
Abstract
Ulcerative Colitis (UC) is an inflammatory bowel disease typically affecting the colon. Patients with active UC have elevated tumor necrosis factor (TNF) concentrations in serum and colonic tissue. Infliximab is a monoclonal antibody directed against TNF and binds with high affinity. Target-mediated drug disposition (TMDD) is reported for monoclonal antibodies meaning that their pharmacokinetics are affected by high target affinity. Here, a TMDD model is proposed to describe the interaction between infliximab and TNF in UC patients. Data from 20 patients with moderate to severe UC was used. Patients received standard infliximab induction therapy (5 mg kg−1) at week 0, followed by infusions at week 2 and 6. IFX, anti-drug antibodies and TNF serum concentrations were measured at day 0 (1 h after infusion), 1, 4, 7, 11, 14, 18, 21, 28 and 42. A binding model, TMDD model, and a quasi-steady state (QSS) approximation were evaluated using nonlinear mixed effects modeling (NONMEM). A two-compartment model best described the concentration–time profiles of infliximab. Typical clearance of infliximab was 0.404 L day−1 and increased with the presence of anti-drug antibodies and with lower albumin concentrations. The TMDD-QSS model best described the pharmacokinetic and pharmacodynamics data. Estimate for TNF baseline (Bmax was 19.8 pg mL−1 and the dissociation constant (Kss) was 13.6 nM. This model could eventually be used to investigate the relationship between suppression of TNF and the response to IFX therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ulcerative colitis (UC) is a chronic auto-immune disease of the colonic mucosa [1]. Patients with active UC suffer from episodes of bloody diarrhea, often accompanied with abdominal pain. Although the precise pathophysiology of UC has not been unraveled yet, tumor necrosis factor (TNF) plays an important role in mucosal inflammation.
TNF is a pro-inflammatory cytokine and has been found in increased concentrations in blood, stool, and epithelial tissue of UC patients [2,3,4,5,6]. In patients with inflammatory bowel disease (IBD) (i.e. UC or Crohn’s disease), serum TNF concentrations are significantly higher compared to healthy controls [3]. Also, higher TNF concentrations are present in inflamed tissue samples of IBD patients, compared to uninflamed tissue samples, suggesting a local higher inflammatory TNF load in inflamed epithelial tissue [6].
TNF is produced as a transmembrane cytokine (mTNF) and released in its soluble form (sTNF) after conversion by TNF converting enzyme (TACE) [7]. Infliximab (IFX) is an intravenously administered monoclonal antibody directed against TNF and has demonstrated to induce and maintain remission in patients with moderate to severe UC [8]. IFX can bind to both monomeric and trimeric (i.e. biologically active) sTNF with high affinity, thereby preventing binding of sTNF to TNF-receptors, receptor activation, and the subsequent inflammatory processes [9, 10].
The pharmacokinetics of IFX in IBD patients have been described extensively [11,12,13,14,15,16]. Notably, the presence of detectable anti-IFX antibodies and lower serum albumin concentrations are associated with an increased clearance of IFX. Clearance of IFX is increased up to fourfold in patients with detectable anti-IFX antibodies, often accompanied by undetectable IFX trough concentrations and clinical loss of response to IFX.
Monoclonal antibodies exhibit different pharmacokinetic properties compared to small molecules. Due to their high molecular weight and hydrophilicity, distribution to peripheral tissue is limited. Also, unlike small molecules, monoclonal antibodies are not cleared via the kidneys or liver, but via alternative pathways, primarily via proteolytic catabolism after receptor-mediated endocytosis in the reticuloendothelial system. Monoclonal antibodies are designed for a specific target, which they bind to with high affinity. Target-mediated drug disposition (TMDD) is described for monoclonal antibodies that bind with high affinity to their target, to such an extent that it affects the pharmacokinetics of the drug [17]. At low concentrations the monoclonal antibody is eliminated via lysosomal degradation through binding to its target, while at high concentrations, this elimination route becomes saturated and elimination occurs via a linear, non-saturable proteolytic pathway [18]. As a result, clearance is higher at low monoclonal antibody concentrations.
