
Abstract
The future is the recovery of the filler and its reuse in hybrid materials. It is important that as a result of recycling, the filler does not lose its properties, but acquires new ones. The aim of this research work was to investigate the effect of filler recovered by pyrolysis on the flammability of poly(ethylene terephthalate) and recycled PET. It was important to obtain a flammability class higher or equal to the pure PET and RPET matrix. Flammability tests carried out using the UL94, LOI, and PCFC methods allowed perform a first characterization of the properties of materials during their combustion. These studies show that it becomes possible to give specific functional properties to recycled fillers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Poly(ethylene terephthalate) (PET) still plays an important part in many areas, including textiles, packaging, carpeting, and single-use beverage bottles [1, 2]. However, the flammability and the melting drip behavior restrict PET's further applications even in construction [3, 4]. Separating drops of boiling or, what is worse, burning PET alloy can ignite other materials in the room and make the evacuation route difficult for people inside. This should prevent products such as ceiling and wall structural elements from being a source of further ignition points. Therefore, enhancing the flame-retardant properties of processed PET, PET composites, and recycled PET is becoming a crucial problem to solve in recent years [5,6,7]. Importantly, the essence of the problem is to ensure the improvement of physicochemical properties and flammability of PET from recycled bottles. This opens up new ways of waste management and the re-implementation of products made entirely of recycled PET [8,9,10].
The most commonly used methods of poly(ethylene terephthalate) recovery are chemical, thermal, and mechanical degradation. Poly(ethylene terephthalate) is most often recovered in two ways. By chemical method (raw material recycling) using chemical degradation called depolymerization and physical recovery (material recycling). Recovery by depolymerization is only economical for very large amounts of waste. On the other hand, physical recycling is convenient for economic and environmental purposes [11].
Processes such as methanolysis, glycolysis, hydrolysis, ammonolysis, and aminolysis have practical applications for chemical degradation. Of all these methods, methanolysis and glycolysis are the most commonly used on an industrial scale [12, 13].
The above-mentioned methods rely on complete chemolysis, which leads to the formation of polycondensate monomers. Processes are also carried out to obtain oligomers (OET) with shorter chains, most often as a result of partial glycolysis. The obtained oligomers are used to obtain various resins, e.g. polyester, they can also be an additive to elastomers and resins to improve their selected properties. Additionally, they can be used as plasticizers for some plastics, e.g. PVC. Products such as ethylene glycol (EG), dimethyl terephthalate (DMT), di(hydroxyethyl) terephthalate (BHET), and terephthalic acid (TPA) can be successfully used for the production of poly(ethylene terephthalate) [14].
The Lerg company in Poland conducts glycolysis of PET milling from bottles, and the obtained glycolysate is used for the production of polyurethanes, polyurethane foams, and unsaturated polyester resins. Polyester resins obtained with the participation of PET glycolysate acquire new features that are not present in resins without glycolysate. This applies primarily to the improvement of mechanical properties. Thanks to this extension, it is possible to use this type of resin for, for example, the production of polymer concretes [15].
Films, fibers, and most often PET bottles are subject to mechanical recycling due to their mass production, which exceeds the possibility of selective collection and the relatively short time of use by consumers. In the first stage of recycling, post-consumer PET bottles are ground into flakes. Then, water washing of post-consumer PET is used to remove surface contamination and wash off dirt, labels, and glue. Sodium hydroxide with a concentration of 2–3% and detergents are usually used as washing additives. Sodium hydroxide is a substance chemically aggressive towards PET, which helps to clean the surface of PET products. However, the temperatures used and the concentration of sodium hydroxide are not high enough to depolymerize the polyester. The cleaning efficiency of washing processes depends on the residence time of PET flakes in the washing process, water temperature, or washing additives. Ordinary washing processes are able to remove contaminants from the surface of polymers, but they are not able to remove organic substances that have been adsorbed by the polymer. Post-consumer compounds such as flavorings from soft drinks (e.g. limonene) are still detectable in conventionally recycled PET materials. Therefore, the purity of washed PET flakes is usually not suitable for the application of recyclates for direct contact with food [16].
In most cases, the washing step is combined with a separation step where materials other than PET, such as polyolefins, are removed from the mainstream due to the difference in density between the polyolefins and PET. In addition, metal detectors, triboelectric dividers, or devices equipped with infrared detectors are used in the technological lines, which enable the purification of PET from poly(vinyl chloride), which may cause the degradation of poly(ethylene terephthalate). The flakes obtained in this way are directed to the extrusion process using screw extruders equipped with a head for forming thin strands. The obtained strands are cooled in cooling tubs and then directed to the granulator to obtain PET regranulate [16, 17].
The problem that significantly hinders the recycling of PET by the mechanical method is the susceptibility of the polymer to hydrolysis, which breaks the chains of macromolecules, which in turn leads to a decrease in molecular weight, and thus deterioration of mechanical properties. Apart from water, PVC and NaOH are also precursors of hydrolysis [5, 18]. In the processing process, i.e. around 260—280 °C, a hydrolysis reaction occurs between water and PET, which results in the shortening of the polymer chains, which end with hydroxyl and carboxyl groups. In addition, the high temperature used in PET processing breaks the ester bonds in the polymer chains, which also results in shortening the length of macromolecules and the formation of chains with carboxylic and vinyl-ester end groups. Studies on the degradation of PET have shown that increasing the content of carboxyl groups in the polymer reduces the stability of PET (these groups are a catalyst for further decomposition), leading to significant degradation. The occurrence of these reactions has a significant impact on the material's processability and mechanical strength, which contributes to the deterioration of the properties and quality of products obtained from such a material [19].
Despite the described problematic issues related to the recycling of PET material, such methods and recovery parameters have been developed that successfully use waste poly(ethylene terephthalate) on the Polish and global markets. An example of application is to obtain elastic composite materials obtained in the process of pressing recyclate in the form of flakes that have been wetted with a polyurethane elastomer. The obtained material has the ability to dampen or dissipate energy and is also characterized by the ability to elastic deformation, which results in the disappearance of thermomechanical stresses in the product. Such boards are waterproof and water-permeable. Produced under the name IZOPET—R and are used for drainage of sports fields and gardens, and for drainage of foundations. Thanks to good thermal insulation properties, this material is ideal for insulating screeds, e.g. floors in industrial halls [19,20,21,22].
During the last decade, many types of flame retardants, and methods its deposition have been developed [23,24,25]. Different types of nanofillers such as clays and silicas have been incorporated into a polyester matrix. The effect of achieved flame-retardancy usually depends on the type and the contents of the powder particles used most of which would decrease the mechanical properties and vice versa [3, 26,27,28].
So far, no one has tried to use aluminosilicate filler recovered by pyrolysis from other types of nanocomposites. This article presents the effect of pyrolyzed aluminosilicate on the flammability of PET and recycled PET (RPET) composites. These studies show that it becomes possible to give a second life and specific functional properties to powder fillers subjected to appropriate physical modification.
Materials
PET (Tex Pet® CP) was purchased from TK Chemical Corp. (Seoul, Korea).
Recycled PET (RPET) was obtained by collecting and crushing into flakes of PET beverage bottles. The bottles were fed into a plastic cutter (Rapid-2b model, SRS Elektrykow, Poland) for grinding into flakes of approx. 3–5 mm. In the next step, the obtained flakes were washed in an aqueous solution of triethylenetetramine (Emulamine L60, Poland). This agent was used to remove dried beverage residues and glue, and dissolve any paper residues present. For even more thorough cleaning, in the next step, the flakes were washed in a 3% NaOH aqueous solution. The last step of purification was rinsing the obtained flakes three times in hot water at 90 °C. The thus obtained RPET flakes were allowed to dry. The flakes were dried for 7 days at 50 °C.
