
Abstract
In this report, the interaction of monoamide/diamide and monoamide/diglycolamide mixtures with \({\text{UO}}_{2}^{2 + }\) are investigated in pH = 1 methanolic nitric acid media. These monoamides include N,N-dimethylacetamide (DMAA), N,N-diethylacetamide (DEAA), N,N-dibutylacetamide (DBAA) and N,N-dibutylbutanamide (DBBA). N,N,N′N′-tetraethylmalonamide (TEMA) and N,N,N′,N′-tetraethyldiglycolamide (TEDGA), which were chosen as model diamides and diglycolamides, respectively. Complex stability constants for each ligand were modelled using the Stability Quotients Using Absorbance Data program using UV–visible data. Complex stoichiometry of ligand mixtures was determined using Job plots and UV–Vis spectrometry. Monoamides were confirmed to produce only disolvate complexes with \({\text{UO}}_{2}^{2 + }\) in solution. The log10(K) values for monoamides were found to be independent of amine-side chain length, but were slightly dependent on the carbonyl-side chain length. TEDGA was found to produce multiple uranyl complexes in solution. Job plot data indicated that the uranyl cation strongly prefers to bond either only with the monoamide or diamide in ternary monoamide–diamide–UO2 systems. Monoamide–diglycolamide–UO2 systems were more complicated, with Job plot data indicating the potential for multiple ternary species being present is dependent on the monoamide structure.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
A significant limitation in the development of advanced uranium nuclear fuel cycles is the efficient and clean treatment of irradiated nuclear fuel (INF) [1]. INF becomes unusable in conventional nuclear power reactors due to the accumulation of neutron-absorbing nuclei [2]. Reprocessing this waste would not only allow for MOX fuel production from recovered U and Pu, but it would also substantially reduce the volume and heat load of INF waste to be stored in a geological disposal facility (GDF) [3], as well as the time required to store the waste. This would ease GDF site selection and simplify construction.
The current industry standard process for reprocessing INF is the Plutonium Uranium Reduction EXtraction (PUREX) process [4]. Generally, this process exploits the selectivity of tri-N-butyl phosphate (TBP) for tetra- and hexavalent cations to separate U(VI) and Pu(IV) from INF liquors, followed by Pu reduction to separate U from Pu. However, the use of TBP suffers many disadvantages [5]. These include undesired cation and anion extraction [6,7,8,9], and the formation of third phases, stabilized by TBP degradation products that retain radionuclides [10, 11]. The incineration of the degraded solvent at end-of-life results in the formation of an active phosphate residue [12, 13]. These disadvantages result in the need for extra process steps to refine the U and Pu product streams, and in the formation of radioactive phosphate secondary wastes that are difficult and expensive to process.
These issues could be eliminated, or somewhat mitigated, through the exclusive use of CHON extractants; ligands that only incorporate carbon, hydrogen, oxygen and nitrogen in their molecular structure. Such ligands are completely incinerable, thereby any active residue should be readily incorporated in the high level liquid waste raffinate from PUREX. Linear N,N-dialkylamides have long been studied as potential TBP replacements within PUREX chemistry since the work of Siddall [14]. Their degradation products are relatively innocuous to the solution chemistry (mainly carboxylic acids and secondary amines [15]) which, if small enough, may even leave the reprocessing flowsheet in the aqueous raffinate rather than remain in the organic phase to be removed. After many studies, N,N-dihexyloctanamide (DHOA) has been presented as one of the best potential TBP replacements [16,17,18,19]. It boasts excellent Pu recovery; however, U recovery is less efficient than with TBP [17]. This has largely been attributed to the low solubility of the uranyl–nitrato–monoamide complex in the organic phase [20]. While researchers have focused on DHOA’s affinity for tetravalent cations in the developing THOREX process, there has currently been no work to attempt to understand synergies in DHOA to improve U(VI) affinity with other CHON ligands.
Synergism is the phenomenon whereby extraction of a metal ion with two ligands is greater than the summed extraction by the two ligands separately [21]. Classic ligand synergism is usually achieved in similar solvent extraction (SX) processes by combining neutral extractants with lipophilic Brønstead acids [22]; however, these lipophilic acids are almost always organophosphorus compounds, which negates the benefits of using CHON ligands; for example, DMDOHEMA and HDEHP in the DIAMEX-SANEX process [23], or TODGA/TEHDGA and HEH(EHP) in the ALSEP process [24]. The addition of the acidic extractant in these cases is mainly to facilitate the pH dependent separation of lanthanides and actinides. However, as the main issue identified with U–monoamide systems is poor solubility of the uranyl complexes in the organic diluent, it follows that if the U–DHOA complex is made more hydrophobic through interactions with larger amide-based species, U recovery could potentially be increased to the point whereby a CHON-based reprocessing flowsheet becomes feasible.