IFX binds with high affinity to its target sTNF, resulting in the formation of stable IFX-TNF complexes [9, 19].
Based on this mechanism of action of the monoclonal antibody IFX and measured sTNF concentrations, a TMDD model could be used to describe the interaction between IFX and TNF. This would provide more insight into IFX response of IBD patients with regard to not just IFX exposure, but also their TNF concentrations.
In this study, we aimed to quantify the binding of IFX to its biological target, TNF, by means of the development of a TMDD model in patients with moderate to severe UC.
Methods
Data and study design
Data was used from a prospective cohort study with 20 anti-TNF naive patients with moderate to severe UC, as previously reported [12]. Patients received IFX (5 mg kg−1) induction therapy at week 0, 2, and 6. One patient received an additional IFX administration at day 5. Serum IFX concentrations, anti-IFX antibody status, free TNF serum concentrations, C-reactive protein (CRP) and albumin concentrations were collected at day 0 (1 h after infusion), day 1, 4, 7, 11, 14 (before and 1 h after infusion), 18, 21, 28 and 42 (before infusion). Patient characteristics are summarized in Table 1. This study was approved by the local ethical committee and all patients signed informed consent before start of the study.
Serum measurements
Serum IFX concentrations and anti-IFX antibodies were measured with a homogenous mobility shift assay (HMSA) (Prometheus Laboratories, San Diego, CA) [20, 21]. With HMSA, anti-IFX antibodies can be measured in the presence of IFX (i.e. a drug-tolerant or drug non-sensitive assay). Lowest level of quantification (LoQ) for IFX measurements was 0.06 mg L−1 with coefficient of variation (CV) of 12%. Free serum TNF concentrations were measured using an ultrasensitive immunoassay (Singulex, Prometheus Laboratories, San Diego, CA) and LoQ for TNF measurements was 10.0 fgs mL−1, with CV of 15% [22, 23].
Pharmacokinetic/pharmacodynamic analysis
Model development
IFX and TNF concentrations were converted to nanomolar (nM) using their molecular weights of 149 kDa (IFX) and 52 kDa (TNF) [9, 24]. A population pharmacokinetic model for IFX has been developed previously, and was used as the starting point for this study [12]. For this model, the following covariates were evaluated: anti-IFX antibody status, albumin, CRP, and body weight. During forward inclusion, covariates were included when objective function value (OFV) decreased > 3.84 point (p = 0.05). For backwards elimination a more stringent p value of 0.01 was used (OFV decrease > 6.63 points). Continuous covariates were modeled according to the general equation:
where PTV is the typical value of the parameter P in a patient with median covariate value (COV) and θ is the fractional change in P with each unit of deviation from the median covariate. Categorical covariates were modeled according to the general equation:
where PTV is the typical value of the parameter P and θcov is the fractional difference in P between categories.
Several structural models were evaluated to describe the data: a binding model, a full TMDD model, and a TMDD model with quasi-steady state (QSS) approximation [25]. The structural model was selected based on OFV, precision of parameter estimates, and visual inspection of goodness-of-fit plots. Inter-individual variability (IIV) was parameterized assuming exponential models. Concentrations were log-transformed and an additive error model (i.e. proportional error with log-transformed data) was used to capture residual variability.
For the binding model, serum concentrations of TNF bound to IFX were expressed in the $ERROR block as follows:
where Bmax denotes the baseline serum TNF concentration, Ac represents the total IFX amount in serum, Vc the central volume of distribution of IFX and KD the equilibrium dissociation rate constant.
In the TMDD model, synthesis is represented by the zero-order rate constant ksyn (nM day−1) and degradation of TNF is represented by the first-order rate constant kdeg (day−1). TNF forms a complex with IFX with binding rate constant kon (nM−1 day−1) and the complex dissociates with dissociation rate constant koff (day−1), see Fig. 1 (adapted from Mager and Jusko [26]).