The pyrolysis–filler recovery process has been described in detail in references [29]. Briefly, the obtained polymer/clay nanocomposites were pyrolyzed in an in–house designed pyrolysis unit [30] with a horizontal tubular reactor. After purging with nitrogen, the reactor was heated up to 450 °C, and pyrolysis was carried out for 60 min, then the temperature was raised to 500 °C, and carbonization was carried out for 30 min. The obtained pyrolyzed filler (PF) was put in a mortar and then minced.
Samples preparation
In the preparation process (Fig. 1), a processing line consisting of a Brabender DR20 feeder (RHL-Service, Poznań, Poland), twin screw extruder Haake Rheomex OS PTW 16/25 (RHL-Service, Poznań, Poland), cooling bath Zamak W1500 (Zamak Mercator, Skawina, Poland) and pelletizer Zamak G–16/325 (Zamak Mercator, Skawina, Poland) was used. Using this processing line, PET/PF and RPET/PF composites containing 1, 3, 5, and 10 wt.% of PF, were obtained. Standard samples for LOI and UL94 testing were obtained using a P-200 press (Zamak Mercator, Skawina, Poland). The processing conditions have been shown in Table 1.

Samples preparation procedure
Methods
The FTIR tests were carried out on moldings made of composite materials, obtained using a Zamak WT12 ZMK/102/09 piston injection molding machine. A Perkin Elmer Spectrum 65 spectrometer equipped with an ATR attachment and ZnSe crystal was used for the analysis. The tests were performed using the spectral range of 4000—500 cm−1 with a number of 32 scans.
The XRD measurement was made using a D2 Phaser X-ray powder diffractometer by Bruker. The copper lamp emitted radiation of 1.54 Å (0.154 nm). Measurements were made in the range of 1—37° of the 2θ angle using a 0.1 mm gap and a 0.5 mm diaphragm. Samples of composites in the form of injected bars were tested.
The obtained composites were examined for thermal changes using differential scanning calorimetry (DSC) on a Mettler Toledo DSC823e apparatus. Samples weighing about 5 mg were placed in an aluminum dish closed in a press. The test was performed in an inert atmosphere according to the following temperature programs:
-
heating 25 - 300 °C at a rate of 10 °C/minute
-
cooling 300 - 25 °C at a rate of 10 °C/minute
-
heating 25 - 300 °C at a rate of 10 °C/minute.
The SEM analysis was carried out by a JEOL JSM-6010LA Analytical Scanning Electron Microscope (JEOL Ltd., Tokyo, Japan). The images were acquired at an accelerating voltage of 5 kV and 10 mm working distance. Before the investigation, each sample was coated with a 4 nm thick layer of gold.
Pyrolysis-combustion flow calorimetry (PCFC) was performed using the Fire Testing Technology apparatus (FTT, East Grinstead, Great Britain) according to ASTM D7309. Composite tests were performed in the temperature range of 100–750 °C at a heating rate of 1 °C/s. Before the test, crucibles were weighed, in which samples weighing about 5 mg were placed. After the measurement, the crucibles were weighed again in order to calculate the constant residue after the process.
The limiting oxygen index (LOI) test was carried out using the Fire Testing Technology Oxygen Index (FTTOI, East Grinstead, Great Britain) apparatus according to ISO 4589–2:2017.
The UL94 flammability test was carried out in the UL94 HB (horizontal burning) and UL94 V (vertical burning) systems according to IEC 60695–11-10:1999/Amd 1:2003. In both systems, the test was carried out for 5 samples.
Thermogravimetric analysis (TG) was performed on the NETZSCH TG 209F1 Libra apparatus. The test was carried out in an oxidizing atmosphere under the following conditions: temperature range from 30 to 605 °C; heating rate—10 °C/min. Each sample was tested in an open measuring cell made of Al2O3, the weight of the sample was about 5 mg.
Results and discussion
FTIR analysis
Figure 2 shows the FTIR spectra of PET and RPET compositions containing PF. In all the presented graphs, the presence of characteristic absorption bands corresponding to the vibrations of the appropriate functional groups in the composite structures was observed.

FTIR spectra for PET and RPET composites with PF
In the IR spectra of polyester materials, there are strong bands characteristic of esters, corresponding to vibrations of bonds in the ester group. The presented spectra show stretching vibrations (ν) C = O at wave number 1720 cm−1, and νC-O vibrations at wave number 1243, 1100, and 1015 cm−1 originating from ester bonds present in the PET polymer. In the structure of the polymer at the ends of the macromolecular chains, there are hydroxyl and carboxyl functional groups. The analyzed spectra at wave number 3351 cm−1 show stretching vibrations originating from the hydroxyl group -OH in PET chains and vibrations of the ν-OH group present in the carboxyl-terminal functional groups at wave number 2922 cm−1. Poly(ethylene terephthalate) macromolecules are built of aromatic rings, from which the stretching vibrations of the C = C bonds and the = C–H groups originate at wave numbers of 1425 cm−1 and 872 cm−1, respectively. Vibrations vC-H in the ring are present at wave numbers 1348 and 709 cm−1.
The spectra of the PET/PF and RPET/PF composites had the same characteristic absorption bands. It can be concluded that the recycling process did not cause changes in the chemical structure of the polyester. What is important, the use of the coagulation procedure and the ultracentrifugation operation after the filler pyrolysis did not affect on structure of polyester/pyrolyzed filler composites after high-temperature processing.
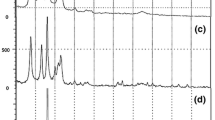
X-ray diffraction
The quantitative method for determining the degree of crystallinity by X-ray diffraction is basically a comparison of the relative amounts of energy diffracted by the crystalline and amorphous regions. The analysis of X-ray diffraction results allowed us to obtain significant information on the structure of the obtained compositions. Unreinforced poly(ethylene terephthalate) can crystallize in three crystalline forms giving signals at 2θ = 16.5; 17.3; 22.7; and 25.6°, corresponding to the crystal planes (011), (010), (011), (100) [31]. Figure 3 shows the diffractograms of PET and RPET and their composites with PF. In the range of 16—26° of the 2θ angle, reflections from the above-mentioned PET crystalline phases can be observed.

XRD patterns for PET and RPET composites with PF
Using the Bragg equation, the interplanar distances were calculated for the signal vertices observed in Fig. 3:
where: n – reflex order d – interplanar distance [nm] λ – X-ray wavelength [nm] θ – angle of reflection [°].
Calculated interplanar distances (dhkl) for angles 16.3; 17.3; 22.7; 25.8° are respectively 0.55; 0.52; 0.40; 0.35 nm.
Literature [32] states that the reflections coming from the clay occur in the vicinity of 2 – 4° of the 2θ angle. In the cited literature, the signal from the used organoclay is present at the value of 3.32° and the interplanar distance (dhkl) is equal to 2.66 nm, and the composites with pyrolyzed filler show a shift of this signal to lower values of the angle of 2.74° and the interplanar distance equal to 3.22 nm. Reinforcement of the matrix with pyrolyzed clay resulted in layer separation. The visible sharp reflections suggest obtaining a composite with an intercalated structure only.