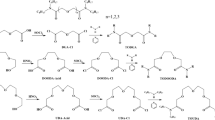
The present study is a first step towards investigating the possibility of utilising amide-based ligand mixtures to improve monoamide recovery of uranium from spent fuel liquors. Monoamide–diamide and monoamide–diglycolamide mixtures have been used to see how these ligand mixtures interact with U(VI). Ultraviolet–visible (UV–Vis) spectra have been analyzed to assess what complexes form between uranyl cations and several amide-based ligands in pseudo-aqueous media. The pseudo-aqueous media were used to aid the dissolution of some of the more hydrophobic ligands, and have also been reported to give clearer absorbance readings [25]. The effect of increasing monoamide size on these interactions has been investigated. The tested ligands, shown in Scheme 1, are N,N-dimethylacetamide (DMAA), N,N-diethylacetamide (DEAA), N,N-dibutylacetamide (DBAA) and N,N-dibutylbutanamide (DBBA) with N1,N1,N3,N3-tetraethyl malonamide (TEMA) or N1,N1,N5,N5-tetraethyl diglycolamide (TEDGA). Small ligands were used as the focus of this study in order to investigate the fundamental interactions between monoamide, diamide and diglycolamide species; as such, using larger ligands such as DHOA was deemed unnecessary. Spectrophotometric titrations of ligands were conducted to assess complex stoichiometry and stability. Job plots were used to assess the dominant complex in solutions of amide ligand mixtures.

Structure and abbreviation of the monoamides (left), diamide (middle) and diglycolamide (right) used in this study
2 Experimental Methods
2.1 Materials
All reagents were ACS reagent grade or higher purity. Nitric acid (70%, Merck), methanol (99.8%, Merck) DMAA (99.8%, Sigma–Aldrich) and DEAA (95%, Fluorochem) were used as received. Uranyl nitrate hexahydrate was supplied by the Immobilisation Science Laboratory at Sheffield University, and was dissolved in pH = 1 nitric acid to generate a concentrated mother solution of uranyl nitrate. Aliquots from this solution were used to generate uranyl nitrate working solutions of the required concentration in methanolic nitric media (50:50 vol% MeOH:deionized water). All tests were conducted at pH = 1 and the 0.2 mol·L−1 ionic strength was controlled by addition of NaNO3.
2.2 Ligand Synthesis
DBAA and DBBA were synthesized through the reaction of acetyl chloride (98%, Alfa Aesar) or butyryl chloride (99%, Acros Organics), respectively, in chloroform with equimolar dibutylamine (99.5%, Sigma–Aldrich) in the presence of equimolar triethylamine base (99%, Acros Organics) in a stirred ice bath. The solution was then heated to reflux at the boiling point of chloroform for at least 2 h. The organic product was washed with deionized water, 10 wt% Na2CO3 solution, 1.2 mol·L−1 HCl solution and a final deionized water wash. The organic layer was dried over anhydrous Na2SO4, filtered, and the solvent was removed under reduced pressure. Both ligands were > 95% pure. The DBAA yield was 76.5%, and the DBBA yield was 81.9%. DBAA 1H NMR (500 MHz, CDCl3): δ 0.68 (m, 6H), 1.07 (m, 4H), 1.27 (m, 4H), 1.82 (s, 3H), 2.98 (t, 2H), 3.05 (t, 2H). DBBA 1H NMR (500 MHz, CDCl3): δ 0.82 (m, 9H), 1.12 (m, 4H), 1.40 (m, 4H), 1.55 (m, 2H), 2.15 (t, 2H), 3.10 (t, 2H), 3.19 (t, 2H).
TEMA and TEDGA syntheses were carried out and supplied by Reading University, UK. TEMA was synthesized by the addition of 2 mol. equivalents of hydroxybenzotriazole (HOBt), triethylamine, diethylamine and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl) to a suspension of malonic acid in chloroform. TEDGA was synthesized by the addition of 2 mol. equivalents of HOBt, triethylamine, diethylamine and EDC·HCl to a suspension of 2,2′-oxydiacetic acid in chloroform. Both reactions were stirred at room temperature for 18 h. The crude reactions were washed with 1 mol·L−1 HCl and 1 mol·L−1 NaOH. The combined organic layers were separately dried over MgSO4, filtered, and the solvent was removed under reduced pressure. The TEMA yield was 86%. The TEDGA yield was 95%. TEMA 1H NMR (400 MHz, CDCl3) δ 3.46–3.37 (m, 10H), 1.20 (t, J = 6.8 Hz, 6H), 1.14 (t, J = 7.2 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 166.4, 42.6, 40.6, 40.3, 14.1, 12.8; (FTMS + pESI) calculated: C11H22N2Na [M+Na]+: 237.1573 g·mol−1; observed: 237.1578 g·mol−1. TEDGA 1H NMR (400 MHz, CDCl3) δ 4.30 (s, 4H), 3.35 (app dq, J = 27.8, 7.1 Hz, 8H), 1.16 (app dt, J = 17.6, 7.1 Hz, 12H); 13C NMR (101 MHz, CDCl3) δ 167.9, 77.5, 77.4 77.2, 76.9, 69.4, 41.0, 40.0, 14.2, 12.9; (FTMS + pESI) calculated: C12H24N2O3Na [M+Na]+: 267.1679 g·mol−1; observed: 267.1687 g·mol−1.
Ligand solutions were prepared by diluting the required mass of ligand with pre-prepared pH = 1 methanolic nitric acid at 0.2 mol·L−1 ionic strength.
