
Abstract
This study reports the synthesis of magnetic nanoparticles (MNPs) using the co-precipitation method with the support of Vernonia Amygdalina (VA) (bitter leaf) extract for the efficient sequestration of rhodamine 6G dye (Rhd 6G) from a water-soluble solution. The prepared MNPs were characterized using Transmission electron microscopy (TEM,) X-ray Diffraction (XRD), Fourier-transform infrared (FTIR) and Ultraviolet–visible (UV–Vis) spectrometry. The average particle sizes from TEM and XRD analyses were 4.81 ± 2.2 nm and 5.61 nm respectively. The sorption of Rhd 6G dye to the MNPs was pH-dependent, with ideal confiscation of dye molecules observed at pH 10 (\(\sim 91.4\%)\). The sorption process of Rhd 6G dye was reported to follow the Freundlich (FRH) isotherm model, while the pseudo-first-order model (PFOM) best depicted the sorption process of Rhd 6G dye to the MNPs from the non-linear modelling. The determined sorption capacity of the MNPs was established to be 454 mg.g−1. Also, the sorption of Rhd 6G dye to the MNPs was observed to be thermodynamically spontaneous, endothermic and with increasing randomness between the interface of the water-soluble solution and the MNPs. Hence, the prepared MNPs have proven to be an effective potential magnetic sorbent for the removal of cationic dyes from an aqueous solution.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The unwarranted dye utilization in contemporary times has been a comprehensive concern owing to dye entry into the ecosystem. It is estimated that nearly 7 million tons of dye are globally used yearly, with around 10–15% of dyes being discharged into the marine ecosystem without any control [1]. Synthetic dyes are created to meet various industrial requirements such as adhesives, glass for construction, paints, paper and pulp, soap, polymers, cosmetics, beverages, art supplies, food, and wax biomedicine. Due to dye’s chemical stability, ease of synthesis, and colour variety, they are generally used in fabric production [2, 3].
Rhd 6G dye is an azo dye with a single or added –N = N– groups affixed to an aromatic structure (Fig. 1). Rhd 6G dye is a xanthene dye derivative, which is extensively used in textile, paper, printing industries, and fluorescent labelling owing to its extreme pink pigment. Owing to their chromophoric features, they are chemically stable and persistent. They are tremendously noxious and dangerous to animals and humans. They can cause skin and respiratory tract inflammation and initiate allergic and asthmatic reactions. However, due to their low-slung cost and colour reinforcement, this dye is regularly combined unlawfully with food matrices as colourants to improve the graphical aesthetics and stimulate sales [4, 5].

Chemical structure of Rhd 6G dye [6]
Their unwarranted releases into the environment produce a further source of probable hazards like bioaccumulation, tumour by chlorination results (SPC), and environmental persistence. Discharging various dyes into the ecosystem causes harmful effects like hypersensitive reactions, hazardous impacts, dermatitis, rhinitis, industrial asthma, and hypersensitive pink eye in living organisms [3]. As the diversity of industrial goods grows, the composition of various dye effluents is becoming progressively intricate. Hence dye effluent treatment has become an enormously complicated undertaking [7] by employing the various available techniques [8]. An extensive array of industrial methods used to treat dye wastes include reverse osmosis, coagulation, electrolysis, adsorption, ion-exchange, and ultrafiltration [9]. Nevertheless, most of these traditional techniques suffer from field-scale pertinence from the high cost of equipment, colossal time utilization, and low technical viability of chemical sludge creation [10, 11]. The sorption method has been a critical research motivation owing to its simplicity, ease of operation, general use, minimal construction and cost of maintenance, ample sorbent resources, effortless reusing, and elevated productivity [3].