The addition of PF to the recycled matrix resulted in the appearance of a signal at 6.9° of the 2θ angle with an interplanar distance of 1.28 nm only for the composite with 5% of pyrolyzed filler. Very high-intensity curves at about 3° were present for each of the materials and most likely originated from the radiation-emitting lamp. Based on the distance between the silicate layers, composites may show an intercalated structure, when the distances between the layers are approx. 1.5 nm, and an exfoliated structure, where the distances are approx. 8 nm [32].
In the case of the analyzed diffractograms, the introduction of PF to recycled polyester matrix mostly resulted in the formation of dual natures: intercalated and exfoliated structure. The presence of the exfoliated structure, defined by the literature as the best possible structure that can be obtained, results in an increase in the physical and mechanical properties of the nanomaterial, such as tensile strength, modulus of elasticity, or impact strength. In addition, such composites are characterized by improved barrier properties and reduced flammability [33, 34].
Differential scanning calorimetry
Figure 4 shows the DSC curves of measurements made in the first and second heating systems with a step of 10°/min for composites with a pure PET matrix. On the basis of the analysis of DSC thermograms from the cooling temperature program, the crystallization temperatures (Tc) and the change of crystallization enthalpy (ΔHc) were determined. On the other hand, from the temperature program of the second heating, the maximum corresponding to the melting temperature (Tm) and the change in the enthalpy of melting (∆Hm) were determined. These indices are summarized in Table 2.

DSC curves for the temperature program of the first and second heating of PET/PF composites
All thermograms showing the amount of heat released as a function of temperature for the heating process in the temperature range of 25 – 300 °C were characterized by the dominant phenomenon of energy absorption by the system. Throughout the entire test cycle, the samples showed a negative energy effect, as evidenced by the slope of the base curves.
Considering the DSC curves for the temperature program of the first heating, at the temperature of 70 – 80 °C, the midpoints of inflection of the curves for PET/PF composites were noted, which inform about the occurrence of glass transformation. The temperature value of the observed glass transition depends on the cooling rate and the thermomechanical history of the sample. The addition of the filler had practically no effect on shifting the transition point from the glassy state to the plastic state. In the temperature range of 95 – 145 °C, during the first heating, a characteristic exothermic peak appeared with a maximum of 126 °C, indicating the occurrence of an exothermic crystallization process called "cold crystallization". This effect is observed during the heating of the sample, which was previously cooled very quickly, and proves the presence of the amorphous structure of the polymer (∆Hcc) in the vicinity of 20 – 23 J/g). The presence of this effect can be explained by rapid changes in the physicochemical transformations of the matrix observed during processing, where at the beginning the granulate was melted and pressed at a temperature of approx. 260 °C and then the resulting stream of PET melt was rapidly cooled in a cooling bath at a temperature of 20 °C.
By analyzing the obtained curves of the first and second heating in the temperature range of 30 – 300 °C, the melting effect of individual amorphous and crystalline phases was observed. However, in the case of the first heating, the appearance of the endothermic curve originating from the glass transition and its location on the heat flux axis, as well as the intensity of the exothermic peak originating from the "cold" crystallization, suggested the presence of interpenetrating amorphous and crystalline phases, which proves the presence of a semi-crystalline structure warp. On the other hand, the analysis of all DSC curves for the second heating temperature program indicated the presence of only the PET crystalline phase [35]. In the considered cases, the peak from the more thermally stable PET crystalline phase was dominant, which proves the tendency of PET to form a stable dominant crystalline phase, regardless of the amount and type of PF used.
When considering the DSC thermograms for the second heating temperature program, it is worth noting that just before the onset point starting the melting process, in the case of PET/PF composites, the curves were slightly bent, defined as the existence of the baseline flow range. The starting point of this transformation was taken for PET at 232 °C. The shape of the maximum peak was sharp, indicating that the transformation had taken place at a constant temperature of 249 °C. The maximum peak intensity was -4.96mW for PET/10PF composite. For PET/PF composites, the transformation start temperature was assumed to be 237 °C, respectively. The slope of the curves within 230 – 260 °C was very similar. The angle of inclination of the arms of the endothermic peak in relation to the baseline allowed us to assess that the faster stage of the process was the one after reaching the maximum intensity in the melting process.
Figure 5 shows the DSC thermograms of composites on the recycled RPET matrix, also taking into account the temperature program of the first and second heating. On the presented DSC thermograms of the obtained RPET composites, for the temperature program of the second heating, in the temperature range of 70 – 80 °C, it became impossible to determine the glass transition temperature of these materials. It should be noted that an additive in the processing step above 1 wt. PF to the RPET matrix clearly indicates the formation of a semi-crystalline structure [36].

DSC curves for the temperature program of the first and second heating of RPET/PF composites
When considering the DSC thermograms for the second heating temperature program, in the temperature range of 220 – 260 °C, the melting effect was also observed, probably originating from individual amorphous structures and crystalline. The starting point of the second transformation was taken for RPET at 236 °C. The shape of the maximum peak was also sharp, indicating that the transformation occurred at a constant temperature of 249 °C. The intensity of the peak was -4.86mW. For the RPET/PF composites, the transformation start temperature was assumed to be 236 °C, respectively. The slope of the curves within 230 – 260 °C was very similar. The angle of inclination of the arms of the endothermic peak in relation to the baseline allowed us to assess that the rate of the process was the same before and after reaching the maximum melting point.
XRD vs DSC
Diffraction bands of semi-crystalline polymers are wide and consist of an amorphous phase and reflections from the crystalline phase. The distribution of the crystalline phase analyzed in this case is very similar to that described in the literature. The intensity of the observed curves can be separated into the contribution of individual crystal planes and the amorphous phase. The intensity of the observed curves can be separated into the contribution of individual crystal planes and the amorphous phase. Figure 6 shows an illustrative separation of these planes, which confirms the assumption that each of the peaks can be represented by a Gaussian curve [31].

Decomposition of the PET diffraction pattern into components derived from the amorphous and crystalline phases
Using Eq. 2, the degree of crystallinity of the PET and RPET composites was determined from the diffractograms of each material. The obtained results are presented in Table 3 in comparison with the crystallinity determined by the DSC method [37]:
where:
XC – degree of crystallinity.
AC – area of the crystalline phase curve.
Aa – area of the amorphous phase curve.
The degree of crystallinity of PET, RPET, and the corresponding composites was also determined from the DSC curves of the second heating temperature program using the formula:
where ΔHm0 is the melting enthalpy of PET with 100% crystallinity. The value of 135 [J/g] was used in the calculations [22].
From the results presented in Table 3, it can be observed that the greater the intensity of the reflections obtained, the lower the calculated crystallinity of the obtained materials. The increase in the overall intensity and surface area of the reflections obtained is most likely caused by the enlargement of the amorphous "halo" defining the content of the amorphous phase. The samples in the form of bars obtained by injection were subjected to XRD testing, where the material heated to 250 °C was introduced at high speed into a mold with a temperature of 120 °C, then pulled out and left to cool at room temperature. Fast and significant changes in the temperature of the melt and the formed material could have had an impact on the reduction of the crystallinity of the obtained composites.
According to the DSC analysis performed, there is a relationship between the presence of the cold crystallization peak and the degree of crystallinity. The higher the Tcc peak, the more amorphous phase in the polyester. According to this thesis, PET/1PF and PET/3PF samples should have more crystalline phase than PET/5PF and PET/10PF samples. Similarly, the RPET/1PF sample should contain more crystalline phase than the samples containing 3, 5 and 10 wt% PF. When analyzing the Xc results presented in Table 3, this relationship is valid only for the results from DSC measurements.