2.3 Spectrophotometric Titrations and Stability Constants
Incremental additions of 0.125 mol·L−1 ligand solution were made via a burette into a stirred 0.025 mol·L−1 uranyl nitrate solution. After each addition, a small aliquot was taken and its absorption spectrum recorded in a 1 cm path length quartz cuvette. The aliquot was then reintroduced into the experiment. Volume losses were minimal. Spectra were recorded on a VWR UV-6300PC Double Beam Spectrophotometer calibrated between 190 and 1100 nm. Absorption across this entire range was recorded; however extreme absorption from the nitrate anions prevented reliable readings below ~ 330 nm. Concentrations of the initial uranium solution were confirmed against a calibration curve. It was assumed that the pH and ionic strength remained constant throughout the experiments. For each test, 24 samples were taken as this was the upper limit of sample inputs for the MODSQUAD version of the SQUAD (Stability Quotients Using Absorbance Data) program. It was found that above a ligand molar excess of ~ 4.6, dilution effects dominate the spectral changes which reduces the accuracy of SQUAD. To that end, the ligand was added in 0.2 molar steps to uranyl nitrate. All tests were carried out in triplicate at room temperature (19 ± 2 °C).
Stability constants were determined using the MODSQUAD version of the SQUAD program considering metal dilution [26]. SQUAD utilizes a nonlinear least-squares approach to calculate the best values for complex stability constants in a given model by reducing the standard deviation of the absorbance data, refined stability constant and of each spectrum through an iterative process. SQUAD convergence confirmed the validity of each tested model, but this validity was also checked through comparison of the outputted molar absorptivities and concentrations of each model component with the experimentally determined spectra. All stability constants reported in this work were calculated over the range 350–499 nm. The Gibbs energy of complex formation \(\Delta G^{\theta }\) was calculated from the determined stability constants using Eq. 1:
where R is the gas constant (8.314 J·mol−1·K−1), T is the absolute temperature, and Kxyz is the stability constant; x, y and z denote the number of metal ions, protons and ligands involved in the complex, respectively.
2.4 Job Plots
Job’s method is an established method for the determination of metal complex stoichiometry [27]. By varying the concentration of metal to ligand, or ligand to ligand, the stoichiometry of the dominant species can be determined by monitoring the absorbance of the solution. After accounting for the absorbance of the individual solution components, the remaining absorbance is due to the complexed species. When plotting this absorbance against mole fraction of ligand, a peak in this absorbance (and hence species concentration) describes the stoichiometry of the dominant species. For example, a peak at 0.5 mol fraction ligand indicates a 1:1 species; a peak at 0.67 indicates a 2:1 species, etc.
For single-ligand Job plots, separate stock solutions of uranyl nitrate, DMAA, DEAA, DBAA, DBBA, TEMA and TEDGA were generated at equal concentration. Varying volumes of uranyl nitrate and ligand solutions were mixed in separate vials to result in the required mole fraction of metal and ligand. Spectra were recorded as above. All tests were carried out in triplicate at room temperature (19 ± 2 °C).
For mixed ligand Job plots, U–ligand stock solutions were prepared such that the ligand was in 4.6 fold molar excess to uranium, mimicking the end point of the spectrophotometric titrations. Varying volumes of U–monoamide/U–diamide or U–monoamide/U–diglycolamide solutions were mixed in separate vials to result in the required mole fraction of diamide or diglycolamide, respectively. Uranium concentration was assumed to be constant throughout the experiment. Spectra were recorded as above. All tests were carried out in triplicate at room temperature (19 °C ± 2 °C).
3 Results
3.1 Uranyl Speciation in Pseudo-Aqueous Media
Figure 1 shows the molar absorptivity of uranyl nitrate in pH = 1 nitric acid in both aqueous and pseudo-aqueous media at 0.2 mol·L−1 total ionic strength. This test was to confirm that no uranyl hydrolysis has taken place when introduced into the methanolic nitric acid media. The retention of the distinct spectral “fingerprint” in the ligand-to-metal charge transfer band (LMCT, ~ 350–500 nm) indicates there is no hydrolysis of the uranyl species.

Molar absorptivity of uranyl nitrate in both pH = 1 nitric acid and pH = 1 methanolic nitric acid (50:50 vol% methanol:deionized water) at 0.2 mol·L−1 total ionic strength
3.2 Single-Ligand Complex Speciation and Stability
Figure 2 shows the results of the spectrophotometric titrations within the range 350–500 nm. This spectral region is dominated by a LMCT band [28], and as such, it is sensitive to the ligand environment in the uranyl equatorial plane. As this band is not the result of f–f electronic transitions, it is not subject to selection rules. Therefore, “silent” complexes with linear actinyl cations identified by Tian et al. [29] and Rao and Tian [30] need not be considered in the modelling process. The initial and final solutions are denoted by the thick and dashed black lines, respectively. The grey lines denote 0.4 molar steps of ligand to uranium.