Metal oxide nanostructure is exclusively getting huge consideration owing to their multi-functionality and production ease [12,13,14]. In this background, nanoparticles (NPs) are considered as an alternative for the sorption of noxious or refractory effluents such as dyes owing to their physicochemical properties. NPs, due to their morphological characteristics like extraordinary reactive sites, controlled size, and superior surface area, have become a preferred sorbent for dye over time. Despite the outstanding sorptive properties of NPs for dye molecules, a general concern has been their toxicity [3, 8]. There has been an increased demand for the production of non-noxious, eco-friendly NPs with production expertise. Recently, various informal, reliable, price-efficient, reproducible, and influential green fabrication techniques for NPs have been carried out, and the idea of safety by design inspires this. As an outcome, various biological approaches and plant extracts are presently utilized in the organic fabrication techniques to create NPs [15].
Over the past few years, there has been a swift increase in the fabrication of NPs to produce innovative smart materials at the nanoscale with distinctive features [16]. The most widely studied NPs owing to its characteristic properties is magnetite (Fe3O4). Fe3O4 has been applied overwhelmingly in multipurpose fields such as aerospace and defence, biomedical, environmental redress, construction, textiles, automotive, electronics, sensing and detection of contaminants, and agriculture and food. Fe3O4 shows the sturdiest magnetization of all the transition metal oxides and signifies one of the utmost remarkable crystallographic phases of iron oxide (IO). Fe3O4 has elevated magnetization which is influenced by the magnetic field and is size dependent. To make NPs with specific features, an effective technique for Fe3O4 fabrication is essential [17,18,19,20].
The key drawback of using chemical and physical fabrication techniques for Fe3O4 synthesis is that they are time-intensive, have low fabrication rates, the cost of fabrication is staggering, defective surface creation, enormous energy requirements, creation of a massive quantity of resulting waste materials and the utilization of noxious reducing agents. To decrease the noxiousness, time and cost of production of NPs, NPs fabrication using the biological procedure from the plant extracts has been shown as a superior substitute in comparison to the chemical and physical techniques [16, 21]. Employing biosynthesis methods using extracts of plants has gained enhanced attention. It is now seen as a gold specification amongst the biological method owing to their simplicity, effectiveness, cost efficiency, plant multiplicity and viability of this method in NPs fabrication [22, 23].
However, there has been recently increased use of plants, mainly plant extracts, in the fabrication of various NPs, owing to these extracts having the capability to create NPs with specified composition, shape, and size. Also, a vast collection of phytochemicals in the extracts of plants may act as reducing agents or organic stabilizers for the fabrication of NPs [24]. Plant extracts also contain polyphenols (secondary metabolites), which have a massive value of standard reduction capability sufficient for metals reduction to their corresponding NPs. They also serve as capping agents, thereby reducing/increasing the accumulation/stability of NPs. This characteristic excludes the distinct addition of stabilizing agents during NPs fabrication [17, 25, 26].
Diverse plant parts such as barks, extracts, exudates, and tissues have emerged as efficient substitutes for NPs fabrication [20]. Various studies based on the fabrication of Fe3O4 NPs using different extracts of plants were reported in the works of Elizondo-Villarreal et al., [27], Vinayagam et al. [28], Das et al. [29], Sivakami et al. [30], Parajuli et al. [31], and Mohammadi et al. [32]. Plants-derived NPs show a superior natural capability in environmental issues, agriculture, bio-engineering, nanomedicine, human health safety, and food science and technology [24]. Plants have a broad diversity of genetic variants in biomolecules and metabolites (proteins, carbohydrates, vitamins, phenols, flavonoids, sterols, alkaloids, and intermediates), with these metabolites having various functional groups (hydroxyl (OH), carbonyl (C═O), and amine (NH2)) that reacts with metal ions to reduce their size to the nanoscale [33].