Based on the obtained XRD results, it was observed that the addition of PF to the PET matrix increased the degree of crystallinity of the PET/10PF material, which suggests that these fillers may act as crystallization nucleates. According to literature [38], the dispersion of pyrolyzed layers in a polymer matrix provides a large interfacial area and promotes heterogeneous crystallization as filler particles act as a strong nucleating agent.
However, the same effect of fillers was not observed for the PET matrix. These differences may be related to the molecular weight of the composites. It is possible that shorter RPET chains promote greater mobility of macromolecules, which facilitates their reorganization and crystal structure formation. Only the RPET samples containing 3, 5, and 10 wt% PF display a Tcc in comparison to the sample without PF because the required energy for crystallization increases as the chains become more oriented [39]. The orientation of molecular segments may increase the nucleation rate of crystallization. The homogeneous nucleating effect of PF particles also results in the reduction of the crystallization temperature of the PET and RPET composites, in comparison to the neat matrices.
An increase in the degree of crystallinity may be beneficial from the point of view of the barrier properties of composites (as well as mechanical properties) because crystallites are mostly impermeable to gases.
SEM analysis
SEM analysis was performed on PET and RPET samples containing 5 and 10 wt% PF. PET composites containing 5 and 10% PF (Figs. 7A and B) were characterized by a highly porous structure. In the photo of the surface of the PET/5PF composite, pores (~ 0.5–5 μm diameter) in which PF particles were located were observed. The dimensions of the diameter of the filler particles visible on the analyzed surface of the PET/5PF samples ranged from 0.5 to 15 μm, with those with a diameter of ~ 6 μm prevailing. In contrast, the surface of composites with twice as much PF content appeared to be less porous than the PET/5PF sample. Smaller particle sizes of the pyrolyzed filler were also observed, with dimensions ranging from 0.2 to 11 μm. However, in this case, finer structures with a diameter of ~ 0.5 μm dominated.

SEM photographs of: A PET/5PF B PET/10PF C RPET/5PF and D RPET/10PF
The surface of the RPET composites had a much more pronounced melt flow direction than the PET composites, which may confirm the observed differences in the degree of crystallinity of the polymer matrices themselves. Nevertheless, long-term SEM analysis of the surface of samples containing recycled PET does not destroy its structure, as is the case with other polyesters and biopolyesters [40,41,42]. This stability under measurement conditions confirms the advantage of recycled synthetic polyester over synthetic biopolyesters. The sample containing 5 wt% PF also had pores (~ 0.2–4 μm) in which fine PF particles with a diameter ranging from 0.05 to 3 μm were located. The doubling of the pyrolyzed filler share confirmed the trend observed for PET composites. The number of pores visible on the surface was reduced at the expense of increasing particle agglomeration to larger dimensions of ~ 6 μm. Although irregular structures were still dominant, the agglomerated larger PF particles took the form resembling rice grains.
PCFC analysis
The conducted PCFC study allowed obtaining Heat Release Rate (HRR) curves as a function of time and temperature (Fig. 8). The indicators read from the HRR curve for the PET and RPET samples clearly showed that the maximum value of the released heat (PHRR) in both cases had identical values of 457 W/g, achieved after nearly 400 s of measurement around 450 °C. This may mean that the conducted process of preparing RPET for high-temperature processing, by prior washing in a solution of detergent and lye, did not affect not only the processing of RPET but also its flammability in relation to the PET sample. This is also confirmed by the shape of the analyzed HRR curve.

HRR vs. temperature with the given HRR indices for the following composites: A PET/PF; B RPET/PF
Reference samples as well as samples of PET/PF composites burned practically at the same time. The addition of 1 and 3 wt% of PF to PET did not affect both the ignition and extinction times of the samples, as well as the values of the ignition and flame extinction temperatures. After exceeding 5 wt% PF, the ignition and extinguishing time of the composites was shifted. Thus, the PET/10PF sample ignited and extinguished 12 s faster than neat PET. Also, with the increase in the mass fraction of PF in the matrix, the PHRR values decreased. Nearly 20% reduction in flammability was obtained for a sample with 10 wt% of PF. This may be due to a decrease in the degree of crystallinity of samples containing 5 and 10% by mass of PF in relation to the reference sample, which was confirmed by DSC and XRD tests.
Different behavior was observed for RPET/PF composites. The sample containing 1 wt% burned the shortest and the longest sample with 5 wt%. In addition, the sample with RPET/1PF not only ignited the slowest but also achieved the highest PHRR among all tested composite materials. Interesting results were obtained for RPET samples containing 5 and 10 wt% PF. Both of these samples achieved a 23% flammability reduction relative to both the RPET and PET samples. However, RPET/5PF ignited and extinguished the fastest among all tested materials. This behavior is consistent with the thesis presented above. With the increase in the amount of pyrolyzed filler, the share of the amorphous phase of RPET increased, confirmed by DSC tests. The high proportion of the amorphous phase in both PET and RPET shifts the TTI and TOF points toward lower temperatures.
The presented considerations show that regardless of the matrix used, exceeding 5 wt% of the pyrolyzed filler share allowed us to obtain the best results of flammability reduction. In addition, the use of RPET as a matrix allowed for much better HRR results than when using pure PET, although all tested materials burned in the same way. Unfortunately, the use of the RPET matrix resulted in faster ignition of the composites. RPET was characterized by better susceptibility to the dynamic action of high temperatures, due to the thermal history leading to partial cracking of the polyester chains.
Thermogravimetry
Figure 9 shows the thermogravimetric curves of mass loss as a function of temperature for the tested composite samples. Among them, the curve coming from the PET/5PF sample stands out the most. In the range of 320–400 °C, this curve most protrudes towards higher temperatures in comparison with the courses of other curves.

TG curves obtain in oxygen atmosphere for the following composites: A PET/PF; B RPET/PF
Table 4 summarizes the most important indicators of thermogravimetric analysis, i.e. 5%, 10%, 20%, 50%, and maximum mass loss, as well as an additional indicator, integral decomposition temperature. The Integral Procedural Decomposition Temperature (IPDT) proposed by Doyle [22, 36] takes into account the slope as well as the shape of the TG curve. The IPDT value corresponds to the temperature at which half of the gaseous products released during the decomposition process of the polymer material evaporate. For the tested range of 30—605 °C, it was calculated on the basis of Eq. 5.
where: A*—fraction obtained by dividing the area under the TG curve by the area of the entire graph rectangle; K*—the ratio of the area under the thermogravimetric curve limited to the temperature TA by the area of the rectangle limited to this temperature; TA – the conventional temperature at which all gaseous decomposition products are released, calculated according to Eq. 4.
From the results presented in Table 4, it can be seen that the largest increase in the decomposition onset temperature was observed in the case of introducing 5% by mass of pyrolyzed filler into PET and it was 22 °C.
In the case of RPET, such a significant effect of fillers on the thermal stability of composites was not noticed. However, it should be noted that a weight loss of 5% occurs at 384 °C for an unfilled RPET matrix. This value is 18 °C higher than for the pure PET matrix and is close to the decomposition temperatures of PET/PF composites. This may be due to the absence of the cold crystallization effect, and thus the low content of the amorphous phase, which was proven by DSC tests.