Spectrophotometric titrations with incremental additions of 0.125 mol·L−1 ligand into \({\text{UO}}_{2}^{2 + }\) in pH = 1 methanolic nitric acid (50:50 vol% MeOH:deionized water) at 0.2 mol·L−1 ionic strength. The black solid and dashed lines denote the start and end points, respectively. Results for a DMAA into 0.0251 mol·L−1\({\text{UO}}_{2}^{2 + }\) up to [M]:[L] = 1:4.6, b DEAA into 0.0250 mol·L−1\({\text{UO}}_{2}^{2 + }\) up to [M]:[L] = 1:4.6, c DBAA into 0.0247 mol·L−1\({\text{UO}}_{2}^{2 + }\) up to [M]:[L] = 1:4.7, d DBBA into 0.0243 mol·L−1\({\text{UO}}_{2}^{2 + }\) up to [M]:[L] = 1:4.7, e TEMA into 0.0249 mol·L−1\({\text{UO}}_{2}^{2 + }\) up to [M]:[L] = 1:4.6, f TEDGA into 0.0254 mol·L−1\({\text{UO}}_{2}^{2 + }\) up to [M]:[L] = 1:4.5
The stability constants for the U(VI) complexes with the tested ligands calculated by SQUAD are shown in Table 1. The Gibbs energies for these complexes at 19 °C are given in Table 2. The molar absorptivities of the SQUAD-proposed complexes are shown in Fig. 3.

Molar absorptivities of the modelled uranyl complexes in pH = 1 methanolic nitric acid (50:50 vol% MeOH:deionized water) at 0.2 mol·L−1 ionic strength given by SQUAD. a DMAA, b DEAA, c DBAA, d DBBA, e TEMA, f TEDGA
Speciation of the dominant single ligand complex with uranyl nitrate and each of the tested ligands is shown in the Job plots in Fig. 4. These were conducted to confirm the validity of reported species from MODSQUAD. It is difficult to infer any kind of trend for the monoamide systems shown in Fig. 4a–d; due to the relatively small amount of measurable interaction, even small amounts of error dominate any perceivable trend in the data.

Job plots of tested ligands with uranyl nitrate in pH = 1 methanolic nitric acid (50:50 vol% MeOH:deionized water) at 0.2 mol·L−1 ionic strength. a DMAA (419 nm), b DEAA (419 nm), c DBAA (419 nm), d DBBA (419 nm), e TEMA (418 nm), f TEDGA (417 nm)
3.3 Mixed-Ligand Complex Speciation
Figure 5 shows the Job plots for the tested monoamide–TEMA systems, and Fig. 6 shows the Job plots for the tested monoamide–TEDGA systems, both at 0.2 mol·L−1 ionic strength. The ligand is in 4.6 × molar excess to uranium, so these conditions mimic the end point of the spectrophotometric titrations. Absorbance has been corrected through multiple steps; (1) subtracting the effects of the uranyl cation and both ligands on absorption, (2) zeroing at 700 nm to minimize the effects of any baseline deviation, (3) baseline corrected to ensure absorption is zero when the mole fraction of TEMA/TEDGA is 0 and 1. Step 3 is a crude but necessary step to take account of absorption change resulting from uranyl complexation with the single ligands (i.e. U–monoamide, U–diamide or U–diglycolamide species).

Job plots of the tested monoamide–diamide systems in pH = 1 methanolic nitric acid (50:50 vol% MeOH:deionized water) at 0.2 mol·L−1 ionic strength, [\({\text{UO}}_{2}^{2 + }\)] held constant at 0.013 mol·L−1 and [M]:[total L] = 4.6. a DMAA–TEMA (416 nm), b DEAA–TEMA (416 nm), c DBAA–TEMA (419 nm), d DBBA–TEMA (419 nm)

Job plots of the tested monoamide–diglycolamide systems in pH = 1 methanolic nitric (50:50 vol% MeOH:deionized water) at 0.2 mol·L−1 ionic strength, \({\text{UO}}_{2}^{2 + }\) = 0.013 mol·L−1; a DMAA–TEDGA (420 nm), b DEAA–TEDGA (420 nm), c DBAA–TEDGA (420 nm), d DBBA–TEDGA (420 nm)
4 Discussion
4.1 Uranyl Speciation
It has previously been reported that the uranyl cation exists as a hydrolyzed (UO2)2O2+ dimer species in methanolic media [31, 32]. In the case of this work, nitric acid prevents the formation of such species, as seen from the retention of the distinct spectral fingerprint in Fig. 1. As such, all models tested with SQUAD consider a single UO22+ at the center of the complex.
Due to the uranyl cation existing in solution as a linear dioxo cation [33], any bonding is restricted to the equatorial plane. It is unusual for bonding/coordination number of uranium(VI) to exceed 8 (6 + 2 from covalently bonded O) [34]. However, it has been reported that di- and tri-solvate uranyl nitrate species are produced with TODGA in acidic nitric media, potentially indicating the formation of a higher coordinated uranium species [35,36,37,38]. Therefore, assuming charge neutralization by two nitrate anions each via two oxygen donors, it follows that the formed complexes can contain no more (and probably less) than four coordination bonds from adduct-forming ligands. To this end, this was the limiting factor for models considered within SQUAD.