Schematic overview target-mediated drug disposition model (adapted from Mager and Jusko [26]). Symbols are defined in Table 2. AC amount drug central compartment, AP amount drug peripheral compartment, k12 first-order rate constant from the central to the peripheral compartment, k21 first-order rate constant from the peripheral to the central compartment, ke internalization rate drug, koff dissociation rate constant, kon binding rate constant, P complex compartment, R receptor compartment
In the full TMDD model, central IFX (Ac) and peripheral IFX (AP) amounts, expressed in nmol, are represented by Eqs. 4 and 5. Free TNF (R), and complex (P) concentration over time (t), expressed in nM, are represented by Eqs. 6 and 7:
where ke represents the first-order elimination rate constant of IFX and ke(P) represents the first-order elimination rate constant of the complex. Free IFX is distributed by first-order processes to the peripheral compartment (k12 and k21).
A QSS approximation was evaluated to improve parameter estimation in the TMDD model [25]. The main assumption of a QSS approximation is that the drug, target, and complex are in QSS, where the binding rate is balanced by the sum of dissociation (koff/kon) and internalization rates (ke(P)/kon). In contrast to a quasi-equilibrium (QE) approximation, the QSS approximation does not assume that the rate of elimination of the complex (ke(p)) is negligible. Instead, the dissociation constant (KSS) is expressed as follows:
where KD denotes the equilibrium dissociation rate constant (koff/kon).
Total central IFX concentration (Ctot), free IFX concentration (C), total central IFX amount (Atot) and the peripheral IFX amount (AP), are represented by Eqs. 9–12, and total TNF concentration (Rtot), is represented by Eq. 13:
Due to a large difference reported kdeg values over multiple orders of magnitude (0.042 day−1 to 39.6 day−1) and an inability to achieve satisfactory model fit with either of these values, a sensitivity analysis was performed to assess the most likely value at which to fix kdeg to ensure reliable and accurate estimation of all remaining parameters [27, 28]. The sensitivity analysis was performed in NONMEM by assessing precision of parameter estimates and OFV values at each value of kdeg. The fixed kdeg values ranged from 0.02 to 40.28 day−1, with a two-fold increase between each value and the next.
Model evaluation
The final model was evaluated using a visual predictive check (VPC), using 1000 simulations and the plots were stratified by compartment. Bootstrap analysis (2000 runs) was performed to test the stability and robustness of the final model parameter estimates.
Software
Pharmacokinetic/pharmacodynamic modeling was performed using nonlinear mixed effects modeling (NONMEM) (Icon, Dublin, Ireland, software version 7.4) with first-order conditional estimation with interaction (FOCE + I). Pearl-speaks-NONMEM (version 4.8.1, Uppsala, Sweden) and R (version 3.5.2, Vienna, Austria) were used to visualize and evaluate the model outcomes.
Results
Serum samples
The dataset included 214 IFX serum concentrations, and 214 TNF serum concentrations from 20 UC patients. Unbound TNF concentrations appeared inversely related to IFX serum concentrations. Directly after IFX infusion, TNF concentrations ranged from 0.27 to 2.1 pg mL−1 and increased to 3.5–31 pg mL−1 at day 42 right before the next IFX administration. Antibodies-to-infliximab were detected in 7/20 patients.
Final model
Supported by the available data and the assumption that degradation of the IFX-TNF complex is not negligible, a TMDD model with QSS approximation best described the interaction between IFX and TNF serum concentrations. The TMDD-QSS approximation allowed the estimation of the steady state equilibrium constant (Kss), the complex internalization constant (ke(P)) and of the degradation constant TNF (kdeg).
Population steady state dissociation constant (Kss) was 13.6 nM, ke(P) was estimated to be 0.984 day−1 and kdeg was fixed to 5.12 day−1 based on a sensitivity analysis (see Supplementary Table 1). Median baseline TNF value (Bmax) was estimated to be 0.38 pM, which is equal to 19.8 pg mL−1 and comparable to literature [5]. IIV was identified for CL, Vc, Vp, and Bmax. Simulated total TNF concentrations are depicted in Fig. 2.