In the case of samples containing PF, the shift of the decomposition temperatures towards higher values may be caused by the fine dispersion of the filler in the polymer matrix, in which the aluminosilicate particles in the presence of carbon black effectively inhibit the diffusion of oxygen to the surface of the material and into it.
Based on the XRD, DSC (1st heating) and SEM results the presence of filler increases the complex viscosity of the samples, which generates larger stresses during extrusion and, consequently, we may expect more oriented chains for the fiber containing the chain extender [38].
Qureshi et al. [43] reported that gas permeability decreases with the increase in the crystallinity degree of PET owing to the lack of free volume in the crystalline structure. However, crystallinity is not a desirable property during the processing of the PET bottle, which may explain the low Xc values for RPET samples tested at room temperature.
Impermeable PF layers block gases transport through a polymer matrix, and creating a tortuous pathway force the permeant to travel a longer diffusive path. Hence, a higher level of disorganization is favorable for barrier properties enhancement [44]. The permeability of gases through a polyester matrix depends on the available free volume of the matrix. The crystalline phase increases the barrier properties of polymers as polymer chains are efficiently packed in crystallites, which reduces the unoccupied volume of the matrix essential for gas transport.
After analyzing the data, there is no doubt that the best thermal stability of the composites on the PET matrix was characterized by a sample with 5 wt% pyrolyzed aluminosilicate content, while in the case of the RPET matrix, the composite obtained with 10 wt% PF content.
According to the course of the TG curves presented in Fig. 3A, the PET/5PF sample slightly showed the best thermal stability among the tested PET/PF composites from the beginning of the measurement. Increased thermal stability began to dominate especially after the second stage of decomposition, i.e. from temperatures above 450 °C (> T50%). This was reflected in the degradation mechanism leading to the formation of more char than in the other samples.
As shown in the SEM images, the filler particles in the PET/5PF sample had a much larger grain size distribution than in the case of the PET/10PF sample. The shape and presence of larger structures of the pyrolyzed filler also influenced the results of the TG analysis. It can be concluded that in the case of highly hygroscopic polyester matrices, the presence of pyrolyzed filler particles (different dimensions in the range of 5–15 μm) has a positive effect on the thermal stability of the composite.
Different conclusions can be drawn by looking at the TG curves (Fig. 3B) for the RPET/PF samples. In this case, a matrix was used that already had a certain thermal history, which, due to its use in the packaging sector, had a low degree of crystallinity [43]. At the same time, the low viscosity of the melt allows for a much better dispersion of particles in the polymer melt.
The TG results indicate that filling in the range of 1 to 5 wt% of PF practically does not affect the thermal stability of the RPET. This is also consistent with the XRD analysis results. Only a visible difference can be observed after increasing the share of PF to 10% by mass, and only in the second stage of char decomposition and after burning.
This means that the RPET/PF sample composed of a polyester matrix with a lower degree of crystallinity is able to achieve the level of thermal stability of the PET/5PF sample only when its mass fraction is doubled.
TG vs PCFC
Based on the TG and PCFC data, the reaction rate was calculated using the formulas given in [45,46,47], and the obtained data are presented in Fig. 10. The data obtained were used to determine the initial temperature of the decomposition and combustion process.

Comparison of the pyrolysis-combustion flow calorimetry (PCFC) and thermogravimetry (TG) results during the thermo-oxidative decomposition of the PET and RPET composites
Saha and Ghoshal [48] carried out a TG-based kinetic study with multiple heating mass loss data of waste PET obtained from different soft drink bottles. Martín-Gullón et al. [49] predicted the degradation reaction model based on two separate portions of the mass loss curve; the first one where the original PET sample thermally degraded (around 80%) quickly into char and volatiles at around 377–527 °C. In the second part, a further increase in temperature slowly decomposed (6–7%) the char. The char (residue) may be combusted to reduce the residual amount.
Buxbaum [50], established that PET follows a random scission of the ester links under thermal treatment which involves a six-centred cyclic transition state to produce vinyl ester and carboxylic acid. These primary products of pyrolysis then undergo secondary reactions to give various products like CO, CO2, acetaldehyde, aromatic acid, and their vinyl esters.
Also, Villain et al. [51] reported the results of investigations of PET degradation based on thermogravimetric analysis, on the formation of derivatives of volatile carbonyl compounds with 2,4-dinitrophenyl hydrazine and on the monitoring of formaldehyde (FA) and acetaldehyde (AA) formation. At short degradation times, AA was predominantly formed and the curve clearly showed a maximum at 280 °C, whereas only small amounts of FA were detected over the whole temperature range.
The ether links are first affected by thermal oxidation, forming hydroxyl radicals. These radicals react with the aromatic groups yielding di-hydroxyl compounds, which ultimately lead to quinone-type structures incorporated into the PET backbone that are responsible for the changes in color. The observed about 15% weight loss change can be attributed to the formation of low molar mass compounds, such as acetaldehyde [52]. The formation of acetaldehyde can occur through three different routes: a) from the hydroxyl end-group, b) from the vinyl end-group, and c) and from mid-polymer chain-scission, which generates acetaldehyde via newly formed vinyl end-groups. The thermo-oxidative degradation occurs in both, the aromatic and aliphatic parts of the polymeric chain. The formation of quinone species, for example, decreases the proton amount attributed to the aromatic rings, directly affecting the change in the PR value between aromatic and aliphatic groups [53].
Bottle-grade PET is produced using a low concentration of co-monomers to decrease the thermal crystallization rate and improve ductility, processability and clarity. Some of the more common co-monomers used are diethylene glycol (DEG), isophthalic acid (IA) and cyclohexanedimethylene glycol [54]. DEG's ether groups provide more flexibility to the relatively stiff PET backbone, thereby slowing down its thermal crystallization rate. This type of copolymerization reduces the hazing observed upon cooling of blow-moulded objects such as PET bottles [55,56,57,58]. DEG units are known to be reactive sites along the PET chain, leading to the thermal and thermo-oxidative degradation of PET [59]. Studies [60] also showed that the thermo-oxidative degradation occurs preferentially at the ether link of DEG, leading to a possible pathway to colour formation in PET.
The DEG is shown to be the precursor for the formation of chromophore groups responsible for the colour changes in the RPET, where molecular oxygen reacts mainly with the methylenic protons adjacent to the ether oxygen atoms. This behaviour is shown in the thermo-oxidative degradation process, in which the DEG degradation by-products were identified. The DEG degradation causes the release of hydroxyl radicals in the polymeric matrix, producing mono- and di-hydroxyl substituted species [51].
According to the presented results, the temperature at which the maximum decomposition rate occurs should not be exceeded. This comparison proves that despite the fact that the decomposition during the thermogravimetric measurement was two-stage for all samples, the observed maximum values of the decomposition reaction rate coincided with the values obtained for the PCFC measurement. A relationship was observed between the mass fraction of the filling and the overlapping of the maximum points of the decomposition reaction rate for both measurement methods. Thus, regardless of the matrix used, the higher the filling of the pyrolyzed filler, the better the overlap of the decomposition reaction rate curves. The intensity of the maximum points decreased only for RPET composites. Accordingly, for effective pyrolysis, it is not recommended to exceed the temperature of 435 °C for PET composites and 490 °C for RPET composites.
LOI measurements
The LOI is the basic parameter that determines the flammability of polymeric materials by determining the lowest oxygen content necessary for further self-burning of the material. The results of the measurement are presented in Table 5.