4.2 Single-Ligand Complex Speciation and Stability
4.2.1 UO2(NO3)2–Monoamide Species
Monoamide complexes (Fig. 2a–d) all produce very similar responses in the UV–Vis absorption spectra; a slight bathochromic shifting of the peaks, apparent loss in vibrational coupling and the production of at least one isosbestic point at longer wavelengths. As only one set of “new” peaks appear to arise, this indicates that only one complex is formed in solution. The molar absorptivity of these solutions increase slightly when dilution of the metal ion is taken into account, indicating complex formation. Increasing the size of the amine-side chains induces a larger increase in molar absorptivity. However, increasing the carbonyl-side chain appears to reduce it.
The isosbestic points observed in the monoamide spectra are good evidence that at least two species are present in solution [39], including the uranyl cation. The best fitting model indicates that only a 102 complex is produced, as shown in Table 1. It appears that even when the ligand concentration is less than the uranyl concentration, a 102 complex is strongly preferred over a potential 101 complex. These findings also correlate with many studies that use slope analysis to determine complex stoichiometry [5, 40,41,42], recent structural and spectroscopic studies of uranyl–monoamide complexes [43, 44], as well as previous spectrophotometric studies with hexavalent actinides [45, 46] when considering these systems at low acidity ([HNO3] < 4 mol·L−1).
There is no discernible trend in the UO2–monoamide stability constant as the amine-side chain increases in size. However, it is observed that increasing the carbonyl-side chain slightly increases stability. The larger carbonyl-side chain likely provides stability to the amide bond, resulting in a larger donor strength on the carbonyl oxygen. The stability constants determined by SQUAD are comparable to those determined by Prabhu et al. [42], which lends support to the SQUAD methodology for stability constant determination.
4.2.2 UO2–Diamide Species
The spectrophotometric titration results for TEMA indicate that diamide interactions with the uranyl cation are similar to those of monoamide interactions, but there are significant differences. More significant bathochromic shifting indicates the formation of more stable species; this is inferred by the species absorbing a lower energy photon. Like the monoamides, the nature of the peak shift indicates that only one complex is formed in solution. Modelling shows the most likely scenario is the formation of the 102 complex, as seen in Table 1. This is in agreement with the similar monoamide spectra above, and also with literature results [47]. The higher stability of the diamide complex comes from the bidentate nature of the ligands. It should be noted that the 101 models were also seen to be possible, but less likely.
4.2.3 UO2–Diglycolamide Species
Initially in the spectrophotometric results for TEDGA, the “fingerprint” is maintained with a large increase in molar absorptivity. As [M]:[L] increases, peak broadening and significant bathochromic shifting is observed. While this could be a sign of uranyl hydrolysis, the molar absorptivities indicate that the 102 and 104 species both have broadened spectral peaks. This could possibly be due to Jahn–Teller distortions; this implies that fitting of these ligands around the metal center distorts the bond length of the double bonded oxygens, causing band splitting which ultimately results in a peak broadening effect when the spectra are superimposed. The log10(K101) and log10(K102) values reported in Table 1 are roughly twice as large as those reported for the linear \({\text{NpO}}_{2}^{ + }\) with tetramethyl diglycolamide [30]. This difference is likely due to the larger formal charge on the uranyl cation.
The Job plot of UO2 and TEDGA definitively shows the dominant solution complex is a 101 species, which contradicts the slope analysis studies in the literature, which generally report the 102 species being dominant. As mentioned previously, 103 complexes with diglycolamides are also reported by Sasaki et al. [36] and Peng et al. [38], both determined from slope analysis. All models tested with a 103 complex resulted in large amounts of error; the best fitting model of this kind was tested for 101, 102, 103 and 104 complexes. As such, it was decided that these results likely do not suggest the presence of a 103 complex with TEDGA. It is possible that the gradient from the slope analysis was due to a mixture of diglycolamide species, such as the range of species reported in the present work, rather than a definitive 103 complex.
4.3 Mixed-Ligand Complex Speciation
These Job plots were conducted with the aim of determining the ratio of monoamide to diamide/diglycolamide in uranyl complexes in these systems. It should be noted that the diglycolamide solutions used for the Job plot tests already contain multiple uranyl species, as is evident from Fig. 7. This immediately arises a source of error when baseline correcting for the effect of the initial and final complexes in solution. To baseline correct this, a straight line was drawn from TEMA or TEDGA at mole fraction = 0 and 1.0, and this was taken to be the baseline. While the total ligand concentration remained constant throughout the test, the changing concentrations of each individual ligand may cause a change in complex speciation. This may mean taking a linear baseline lends some inaccuracy; however it will at least serve as a good approximation as to what is occurring in solution.

Speciation of the solution components and proposed U(VI) complexes with TEDGA against [M]:[L] calculated by SQUAD in pH = 1 methanolic nitric acid (50:50 vol% MeOH:deionized water) at 0.2 mol·L−1 ionic strength
4.3.1 UO2–Monoamide–Diamide Species
Due to the small absorbance changes observed, it is likely that the following discussion is not entirely representative of the solution chemistry in these systems, but it is presented here for qualitative purposes.