Predicted total TNF concentrations over time. Open circles represent the predicted values for total TNF concentrations. The solid line represents the median of the predicted total TNF concentrations and the dashed lines represent the lower and upper limit of the predicted total TNF concentrations
Initial estimates for the typical pharmacokinetic parameters of IFX were derived from the previously developed pharmacokinetic model, and optimized during model development. The presence of antibodies-to-infliximab increased clearance of IFX by threefold (Fig. 3a). In addition, clearance of IFX ranged from 0.94 to 0.24 L day−1, for albumin concentrations from 23 to 51 g L−1 (Fig. 3b). The final parameters estimates are summarized in Table 2.

Infliximab clearance in relation to albumin concentration (a) and anti-drug antibodies (b)
Goodness-of-fit plots for evaluation of IFX are depicted in Supplementary Fig. 1A and goodness-of-fit plots for evaluation of TNF prediction are depicted in Supplementary Fig. 1B. Population and individual predictions were randomly distributed around the line of identity for both the observed IFX and TNF concentrations. Individual weighted residuals were equally distributed along the zero line relative to individual predictions and conditional weighted residuals were equally distributed along the zero line relative to time after dose.
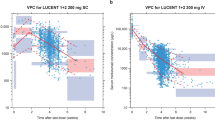
Evaluation of the VPC (Fig. 4) showed that the median and 5th and 95th percentiles of the observed data (both infliximab and TNF data) are situated within the associated 90% confidence intervals of the prediction intervals. This indicated a good qualification of the QSS model. Bootstrap results confirmed the validity of the model (Table 1). Out of 2000 bootstrap runs for model evaluation, 1768 runs (88%) were successful and both parameters and precision were comparable to the final model.

Time after dose course of model-predicted and observed infliximab and TNF concentrations. Simulations were performed (n = 1000) on the entire dataset based on the final TMDD model with QSS approximation, described by Eq. 8–13 and final parameters estimates in Table 2. Individual observations are depicted by the black dots. The solid red line represents the median of the observed data and the dashed blue lines represent the observed 5th and 95th percentiles. The red shaded area represents the 90% confidence interval of the median of the simulated data, with the red dashed lines representing the median of the simulated data per bin. The blue shaded areas represent the 90% confidence intervals of the 5th and 95th percentiles of the simulated data
Simulations
Evaluation of different initial estimates showed that a lower initial estimate for Kss resulted in a higher kdeg estimate, and higher initial estimates for Kss resulted in a lower kdeg estimate. For that reason, different values for Kss and kdeg were simulated and eventually, the degradation constant of TNF (kdeg) was fixed to 5.12 day−1, a biologically plausible value obtained from the sensitivity analysis (Supplementary Table 1).
Discussion
IFX is an anti-TNF agent used in patients with moderate to severe UC. In this present study we analyzed the pharmacokinetics of IFX and TNF within the first 6 weeks after start of IFX therapy in UC patients. IFX binds with high affinity to its target TNF and as a result exhibits TMDD. In this study, application of a TMDD model allowed not only the estimation of the population pharmacokinetic parameters of IFX but also the first-order elimination rate of TNF (kdeg) and the dissociation constant of the complex of IFX and TNF (Kss).
A theoretical pharmacokinetic/pharmacodynamic model in which the degree of inflammation (reflected by the poor surrogate marker Crohn’s Disease Activity Index), changed by the binding of IFX to TNF in patients with CD was previously proposed [29]. More recently, the same conceptual model was evaluated for adalimumab, a subcutaneous anti-TNF agent, administered to CD patients [30]. Although both models could be applied to predict CDAI values in these patients, dependent on the complex formation between the anti-TNF agent and TNF, TNF values were not actually measured but estimated in both studies. A minimal physiology-based pharmacokinetic model with TMDD component also been has proposed to assess the interrelationship between IFX and TNF [31]. In this study, administration of a subcutaneous recombinant human-TNF infusion to boost baseline TNF, enabled the quantification of recombinant-human-TNF in the plasma of rats. A minimal physiology-based model with TMDD component was able to quantitatively describe the time-course of TNF suppression by IFX. To date, no population model has been proposed to describe the pharmacokinetics of IFX and measured sTNF in human patients.