First, LOI values for PET and RPET were determined, which were 22.5 and 22.8% oxygen in the oxygen–nitrogen mixture, respectively. According to the literature [61,62,63], the oxygen index values for PET range from 20 to 25%.
PET filling already with 1 wt% of PF resulted in a decrease in the susceptibility to ignition of the composite by nearly 5%. Increasing the share of PF to 5 wt% in the matrix did not affect the achieved LOI level. Only 10 wt% of PF resulted in a break in the trend, although the LOI values were still higher than for the reference sample.
The efficiency of the pyrolyzed filler, confirmed by the presented results, may be due to the presence of char in the PF structure, which together can create an effective multi-layer thermal barrier for PET, protecting against heat transfer from the fire source to the interior of the sample. In this case, the carbon surrounded by a mineral filler performs a different task than just physically maintaining the spatial structure of the alloy, but synergistically with nitrogen and silicon compounds, it creates a barrier that is not so easily destroyed with increasing temperature.
On the other hand, RPET with a rich thermal history from the point of view of physicochemistry should be more susceptible to ignition than pure PET. The filling effect of the recovered filler is only effective up to 3 wt%, achieving an improvement in ignition resistance of a maximum of only 2% LOI. A further increase in the share of PF in RPET (5 and 10 wt%) resulted in a reverse trend and an increase in the susceptibility to ignition of these composites. This proves the limited effectiveness of the use of pyrolyzed filler in fully recycled polyester.
UL94 HB and VB tests
Regardless of the horizontal (HB) or vertical (VB) version of the UL94 test used, during the combustion of the samples, the material melted, which was accompanied by the formation of droplets, which fell down under the action of gravity, taking them a certain amount of heat energy and a portion of the material. This resulted in a decrease in the local temperature of the material surface, and thus the flammability of the remaining part of the composite.
During the combustion of vertically positioned samples (VB), uniform burning of both sides of the samples was observed. Such a location of the sample and the application of the flame from the bottom led to both sides of the molten and hot material flowing downwards along the walls of the material. The PET sample containing 10 wt% pyrolyzed filler burned more than 4 times slower than the reference sample (Table 6). On the other hand, the RPET sample with 10 wt% PF content burned more than 3 times slower than the RPET and 2 times slower than the PET. In addition, with the increase in the proportion of pyrolyzed filler in PET, the number of detached drops decreased. On the other hand, the addition of 5 and 10 wt% PF to RPET resulted in the occurrence of the phenomenon of hot composite alloy dripping. It is worth noting that none of the dripping melt ignited the cotton placed under the sample. The assessment of the flammability class made in accordance with the standard allowed to assign the V-0 class to all samples, except for the PET/5PF and PET/10PF samples, for which the V-1 class was obtained.
The measurement made in the horizontal mode indicated that when the flame was applied to the sample at an angle of 45°, the composites burned only from the bottom. As a result, smaller amounts of material were melted, while increasing the burning time (Table 7). Regardless of the additive and polymer matrix used, the length of all burnt samples did not exceed 8 mm. A similar trend in the combustion of PET and RPET with the addition of PF was observed. PET/PF composites burned slower than RPET/PF, although, in the case of the latter, it was possible to almost completely eliminate the effect of hot melt detachment already at 5 wt% of filling. All samples except RPET and RPET/1PF obtained the HB flammability class.
The influence of pyrolyzed filler on poly(ethylene terephthalate)
Dispersed layers of pyrolyzed aluminosilicate in the polyester matrix may have an impact on reducing the flammability of composites, which in the polymer matrix create a barrier for the released gaseous products of polymer decomposition, and also cause difficulties in the spread of the flame, thanks to which the mass loss and the amount of released energy are smaller. Another reason may be the reduced mobility of polymer chains located between the layers, which may inhibit the process of destruction and thermal degradation of the material [5]. In the case of a PF, the flammability reduction may be a synergistic effect of the presence of aluminosilicate and carbon black in this filler. Soot is used to modify plastics, e.g. plastics exposed to UV radiation or oxygen. Due to the very well-developed surface, it has high absorption values, which allows the deposition of various substances inside it. In addition, carbon black acts as a filler, and pigment, and is also an effective adsorber in the visible light range. Thus, it acts as a trap for hydroxyl free radicals, as well as radicals formed during the decomposition of polyesters. Additionally, the addition of carbon black to polyesters increases the value of the oxidation induction time, i.e. the time during which a sample kept at a certain constant temperature and subjected to flowing oxygen will not oxidize [64].
In addition, the pyrolyzed filler has been shown to reduce the appearance of cold crystallization even for recycled polyester. Thus, its presence affects the content of the amorphous phase. At low temperatures, the presence of carbon and silicon does not affect the phase transition points. Differences are observed only after exceeding the critical point, i.e. T50% mass loss, i.e. during the second stage of thermo-oxidative decomposition.
In the case of slow heating of the material in static tests, the decomposition mechanism is slightly different than when the heating is over 10 °C/min. The very different particle sizes of the layered filler impede the inflow and outflow of gases during heating. A linear increase in temperature causes decomposition to take place in both the aromatic and aliphatic parts of the polymer chain, forming DEG, capable of breaking down into hydroxyl radicals. As a consequence, this leads to the formation of formaldehyde and acetaldehyde trying to get out of the growing trap. And the emerging labyrinth-like barrier influences the appearance of the porous matrix structure.
The results show a different behavior when heated at rates in excess of 10 °C/min. In this case, the difference lies in the origin of the polyester matrix used. If a matrix without thermal history was used, the addition of PF made it difficult to ignite the material under standard conditions. In the case of using recycled polyester, the PF particles acted as additional heat transfer points, thus reducing susceptibility to ignition in oxygen-deficient conditions. Regardless of the origin of the matrix, its burning time does not depend on the content of the crystalline phase and thermal history. The amount of PF additive affects the rate of combustion of PET and RPET composites. With the increase in the amount of PF, the burning time was extended in two different directions. This proves that with rapid temperature increases, the barrier effect caused by PF particles is stronger the more additive in the matrix. In addition, the 5 and 10% mass increase of the pyrolyzed filler eliminates the hot melt dripping effect. These results may confirm the formation of a spatial network from char, which maintains the geometry of the samples throughout the measurement period.
Conclusions
The obtained results show that the filler recovered by pyrolysis can be successfully used as a flame retardant for polyesters, including recycled ones. The obtained polymeric materials after adding PF were characterized by different degrees of flammability reduction, depending on the type of test. The results especially show a different effect of PF on PET and RPET samples depending on the rate of heating of the composites. If the temperature of the material increased slowly, the composites tended to form a microporous structure. In the case of fast combustion of composites, a significant impact of PF particles on the formation of the barrier effect was observed, especially at higher fillings above 5% by mass. Reducing the susceptibility to ignition in standard conditions, while eliminating melt dripping, justifies the need for a time-consuming process of filling recovery by pyrolysis.
Data availability
The data are contained within the article.