The low absorption change in the monoamide–diamide Job plots (Fig. 5) indicate there is very little mixed interaction between the uranyl cation and both ligands. It appears that the uranyl center strongly prefers to bond with either the monoamide or the diamide. Isosbestic points were observed in the Job plot spectra of DBAA–TEMA and DBBA–TEMA, indicating the presence of multiple UO2 species, likely the 102 complex of both the mono- and diamide. Absorption generally peaks at a TEMA mole fraction of 0.5, indicating the formation of a 1:1 monoamide:diamide complex with uranyl nitrate. This species is certainly possible from a steric perspective, however, the lack of absorption in Fig. 5 suggests that this species is in low concentration.
4.3.2 UO2(NO3)2–Monoamide–Diglycolamide Species
The monoamide–diglycolamide Job plots shown in Fig. 6 indicate this system is much more complex than the monoamide–diamide system. The relatively large increases in absorption indicate the presence of mixed ligand complexes. As the monoamide size increases, an increased absorbance change is observed which indicates an increase in complex concentration, inferring greater stability. Absorbance peaks at 0.5 mol fraction of TEDGA for the DBAA–TEDGA system, indicate the formation of a 1:1 monoamide:diglycolamide complex. However, the other three tested systems all clearly have a peak at 0.4 mol fraction of TEDGA. This could have two interpretations; the predominant complex in solution has a ratio of 3:2 monoamide:diglycolamide, or there is a mixture of 1:1 and 2:1 monoamide:diglycolamide species. From a steric perspective, it can be argued that the latter scenario is more likely. However, EXAFS data would be required to underpin quite how these ligands are arranged. It appears that the stoichiometry of the mixed ligand complex is not based on the stability of the single ligand complexes; if it were the case, it might be expected that the DMAA–TEDGA systems also solely produce 1:1 complexes, which are not seen in the data. Nevertheless, it is suggested that mixed-ligand species are present in solution, and the difference in speciation would undoubtedly result in changes in complex hydrophobicity. A key limitation in the application of monoamides to spent fuel reprocessing is the solubility of the extracted complex in the organic phase. Therefore, the next step will be to see how these systems behave in a solvent extraction setting.
5 Conclusions
There is little information in the literature associated with interactions between monoamides and amide-based ligands. This may be of use for developing more efficient, sustainable nuclear fuel waste treatment. In this study, the interactions between monoamides and diamides or diglycolamides with UO22+ were investigated in pseudo-aqueous media. The effect of changing the monoamide structure on these interactions was also investigated. As expected, monoamides were confirmed to produce 2:1 ligand:metal complexes with the uranyl cation, and diamides followed this trend. Diglycolamides were seen to produce multiple species of uranyl complexes, up to 104, leading to more complex solution chemistry than previously thought. While diamides have similar complex behavior to monoamides, Job plots suggest that the uranyl cation strongly prefers to bond exclusively with only monoamide or diamide species, regardless of the monoamide structure. Diglycolamides were shown to produce stronger complexes with UO2 than monoamides. However, Job plots suggest that multiple mixed-ligand species are produced in the UO2–monoamide–diglycolamide systems, with no clear indication on the dominant species in solution. It is likely that both 1:1 and 2:1 monoamide:diglycolamide species are produced in solution. Future work needs to assess monoamide–diglycolamide interactions with UO22+ in more detail, perhaps with EXAFS data to underpin complex geometry.
References
Taylor, R.: Reprocessing and Recycling of Spent Nuclear Fuel (Preface). Woodhead Publishing, Cambridge (2015)
Choppin, G., Liljenzin, J., Rydberg, J.: Radiochemistry and Nuclear Chemistry, Chap. 21. Butterworth–Heinemann, Woburn (2002)
Paulenova, A.: Physical and chemical properties of actinides in nuclear fuel reprocessing. In: Nash, K., Lumetta, G. (eds.) Advanced Separation Techniques for Nuclear Fuel Reprocessing and Radioactive Waste Treatment, Chap. 2. Woodhead Publishing, Cambridge (2011)
Mckay, H.A.C.: The PUREX process. In: Schulz, W.W., Burger, L.L., Navratil, J.D. (eds.) Science and Technology of Tributyl Phosphate. CRC Press, Boca Raton (1990)