Several structural models were evaluated to describe the pharmacokinetics of IFX and TNF in this population. First, a binding model was evaluated to describe TNF concentration–time curves, in which the binding of TNF to IFX did not influence the pharmacokinetics of the latter. However, when using a binding model to describe pharmacokinetics and dynamics, total receptor (i.e. TNF) concentrations are assumed to be constant over time. As shown in Fig. 2, total TNF concentrations increased after administration of IFX and the binding model was not able to capture this behavior adequately.
A full TMDD model was evaluated, but parameter estimates were highly dependent on initial estimates, indicating poor stability of the model and, subsequently, several approximations of the TMDD model were evaluated. Both QE and QSS approximations of the TMDD model assume an equilibrium between free drug/receptor concentrations and formation of the complex. In contrast to the QE approximation, a QSS approximation does not assume the elimination constant of the complex to be negligible. Because of the assumption that IFX-TNF complex is mainly eliminated, rather than dissociated, this approximation was deemed more applicable.
Evaluation of different initial estimates showed that a lower initial estimate for Kss resulted in a higher kdeg estimate, and higher initial estimates for Kss resulted in a lower kdeg estimate. To stabilize the QSS model, kdeg was fixed to a value which, in the absence of applicable values in literature, was based on a sensitivity analysis. Estimated Kss (13.6 nM) was higher than binding affinities (KD) reported for IFX and TNF, which range from 0.0273 nM to 1.92 nM [19, 31, 32]. Kss was expected to be higher than KD values reported in literature because Kss represents steady-state equilibrium by taking into account not only the equilibrium constant (KD = koff/kon) but also the elimination of the complex (ke(P)/kon). Moreover, equilibrium constants are difficult to measure and are highly dependent on the type of assay performed. Furthermore, differences may exist between the in vitro and in vivo derived affinity values.
With the use of the QSS approximation, the elimination of the complex was assumed to be non-negligible and the proposed model was able to estimate the degradation constant (ke(P)), i.e. 0.984 day−1. Nonetheless, little is known about the fate of the IFX-TNF complex. It is hypothesized that the complex is recycled via the neonatal Fc-receptor (FcRn), as described for free concentrations of monoclonal antibodies. Also, the IFX-TNF complex could be subject to proteolytic degradation.
As described in Eq. 11, it was assumed that IFX is subject to nonlinear elimination. From previous literature it is however known that IFX is mainly cleared via linear elimination [11,12,13,14,15,16]. Removal of the nonlinear elimination term for IFX clearance provided a similar fit of the model to the data, suggesting that the nonlinear elimination route for IFX in the current model is indeed negligible. However, precision of parameter estimates increased indicating decreased precision. In addition, because the main purpose of this study was to describe both the interaction between IFX and TNF and the mass balance, the nonlinear elimination term was maintained in the current model.
TNF is a pro-inflammatory cytokine and after conversion released in its soluble form, i.e. sTNF. We were able to measure free sTNF concentrations in sera of patients. Although measuring free sTNF is challenging, due to the low detection level needed, Song et al. also previously reported measurements of free sTNF in patients with Crohn’s disease and found a similar trend, i.e. decrease of free sTNF concentrations after initiation of anti-TNF therapy [33].Total TNF concentrations were simulated and found increased after IFX administration (Fig. 2). This is due to the slower internalization rate of the complex (ke(P)) compared to the elimination rate of TNF (kdeg), reflecting a prolonged half-life of TNF due to formation of the complex, which has been previously proposed [34]. Hence, binding of TNF to IFX protects TNF from elimination, resulting in accumulation of total TNF in the serum. This finding is in line with a recent publication that showed an increase in total TNF concentrations after start of adalimumab therapy, a subcutaneously administered anti-TNF agent [35]. In addition, it is hypothesized that the administration of IFX might trigger the conversion of membrane-bound TNF to sTNF, which results in an increased production of TNF after administration of IFX [36].