Vernonia amygdalina (VA), generally acknowledged as a bitter leaf (BL) is a herbaceous plant physically located in West Africa and is a part of the Asteraceae species with green leaves, a distinctive odour, and a pungent taste [34]. It is a plant that develops up to 3 m in height in the tropics of Africa and other areas, especially in Nigeria, Zimbabwe, and Cameroon. It is believed to have numerous health advantages [35]. Their leaves are loaded with several functional groups like carboxylic acids, ethers, alcohols, phenol, etc., which are effective in dye molecule sorption [36]. The utilization of the VA leaf extract as a reducing agent to prime the NPs was due to their high possible application as a resource for green procedure processes, improving NPs’ surface area owing to the phytochemical coating and NPs’ biocompatibility. Also, their usage in the NPs fabrication reduces energy requirements, labour, and chemical waste [37]. For Fe3O4 to be utilized in polluted water with different contaminants remediation, it must possess a strong magnetic response, be chemically stable, have low toxicity and be swiftly reused [19]. But published works relating to the green fabrication of Fe3O4 NPs using VA leaf extract and their application in wastewater treatment are very few. The most application of the fabrication of NPs using the extract of VA leaves has been on antimicrobial activity and biomedical performance [38, 39]. The current study aims for the sustainable fabrication of MNPs from the aqueous extract of VA leaf (bitter leaf), their characterization and the study of the impact of the prepared NPs in the treatment of Rhd 6G dye.
2 Materials and Method
The entire chemicals utilized in this study were of analytical grade and were prepared prior to further usage. All chemicals used (ferrous-sulphate (FeSO4.7H2O), ferric-chloride (FeCl3.6H2O), sodium chloride-NaCl, sodium hydroxide-NaOH, Rhd 6G dye, and hydrochloric acid-HCl,) were acquired from Merck (Sigma-Aldrich), South-Africa. Rhd 6G dye (95%) stock-solution (1000 mg L−1) was prepared by dissolving 1 g of Rhd 6G dye in a litre (L) of deionized water. Subsequently, desired concentrations of Rhd 6G dye were prepared from the stock solution by dissolving measured volumes using deionized water.
2.1 VA Leaves Extract Preparation
Fresh VA leaves were collected from a house in Highveld Centurion, Tshwane municipal area (−25,882,195, 28,194,262) and severely washed with deionized water to remove dust and sand. The washed VA leaves were subsequently dried under sunlight for three days. To prepare the aqueous extract, 30 g of the dried VA leaves were weighed and added to 100 mL of deionized water, which was heated at 80 \(^\circ{\rm C}\) for 1 h. The extract from the leaves was allowed to cool down and filtered using the Whatman filter-paper (No. 42). Ultimately, the resulting leaf extract was stored at 4 \(^\circ{\rm C}\) for further usage.
2.2 Green Synthesis of MNPs
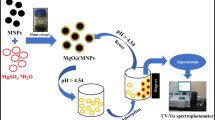
The MNPs were fabricated based on the technique used by Dhar et al. [40] with slight modifications. To fabricate the MNPs, the co-precipitation technique was applied. In a 1:2 ratio, FeSO4.7H2O (2 g) and FeCl3.6H2O (4 g) were dissolved in 100 mL of deionized water under continuous stirring for 30 min at 80 °C using a magnetic stirrer (separation-MR Hei-Tec). Subsequently, 15 mL of the leaf extract was added to the solution dropwise while stirring. 5 M of NaOH (50 mL) was then added dropwise while stirring at 80 °C for 30 min until the solution pH was 11 for a homogenous solution and reaction completion. The obtained solution was centrifuged at 5000 rpm for 20 min utilizing a pro-analytical centrifuge. The obtained material was subsequently washed three times using deionized water and centrifuged under the same process above to remove impurities. The obtained product was dried in an oven at 70 °C for 24 h utilizing a scientific oven. The obtained material was stored in an airtight vial to avoid contact with air moisture. The overall chemical reaction for the preparation of the MNPs is given by Eqs. 1 and 2. Figure 2 shows the schematic diagram for preparing the MNPs using VA extract as reducing and stabilizing agents.

The schematic diagram for preparing MNPs using VA extract as support and reducing agent
2.3 Spectroscopy and Microscopic Investigation of MNPs
The synthesized MNPs were characterized using TEM (JEOL 2010, 200 kV, Tokyo, Japan)) to determine their surface morphology and size distribution. To assess the magnetic properties of the MNPs, an XRD diffractogram was utilized (Bruker D8 advance diffractometer). The functional groups of the prepared MNPs were established using FTIR spectrometry (Perkin Elmer Spectrum Two UATR). While the optical properties of the MNPs were assessed utilizing UV–Vis spectrometry (Perkin Elmer Spectrum spectrometry-SP-UV 500 spectrophotometry).