Gupta, K.K., Manchanda, V.K., Subramanian, M.S., Singh, R.K.: Solvent extraction studies on U(VI), Pu(IV) and fission products using N,N-dihexyloctanamide. Solvent Extr Ion Exc 18(2), 273–292 (2000)
Alcock, K., Bedford, F.C., Hardwick, W.H., McKay, H.A.C.: Tri-n-butyl phosphate as an extracting solvent for inorganic nitrates—I. J. Inorg. Nucl. Chem. 4(2), 100–105 (1957)
Guillaume, B., Moulin, J.P., Maurice, C.: Chemical properties of neptunium applied to neptunium management in extraction cycles of PUREX process. In: Symposium on Liquid–Liquid Extraction Science. I. Chem. E, Dounreay, pp. 31–45 (1984)
Herbst, R.S., Baron, P., Nilsson, M.: Standard and advanced separation: PUREX processes for nuclear fuel reprocessing. In: Nash, K., Lumetta, G. (eds.) Advanced Separation Techniques for Nuclear Fuel Reprocessing and Radioactive Waste Treatment, pp. 141–175. Woodhead Publishing, London (2011)
Phillips, C.: Uranium–plutonium partitioning by pulsed columns in the first cycle of the three cycle thermal oxide reprocessing plant. Waste management’ 92: working towards a cleaner environment: waste processing, transportation, storage and disposal, technical programs and public education, vols. 1 and 2, pp. 1041–1045 (1992)
Sugai, H.: Alternative destruction method for emulsions stabilized by precipitates of Zr and TBP degradation products. J. Nucl. Sci. Technol. 29(1), 92–94 (1992)
Zilberman, B.Y., Fedorov, Y.S., Shmidt, O.V., Goletsky, N.D., Sukhareva, S.Y., Puzikov, E.A., Suglobov, D.N., Mashirov, L.G., Choppin, G.R.: Extraction of transplutonium and rare-earth elements, molybdenum and iron with zirconium salt of dibutyl phosphoric acid. Czech J. Phys. 53, 479–486 (2003)
Manohar, S., Srinivas, C., Vincent, T., Wattal, P.K.: Management of spent solvents by alkaline hydrolysis process. Waste Manage. 19(7–8), 509–517 (1999)
Schulz, W.W.: Macroreticular anion exchange resin cleanup of TBP solvents (No. ARH-SA-129). Atlantic Richfield Hanford Co., Washington (1972)
Siddall III, T.H.: Effects of structure of N,N-disubstituted amides on their extraction of actinide and zirconium nitrates and of nitric acid. J. Phys. Chem. 64(12), 1863–1866 (1960)
Mincher, B.J., Modolo, G., Mezyk, S.P.: Review article: the effects of radiation chemistry on solvent extraction: 1. Conditions in acidic solution and a review of TBP radiolysis. Solvent Extr. Ion Exch. 27(1), 1–25 (2009)
Manchanda, V.K., Ruikar, P.B., Sriram, S., Nagar, M.S., Pathak, P.N., Gupta, K.K., Singh, R.K., Chitnis, R.R., Dhami, P.S., Ramanujam, A.: Distribution behaviour of U(VI), Pu(IV), Am(III), and Zr(IV) with N,N-dihexyl octanamide under uranium-loading conditions. Nucl. Technol. 134(3), 231–240 (2001)
Manchanda, V.K., Pathak, P.N.: Amides and diamides as promising extractants in the back end of the nuclear fuel cycle: an overview. Sep. Purif. Technol. 35(2), 85–103 (2004)
Kumar, J.R., Kim, J.-S., Lee, J.-Y., Yoon, H.-S.: A brief review on solvent extraction of uranium from acidic solutions. Sep. Purif. Rev. 40(2), 77–125 (2011)
Pathak, P.N.: N,N-Dialkyl amides as extractants for spent fuel reprocessing: an overview. J. Radioanal. Nucl. Chem. 300(1), 7–15 (2014)
Musikas, C.: Solvent extraction for the chemical separations of the 5f elements. Inorg. Chim. Acta 140, 197–206 (1987)
Akaiwa, H., Kawamoto, H.: The application of synergistic extraction to analytical chemistry. Rev. Anal. Chem. 6(1), 65–86 (1982)
Nash, K.L., Madic, C., Mathur, J., Lacquement, J.: Actinide separation science and technology. In: Morss, L.R., Edelstein, N.M., Fuger, J. (eds.) The Chemistry of the Actinide and Transactinide Elements, 4th edn. Springer, Dordrecht (2010)
Hérès, X., Baron, P., Hill, C., Ameil, E., Martinez, I., Rivalier, P.: The separation of extractants implemented in the DIAMEX-SANEX process (2008)
Gelis, A.V., Lumetta, G.J.: Actinide lanthanide separation process—ALSEP. Ind. Eng. Chem. Res. 53(4), 1624–1631 (2014)
Rabinowitch, E., Belford, R.L.: Spectroscopy and Photochemistry of Uranyl Compounds, Chap. 2. Macmillan, New York (1964)
Leggett, D.J.: Computational Methods for the Determination of Formation Constants, Chap. 6. Plenum, New York (1985)
Gullekson, B.J., Brown, M.A., Paulenova, A., Gelis, A.V.: Speciation of select f-elements with lipophilic phosphorus acids and diglycol amides in the ALSEP backward-extraction regime. Ind. Eng. Chem. Res. 56, 12174–12183 (2017)
Libuś, Z.: Absorption spectra of uranium(VI) complexes in solution. J. Inorg. Nucl. Chem. 24(61), 9–631 (1962)