First, the study is limited by the relatively small samples size (n = 20), despite rich sampling. The main limitation of our study however is the absence of measured baseline TNF values. Baseline TNF serum concentrations have been found to be higher in responders compared to non-responders before treatment with IFX in patients with fistulizing CD [5]. In addition, TNF as predictor for treatment outcomes would be best supported by measurements of colonic tissue concentrations of TNF, as previously described by Yarur et al. [37]. Colonic tissue concentrations of TNF are significantly higher in inflamed tissue compared to matched uninflamed tissue. Also, in inflamed tissue the ratio of tissue TNF to anti-TNF are elevated compared to uninflamed tissue. With serum and colonic concentrations of both TNF and IFX, a mechanism-based model could be developed to describe the total fate of TNF in relation to IFX in this study population. Also, the potential binding of sTNF to IFX in colonic tissue could then be explored. More extensive knowledge about the total fate of TNF could then be used to investigate the relationship between suppression of TNF and the response to IFX therapy. As a result, IFX response might be predicted based on TNF concentrations and this would potentially support individualized IFX treatment.
In conclusion, we propose a TMDD model with QSS approximation to describe the interaction between IFX and TNF. This model could eventually be used to investigate the relationship between suppression of TNF and the response to IFX therapy.
References
Ordás I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ (2012) Seminar ulcerative colitis. Lancet 380:1606–1619
Murch SH, Lamkin VA, Savage MO, Walker-Smith JA, MacDonald TT (1991) Serum concentrations of tumour necrosis factor in childhood chronic inflammatory bowel disease. Gut 32:913–917
Komatsu M, Kobayashi D, Saito K, Furuya D, Yagihashi A, Araake H, Tsuji N, Sakamaki S, Niitsu Y, Watanabe N (2001) Tumor necrosis factor in serum of patients with inflammatory bowel disease as measured by a highly sensitive immuno-PCR. Clin Chem 47:1297–1301
Braegger CP, Nicholls S, Murch SH, MacDonald TT, Stephens S (1992) Tumour necrosis factor alpha in stool as a marker of intestinal inflammation. Lancet 339(8785):89–91
Martínez-Borra J, López-Larrea C, González S, Fuentes D, Dieguez A, Deschamps EM, Pérez-Pariente JM, López-Vázquez A, De Francisco R, Rodrigo L (2002) High serum tumor necrosis factor-α levels are associated with lack of response to infliximab in fistulizing Crohn’s disease. Am J Gastroenterol 97(9):2350–2356
Yarur AJ, Jain A, Sussman DA, Barkin JS, Quintero MA, Princen F, Kirkland R, Deshpande AR, Singh S, Abreu MT (2015) The association of tissue anti-TNF drug levels with serological and endoscopic disease activity in inflammatory bowel disease: the ATLAS study. Gut 65:1–7
Neurath MF (2014) Cytokines in inflammatory bowel disease. Nat Rev Immunol 14(5):329–342
Rutgeerts P, Sandborn WJ, Feagan BG, Reinisch W, Olson A, Johanns J, Travers S, Rachmilewitz D, Hanauer SB, Lichtenstein GR, de Villiers WJS, Present D, Sands BE, Colombel JF (2005) Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med 353(23):2462–2476
Scallon B, Cai A, Solowski N, Rosenberg A, Song X-Y, Shealy D, Wagner C (2002) Binding and functional comparisons of two types of tumor necrosis factor antagonists. J Pharm Exp Ther 301:418–426