References
Ferdinánd M, Várdai R, Lummerstorfer T et al (2023) Impact modification of PP with short PET fibers: Effect of heat setting on fiber characteristics and composite properties. Compos Struct 311:116810. https://doi.org/10.1016/J.COMPSTRUCT.2023.116810
Haseebuddin MR, Santhosh S, Shandilya AB (2021) Development and characterization of PET flakes reinforced polyester resin composites. Mater Today Proc 46:6075–6082. https://doi.org/10.1016/J.MATPR.2021.05.462
Mashaan N, Chegenizadeh A, Nikraz H (2022) Performance of PET and nano-silica modified stone mastic asphalt mixtures. Case Stud Constr Mater 16:e01044. https://doi.org/10.1016/J.CSCM.2022.E01044
Li J, Zhang S, Hua Y et al (2023) Facile synthesis of accordion-like porous carbon from waste PET bottles-based MIL-53(Al) and its application for high-performance Zn-ion capacitor. Green Energy Environ. https://doi.org/10.1016/J.GEE.2023.01.002
Zaikov GE, Lomakin SM (2002) Ecological issue of polymer flame retardancy. J Appl Polym Sci 86:2449–2462. https://doi.org/10.1002/APP.10946
Zhang W, Zheng C, Zhang Y, Guo W (2019) Preparation and characterization of flame-retarded poly(butylene terephthalate)/poly(ethylene terephthalate) blends: effect of content and type of flame retardant. Polymers 11:1784. https://doi.org/10.3390/POLYM11111784
Wang YH, Wang WH, Zhang Z et al (2016) Study of the glass transition temperature and the mechanical properties of PET/modified silica nanocomposite by molecular dynamics simulation. Eur Polym J 75:36–45. https://doi.org/10.1016/J.EURPOLYMJ.2015.11.038
Ragaert K, Delva L, Van Geem K (2017) Mechanical and chemical recycling of solid plastic waste. Waste Manage 69:24–58. https://doi.org/10.1016/J.WASMAN.2017.07.044
Luu TTA, Baker JR (2021) Exploring consumers’ purchase intention of rPET bottle-based apparel in an emerging economy. J Open Innov Technol Mark Complex 7:22. https://doi.org/10.3390/JOITMC7010022
Dahlbo H, Poliakova V, Mylläri V et al (2018) Recycling potential of post-consumer plastic packaging waste in Finland. Waste Manage 71:52–61. https://doi.org/10.1016/J.WASMAN.2017.10.033
Kráčalík M, Studenovský M, Mikešová J et al (2007) Recycled PET nanocomposites improved by silanization of organoclays. J Appl Polym Sci 106:926–937. https://doi.org/10.1002/APP.26690
Paszun D, Spychaj T (1997) Chemical recycling of poly(ethylene terephthalate). Ind Eng Chem Res 36:1373–1383. https://doi.org/10.1021/IE960563C
Al-Sabagh AM, Yehia FZ, Eshaq G et al (2016) Greener routes for recycling of polyethylene terephthalate. Egypt J Pet 25:53–64. https://doi.org/10.1016/J.EJPE.2015.03.001
Kijeński J, Błędzki AK (2011) Odzysk i recykling materiałów polimerowych. 316
Szostak M (2005) Recykling politereftalanu etylenu – PET. Opakowanie 50:36–41
Webb HK, Arnott J, Crawford RJ, Ivanova EP (2012) Plastic degradation and its environmental implications with special reference to poly(ethylene terephthalate). Polymers 5:1–18. https://doi.org/10.3390/POLYM5010001
Welle F (2011) Twenty years of PET bottle to bottle recycling—An overview. Resour Conserv Recycl 55:865–875. https://doi.org/10.1016/J.RESCONREC.2011.04.009
Merijs Meri R, Zicans J, Maksimovs R et al (2014) Elasticity and long-term behavior of recycled polyethylene terephthalate (rPET)/montmorillonite (MMT) composites. Compos Struct 111:453–458. https://doi.org/10.1016/J.COMPSTRUCT.2014.01.017
Awaja F, Pavel D (2005) Recycling of PET. Eur Polym J 41:1453–1477. https://doi.org/10.1016/J.EURPOLYMJ.2005.02.005
Koszkul J, Mazur P (2003) Kompozyty z recyklatu poli(tereftalanu etylenu) z krótkim włóknem szklanym. Kompozyty R 3(8):349–352
Gawdzik B, Matynia T, Maciąga-Dembińska D, Kurczak M (2000) Zastosowanie recyklatu PET do produkcji polimerobetonów. Przemysł Chemiczny T 79(6):197–198
Slonov AL, Musov IV, Zhansitov AA et al (2018) Investigation of the structure and thermal properties of composites based on polyethylene terephthalate and organoclay. Int Polym Sci Technol 43:43–47. https://doi.org/10.1177/0307174X1604300610
Köklükaya O, Carosio F, Wågberg L (2017) Superior flame-resistant cellulose nanofibril aerogels modified with hybrid layer-by-layer coatings. ACS Appl Mater Interfaces 9:29082–29092. https://doi.org/10.1021/ACSAMI.7B08018/SUPPL_FILE/AM7B08018_SI_001.PDF
Pan Y, Liu L, Song L et al (2019) Durable flame retardant treatment of polyethylene terephthalate (PET) fabric with cross-linked layer-by-layer assembled coating. Polym Degrad Stab 165:145–152. https://doi.org/10.1016/J.POLYMDEGRADSTAB.2019.05.009
Carosio F, Laufer G, Alongi J et al (2011) Layer-by-layer assembly of silica-based flame retardant thin film on PET fabric. Polym Degrad Stab 96:745–750. https://doi.org/10.1016/J.POLYMDEGRADSTAB.2011.02.019
Ni YP, Wu WS, Chen L et al (2020) How hydrogen bond interactions affect the flame retardancy and anti-dripping performances of PET. Macromol Mater Eng 305:1900661. https://doi.org/10.1002/MAME.201900661
Meng X, Qi P, Sun J et al (2020) Fabrication of transparent clay-polymer hybrid coatings on PET film to enhance flame retardancy and oxygen barrier properties. Prog Org Coat 147:105788. https://doi.org/10.1016/J.PORGCOAT.2020.105788
Sarde B, Patil Y, Dholakiya B, Pawar V (2022) Effect of calcined kaolin clay on mechanical and durability properties of pet waste-based polymer mortar composites. Constr Build Mater 318:126027. https://doi.org/10.1016/J.CONBUILDMAT.2021.126027
Majka TM, Bartyzel O, Raftopoulos KN et al (2016) Recycling of polypropylene/montmorillonite nanocomposites by pyrolysis. J Anal Appl Pyrolysis 119:1–7. https://doi.org/10.1016/J.JAAP.2016.04.005
Majka TM, Bartyzel O, Raftopoulos KN et al (2019) Examining the influence of re–used nanofiller—pyrolyzed montmorillonite, on the thermal properties of polypropylene-based engineering nanocomposites. Materials 12:2636. https://doi.org/10.3390/MA12162636
Nunes RAX, Costa VC, CaladoBranco VMDAJRT (2009) Wear, friction, and microhardness of a thermal sprayed PET: poly (ethylene terephthalate) coating. Mater Res 12:121–125. https://doi.org/10.1590/S1516-14392009000200002
Kalkar AK, Deshpande VD, Vatsaraj BS (2014) Isoconversional kinetic analysis of DSC data on nonisothermal crystallization: Estimation of Hoffman-Lauritzen parameters and thermal transitions in PET/MMT nanocomposites. Polymer (Guildf) 55:6948–6959. https://doi.org/10.1016/J.POLYMER.2014.10.068
Okamoto M (2013) Recent advances in polymer/layered silicate nanocomposites: an overview from science to technology. Mater Sci Technol 22:756–779. https://doi.org/10.1179/174328406X101319