Tian, G., Xu, J., Rao, L.: Optical absorption and structure of a highly symmetrical neptunium(V) diamide complex. Angew. Chem. Int. Ed. 44, 6200–6203 (2005)
Rao, L., Tian, G.: Symmetry, optical properties and thermodynamics of neptunium(V) complexes. Symmetry 2, 1–14 (2010)
Ogden, M.D., Hoch, C.L., Sinkov, S.I., Meier, G.P., Lumetta, G.J., Nash, K.L.: Complexation studies of bidentate heterocyclic N-donor ligands with Nd(III) and Am(III). J. Solution Chem. 40(11), 1874–1888 (2011)
Ogden, M.D., Sinkov, S.I., Lumetta, G.J., Nash, K.L.: Affinity of An(VI) for N4-tetradentate donor ligands: complexation of the actinyl(VI) ions with N4-tetradentate ligands. J. Solution Chem. 41(4), 616–629 (2012)
Rabinowitch, E., Belford, R.L.: Spectroscopy and Photochemistry of Uranyl Compounds, Chap. 1. Macmillan, New York (1964)
Cotton, S.: Coordination chemistry of the actinides. In: Cotton, S. (ed.) Lanthanide and Actinide Chemistry, Chap. 11. Wiley, Chichester (2006)
Zhu, Z.-X., Sasaki, Y., Suzuki, H., Suzuki, S., Kimura, T.: Cumulative study on solvent extraction of elements by N,N,N′,N′-tetraoctyl-3-oxapentanediamide (TODGA) from nitric acid into n-dodecane. Anal. Chim. Acta 527(2), 163–168 (2004)
Sasaki, Y., Sugo, Y., Suzuki, S., Tachimori, S.: The novel extractants, diglycolamides, for the extraction of lanthanides and actinides in HNO3–n-dodecane system. Solvent Extr. Ion Exc. 19(1), 91–103 (2001)
Liu, X., Sun, G., Cai, X., Yang, X., Li, Y., Sun, Z., Cui, Y.: Extraction of U(VI) with N,N,N′,N′-tetraoctyl diglycolamide from nitric acid solution. J. Radioanal. Nucl. Chem. 306, 549–553 (2015)
Peng, X., Li, Y., Ma, J., Cui, Y., Sun, G.: Extraction and separation of uranium(VI) and Fe(III) from lanthanide chlorides with N,N,N′,N′-tetrasubstituted alkyl diglycolamide extractants. J. Radioanal. Nucl. Chem. 313, 327–332 (2017)
Smith, N.A.: Speciation and spectroscopy of the uranyl and tetravalent plutonium nitrate systems: fundamental studies and applications to used fuel reprocessing. PhD thesis, University of Nevada (2010)
Gupta, K.K., Manchanda, V.K., Subramanian, M.S., Singh, R.K.: N,N-Dihexyl hexanamide: a promising extractant for nuclear fuel reprocessing. Sep. Sci. Technol. 35(10), 1603–1617 (2000)
Pathak, P.N., Kumbhare, L.B., Manchanda, V.K.: Structural effects in N,N-dialkyl amides on their extraction behaviour toward uranium and thorium. Solvent Extr. Ion Exch. 19(1), 105–126 (2001)
Prabhu, D.R., Mahajan, G.R., Nair, G.M.: Di(2-ethyl hexyl) butyramide and di(2-ethyl hexyl)isobutyramide as extractants for uranium(VI) and plutonium(IV). J. Radioanal. Nucl. Chem. 224(1–2), 113–117 (1997)
Loubert, G., Volkringer, C., Henry, N., Arab-Chapelet, B., Delahaye, T., Loiseau, T.: Structural studies of a series of uranyl alkylacetamides and piracetam complexes obtained in nitric acid aqueous solution. Polyhedron 138, 7–12 (2017)
Verma, P.K., Mohapatra, P.K., Bhattacharyya, A., Yadav, A.K., Jha, S.N., Bhattacharyya, D.: Structural investigations on uranium(VI) and thorium(IV) complexation with TBP and DHOA: a spectroscopic study. New J. Chem. 42, 5243–5255 (2018)
Condamines, N., Musikas, C.: The extraction by N,N-dialkylamides II—Extraction of actinide cations. Solvent Extr. Ion Exch. 10(1), 69–100 (1992)
McCann, K., Sinkov, S.I., Lumetta, G.J., Shafer, J.C.: Inner versus outer sphere metal–monoamide complexation: ramifications for tetravalent and hexavalent actinide selectivity. New J. Chem. 42(7), 5415–5424 (2018)
Ruikar, P.B., Nagar, M.S.: Synthesis and characterisation of some new mono- and diamide complexes of plutonium(IV) and dioxouranium(VI) nitrates. Polyhedron 14(20–21), 3125–3132 (1995)
Acknowledgement
The authors would like to acknowledge the SNUCER group at the University of Sheffield for support through this work. They would also like to acknowledge the Harwood group at the University of Reading for ligand synthesis. Finally, the authors would like to thank EPSRC for funding this work.
Funding
This work was funded through an EPSRC Doctoral Training Grant through the Chemical and Biological Engineering Department at the University of Sheffield.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Canner, A.J., Harwood, L.M., Cowell, J. et al. Spectrophotometric Analysis of Ternary Uranyl Systems to Replace Tri-N-butyl Phosphate (TBP) in Used Fuel Reprocessing. J Solution Chem 49, 52–67 (2020). https://doi.org/10.1007/s10953-019-00942-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10953-019-00942-7