Van Schie KA, Ooijevaar-De Heer P, Dijk L, Kruithof S, Wolbink G, Rispens T (2016) Therapeutic TNF inhibitors can differentially stabilize trimeric TNF by inhibiting monomer exchange. Sci Rep 6:32747
Fasanmade AA, Adedokun OJ, Blank M, Zhou H, Davis HM (2011) Pharmacokinetic properties of infliximab in children and adults with Crohn’s disease: a retrospective analysis of data from 2 phase III clinical trials. CLITHE 33:946–964
Brandse JF, Mathôt RA, van der Kleij D, Rispens T, Ashruf Y, Jansen JM, Rietdijk S, Löwenberg M, Ponsioen CY, Singh S, van den Brink GR, D’Haens GR (2016) Pharmacokinetic features and presence of antidrug antibodies associate with response to infliximab induction therapy in patients with moderate to severe ulcerative colitis. Clin Gastroenterol Hepatol 14(2):251–258
Fasanmade AA, Adedokun OJ, Ford J, Hernandez D, Johanns J, Hu C, Davis HM, Zhou H (2009) Population pharmacokinetic analysis of infliximab in patients with ulcerative colitis. Eur J Clin Pharmacol 65(12):1211–1228
Ternant D, Aubourg A, Magdelaine-Beuzelin C, Degenne D, Watier H, Picon L, Paintaud G (2008) Infliximab pharmacokinetics in inflammatory bowel disease patients. Ther Drug Monit 30(4):523–529
Dotan I, Ron Y, Yanai H, Becker S, Fishman S, Yahav L, Ben Yehoyada M, Mould DR (2014) Patient factors that increase infliximab clearance and shorten half-life in inflammatory bowel disease. Inflamm Bowel Dis 20(12):2247–2259
Buurman DJ, Maurer JM, Keizer RJ, Kosterink JGW, Dijkstra G (2015) Population pharmacokinetics of infliximab in patients with inflammatory bowel disease: potential implications for dosing in clinical practice. Aliment Pharmacol Ther 42:529–539
Levy G (1994) Pharmacologic target-mediated drug disposition. Clin Pharmacol Ther 56(3):248–252
Djebli N, Martinez J-M, Lohan L, Khier S, Brunet Aurélie, Hurbin F, Fabre D (2017) Target-mediated drug disposition population pharmacokinetics model of alirocumab in healthy volunteers and patients: pooled analysis of randomized phase I/II/III studies. Clin Pharm 56:1155–1171
Kaymakcalan Z, Sakorafas P, Bose S, Scesney S, Xiong L, Hanzatian DK, Salfeld J, Sasso EH (2009) Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin Immunol 131(2):308–316
Wang S-L, Ohrmund L, Hauenstein S, Salbato J, Reddy R, Monk P, Lockton S, Ling N, Singh S (2012) Development and validation of a homogeneous mobility shift assay for the measurement of infliximab and antibodies-to-infliximab levels in patient serum. J Immunol Methods 382(1):177–188
Vande Casteele N, Gils A, Singh S, Ohrmund L, Hauenstein S, Rutgeerts P, Vermeire S (2013) Antibody response to infliximab and its impact on pharmacokinetics can be transient. Am J Gastroenterol 108(6):962–971
Yeung D, Ciotti S, Purushothama S, Gharakhani E, Kuesters G, Schlain B, Shen C, Donaldson D, Mikulskis A (2016) Evaluation of highly sensitive immunoassay technologies for quantitative measurements of sub-pg/mL levels of cytokines in human serum. J Immunol Methods 437:53–63
Garcet S, Nograles K, Correa da Rosa J, Schafer PH, Krueger JG (2018) Synergistic cytokine effects as apremilast response predictors in patients with psoriasis. J Allergy Clin Immunol 142(3):1010–1013
Wingfield P, Pain RH, Craig S (1987) Tumour necrosis factor is a compact trimer. FEBS Lett 211(2):179–184
Gibiansky L, Gibiansky E, Kakkar T, Ma P, Gibiansky L, Gibiansky E, Kakkar T, Ma P (2008) Approximations of the target-mediated drug disposition model and identifiability of model parameters. J Pharmacokinet Pharmacodyn 35:573–591
Mager DE, Jusko WJ (2001) General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharm Pharm 28(6):507–532
