). https://eur-lex.europa.eu/eli/dir/2013/51/oj
PROCORAD, Association for the promotion of the quality in radiotoxicologic analyses. http://www.procorad.org/en
BfS (Bundesamt fur Strahlenschutz) Ringversuche (2015) und 2017. https://www.bfs.de
Piguet C, Williams AF, Bernardinelli G, Bunzli JCG (1993) Structural and photophysical properties of lanthanide complexes with planar aromatic tridentate nitrogen ligands as luminescent building-blocks for triple-helical structures. Inorg Chem 32(19):4139–4149. https://doi.org/10.1021/ic00071a029
Brayshaw PA, Bunzli JCG, Froidevaux P, Harrowfield JM, Kim Y, Sobolev AN (1995) Synthetic, structural, and spectroscopic studies on solids containing tris(dipicolinato) reare-earth anions and transition or main-group metal-cations. Inorg Chem 34(8):2068–2076. https://doi.org/10.1021/ic00112a019
Acknowledgements
This work was supported by the Swiss Federal Office of Public Health (contract n_ 14.000461/434.003/-3). L. Pfefferlé is acknowledged for her help in the ligand L1 and resin synthesis. Florence Barraud is acknowledged for her help in the samples analysis.
Funding
Open Access funding provided by Université de Lausanne. The authors have no relevant financial or non-financial interests to disclose.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Froidevaux, P., Pittet, PA., Bühlmann, D. et al. Ion-imprinted resin for use in an automated solid phase extraction system for determining 90Sr in environmental and human samples. J Radioanal Nucl Chem 330, 797–804 (2021). https://doi.org/10.1007/s10967-021-07974-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-021-07974-8