Pavlidou S, Papaspyrides CD (2008) A review on polymer–layered silicate nanocomposites. Prog Polym Sci 33:1119–1198. https://doi.org/10.1016/J.PROGPOLYMSCI.2008.07.008
Richardson JH, Peterson RV (1978) Systematic materials analysis. Volume IV. Academic Press
Demirel B, Yaraș A, Elçiçek H (2011) Crystallization Behavior of PET Materials. BAÜ Fen Bil Enst Dergisi Cilt 13:26–35
Shabafrooz V, Allahkarami M, Bandla S, Hanan J (2016) Measuring thermal crystallinity in PET
Ghanbari A, Heuzey MC, Carreau PJ, Ton-That MT (2013) A novel approach to control thermal degradation of PET/organoclay nanocomposites and improve clay exfoliation. Polymer (Guildf) 54:1361–1369. https://doi.org/10.1016/J.POLYMER.2012.12.066
Matthews RG, Ajji A, Dumoulin MM, Prud’Homme RE (2000) The effects of stress relaxation on the structure and orientation of tensile drawn poly(ethylene terephthalate). Polymer (Guildf) 41:7139–7145. https://doi.org/10.1016/S0032-3861(00)00052-5
Komisarz K, Majka TM, Kurczab M, Pielichowski K (2022) Synthesis and Characterization of Thermally Stable Lignosulfonamides. Molecules 27:7231. https://doi.org/10.3390/MOLECULES27217231
Komisarz K, Majka TM, Pielichowski K (2022) Chemical transformation of lignosulfonates to lignosulfonamides with improved thermal characteristics. Fibers. https://doi.org/10.3390/FIB10020020
Komisarz K, Majka TM, Pielichowski K (2022) Chemical and physical modification of lignin for green polymeric composite materials. Materials 16:16. https://doi.org/10.3390/MA16010016
Qureshi N, Stepanov EV, Schiraldi D, Hiltner A, Baer E (2000) Oxygen‐barrier properties of oriented and heat‐set poly(ethylene terephthalate). J Polym Sci B Polym Phys. Wiley Online Library. https://onlinelibrary.wiley.com/doi/abs/10.1002/1099-0488%2820000701%2938%3A13%3C1679%3A%3AAID-POLB10%3E3.0.CO%3B2-P. Accessed 3 Aug 2023
Bhattacharya SN (2008) Polymeric nanocomposites : theory and practice /. Carl Hanser Publishers, Munich
Berkowicz G, Majka TM, Żukowski W (2020) The pyrolysis and combustion of polyoxymethylene in a fluidised bed with the possibility of incorporating CO2. Energy Convers Manag 214:112888. https://doi.org/10.1016/J.ENCONMAN.2020.112888
Żukowski W, Berkowicz G, Majka TM (2020) Dataset on flue gas composition during pyrolysis of polyoxymethylene in a fluidised bed with the possibility of incorporating CO2. Data Brief 31:105703. https://doi.org/10.1016/J.DIB.2020.105703
Majka TM, Berkowicz-Płatek G, Zukowski W et al (2021) Modeling of the kinetics of polyoxymethylene decomposition under oxidative and non-oxidative conditions. Materials 2281(14):2281. https://doi.org/10.3390/MA14092281
Saha B, Ghoshal AK (2005) Thermal degradation kinetics of poly(ethylene terephthalate) from waste soft drinks bottles. Chem Eng J 111:39–43. https://doi.org/10.1016/J.CEJ.2005.04.018
Martín-Gullón I, Esperanza M, Font R (2001) Kinetic model for the pyrolysis and combustion of poly-(ethylene terephthalate) (PET). J Anal Appl Pyrolysis 58–59:635–650. https://doi.org/10.1016/S0165-2370(00)00141-8
Buxbaum LH (1968) The degradation of poly(ethylene terephthalate). Angew Chem 7:182–190. https://doi.org/10.1002/ANIE.196801821
Villain F, Coudane J, Vert M (1994) Thermal degradation of poly(ethylene terephthalate) and the estimation of volatile degradation products. Polym Degrad Stab 43:431–440. https://doi.org/10.1016/0141-3910(94)90016-7
Samperi F, Puglisi C, Alicata R, Montaudo G (2004) Thermal degradation of poly(ethylene terephthalate) at the processing temperature. Polym Degrad Stab 83:3–10. https://doi.org/10.1016/S0141-3910(03)00166-6
Edge M, Wiles R, Allen NS et al (1996) Characterisation of the species responsible for yellowing in melt degraded aromatic polyesters—I: Yellowing of poly(ethylene terephthalate). Polym Degrad Stab 53:141–151. https://doi.org/10.1016/0141-3910(96)00081-X
Allen NS (2010) Photochemistry and photophysics of polymer materials. https://doi.org/10.1002/9780470594179
Lecomte HA, Liggat JJ (2006) Degradation mechanism of diethylene glycol units in a terephthalate polymer. Polym Degrad Stab 91:681–689. https://doi.org/10.1016/J.POLYMDEGRADSTAB.2005.05.028
Turner SR (2004) Development of amorphous copolyesters based on 1,4-cyclohexanedimethanol. J Polym Sci A Polym Chem 42:5847–5852. https://doi.org/10.1002/POLA.20460
Fakirov S, Seganov I, Kurdowa E (1981) Effect of chain composition of poly(ethylene terephthalate) structure and properties. Die Makromolekulare Chemie 182:185–197. https://doi.org/10.1002/MACP.1981.021820120
Hovenkamp SG, Munting JP (1970) Formation of diethylene glycol as a side reaction during production of polyethylene terephthalate. J Polym Sci A 1(8):679–682. https://doi.org/10.1002/POL.1970.150080310
MacDonald WA (2002) New advances in poly(ethylene terephthalate) polymerization and degradation. Polym Int 51:923–930. https://doi.org/10.1002/PI.917
Ciolacu CFL, Roy Choudhury N, Dutta NK (2006) Colour formation in poly(ethylene terephthalate) during melt processing. Polym Degrad Stab 91:875–885. https://doi.org/10.1016/J.POLYMDEGRADSTAB.2005.06.021
Deans T, Schiraldi DA (2014) Flammability of polyesters. Polymer (Guildf) 55:2825–2830. https://doi.org/10.1016/J.POLYMER.2014.04.050
Tao Y, Liu C, Li P et al (2021) A flame-retardant PET fabric coating: Flammability, anti-dripping properties, and flame-retardant mechanism. Prog Org Coat 150:105971. https://doi.org/10.1016/J.PORGCOAT.2020.105971
Zhang C, Zhang C, Hu J et al (2021) Flame-retardant and anti-dripping coating for PET fabric with hydroxyl-containing cyclic phosphoramide. Polym Degrad Stab 192:109699. https://doi.org/10.1016/J.POLYMDEGRADSTAB.2021.109699
Hayashi S, Naitoh A, Machida S, Okazaki M, Maruyama K, Tsubokawa N (1998) Grafting of polymers onto a carbon black surface by the trapping of polymer radicals. Appl Organomet Chem. Wiley Online Library. https://onlinelibrary.wiley.com/doi/abs/10.1002/(SICI)1099-0739(199810/11)12:10/11%3C743::AID-AOC781%3E3.0.CO;2-N. Accessed 23 Feb 2023
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Confict of interests
The author declares no conflict of interest. The author has no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
Cite this article
Majka, T.M. Flammability analysis of poly(ethylene terephthalate) and recycled PET with pyrolyzed filler. J Polym Res 30, 357 (2023). https://doi.org/10.1007/s10965-023-03737-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-023-03737-z