Kimura K, Takayanagi R, Yokoyama H, Yamada Y (2012) Theory-based analysis of anti-inflammatory effect of infliximab on Crohn’s disease and rheumatoid arthritis. Rheumatol Int 32:145–150
Fallahi-Sichani M, Schaller MA, Kirschner DE, Kunkel SL, Linderman JJ (2010) Identification of key processes that control tumor necrosis factor availability in a tuberculosis granuloma. PLoS Comput Biol 6(5):1–19
Furuya Y, Ozeki T, Takayanagi R, Yokoyama H, Okuyama K, Yamada Y (2007) Theory based analysis of anti-inflammatory effect of infliximab on Crohn’s disease. Drug Metab Pharm 22(1):20–25
Kimura K, Yoshida A, Takayanagi R, Yamada Y (2018) Pharmacokinetic and pharmacodynamic model for analysis of adalimumab administered for Crohn’s disease. Biopharm Drug Dispos 39(6):283–288
Chen X, Dubois DC, Almon RR, Jusko WJ (2017) Interrelationships between infliximab and recombinant tumor necrosis factor-a in plasma using minimal physiologically based pharmacokinetic models. Drug Metab Dispos 45(7):790–797
Kim M-S, Lee S-H, Song M-Y, Yoo TH, Lee B-K, Kim Y-S (2007) Comparative analyses of complex formation and binding sites between human tumor necrosis factor-alpha and its three antagonists elucidate their different neutralizing mechanisms. J Mol Biol 374:1374–1388
Song L, Hanlon DW, Chang L, Provuncher GK, Kan CW, Campbell TG, Fournier DR, Ferrell EP, Rivnak AJ, Pink BA, Minnehan KA, Patel PP, Wilson DH, Till MA, Faubion WA, Duffy DC (2011) Single molecule measurements of tumor necrosis factor α and interleukin-6 in the plasma of patients with Crohn’s disease. J Immunol Methods 372(1–2):177–186
Junghans RP, Carrasquillo JA, Waldmann TA (1998) Impact of antigenemia on the bioactivity of infused anti-Tac antibody: implications for dose selection in antibody immunotherapies. Proc Natl Acad Sci USA 95(4):1752–1757
Berkhout LC, l’Ami MJ, Ruwaard J, Hart MH, Heer PO, Bloem K, Nurmohamed MT, van Vollenhoven RF, Boers M, Alvarez DF, Smith CH, Wolbink GJ, Rispens T (2019) Dynamics of circulating TNF during adalimumab treatment using a drug-tolerant TNF assay. Sci Transl Med 11(477):3356
Hepburn TW, Totoritis MC, Davis CB (2003) Antibody-mediated stripping of CD4 from lymphocyte cell surface in patients with rheumatoid arthritis. Rheumatology 42(1):54–61
Yarur AJ, Kanagala V, Stein DJ, Czul F, Quintero MA, Agrawal D, Patel A, Best K, Fox C, Idstein K, Abreu MT (2017) Higher infliximab trough levels are associated with perianal fistula healing in patients with Crohn’s disease. Aliment Pharmacol Ther 45(7):933–940
Acknowledgements
All IFX and TNF measurements were performed by Prometheus Laboratories (San Diego, California, USA).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10928_2019_9652_MOESM2_ESM.tif
Supplementary material 2 (TIFF 4221 kb). Supplementary Figure 1A: Goodness-of-fit plots infliximab. Goodness-of-fit plots of the final model. The dashed lines represent the line of identity
10928_2019_9652_MOESM3_ESM.tif
Supplementary material 3 (TIFF 4422 kb). Supplementary Figure 1B: Goodness-of-fit plots TNF. Goodness-of-fit plots of the final model. The dashed lines represent the line of identity
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Berends, S.E., van Steeg, T.J., Ahsman, M.J. et al. Tumor necrosis factor-mediated disposition of infliximab in ulcerative colitis patients. J Pharmacokinet Pharmacodyn 46, 543–551 (2019). https://doi.org/10.1007/s10928-019-09652-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10928-019-09652-5