2.4 Point of Zero Charge (pHPZC) Determination
The pHPZC of the MNPs was determined by varying pH (2–12) of 0.1 M NaCl (25 mL) with 0.05 g of the MNPs for 24 h. The final pH of the NaCl solution was assessed after 24 h utilizing a pH meter. The change in pH (\(\Delta \mathrm{pH}\) = pHf-pHi) was determined by deducting the obtained values of the initial pH from the established final pH values. The pHPZC of MNPs was established from the plot of the \(\Delta \mathrm{pH}\) versus pHi.
2.5 Sorption Experimental Investigation
Sorption experiments were carried out to establish the pH effect on the adsorption of Rhd 6G dye to the MNPs, by varying the pH (2–10) of 100 mg.L−1 of Rhd 6G dye solution (50 mL) utilizing 0.1 M of HCl or NaOH and contacting it with 0.05 g of MNPs at a stirring speed of 200 rpm (scientific shaker) for 2 h. At the end of the batch experiment, solutions were withdrawn using a syringe and filtered through a syringe filter of 0.2 µM. The final concentration of dye left in the solution was revealed by employing UV–Vis spectrometry at a wavelength of 525 nm. The percentage of Rhd 6 g dye removed (% removal) and the sorption capacity (qe-mg.g−1) of the MNPs were determined using Eqs. 3 and 4.
Successive experiments were carried-out to test the influence of varying dosage (0.02–0.07 g), initial Rhd 6 g dye concentration (50–400 mg.L−1) and time (20–100 min) on the sorption of the Rhd 6 g dye molecules to the prepared material at a stirring speed of 200 rpm, and pH 10. The influence of temperature was also evaluated by contacting 100 mg.L−1 of Rhd 6G dye with 0.06 g sorbent dosage for 40 min at a stimulating speed of 200 rpm, pH 10 and varying temperature of 25–45 \(^\circ{\rm C}\). After all the experiments, the equilibrium concentration of the dye left was determined using UV spectrometry. The percentage of dye confiscated (% removal), the sorption capacity (qe-mg.g−1), and the quantity of dye sorbed by unit mass of the MNPs at the time (t) was determined by employing Eqs. 3, 4 and 5.
Co, Ce\(,\mathrm{ V},\mathrm{ M} and\) Ct signify the initial and final concentrations of Rhd 6G dye, the solution volume (L), the mass of the sorbent (g) and the dye concentration at time (t) left in the solution.
2.6 Desorption Studies
The recycling of MNPs was considered in successive sorption–desorption experiments. For the desorption experiments, hitherto sorbed Rhd 6 g dye to MNPs were contacted with 50 ml of 0.1 M HCl and NaOH for 1 h at a stirring speed of 200 rpm. At the end of the desorption experiment, the solution was withdrawn using a syringe and filtered. The MNPs left were dried in an oven for 2 h and reused for another sorption process. The desorption-sorption process was repeated for four successive cycles. The % of Rhd 6 g dye desorbed from the MNPs was assessed using Eq. 6, which is the ratio of the quantity of contaminant desorbed to the amount of contaminant sorbed.
2.7 Error Analysis
A critical part of any systematic research is process modelling. The sorption isotherm and kinetic studies give an understanding of the sorbate-sorbent interaction nature and the time dependency of the sorption structure. Owing to the characteristic preference resulting from the linearization, different factor sets are assessed by the non-linear regression. This offers a meticulous statistical technique for assessing factors employing the unique form of the models’ equations. As various forms of the sorption models equations impact the values of R2 during the linear scrutiny, the non-linear study is the key approach to avoiding such errors [41].
Also, a key drawback linked with the linear regression technique is its inadequate applicability in approximating just two variables in an experimental equation. Hence, it is deemed an unsuitable procedure for assessing sorption kinetic and isotherm parameters. The non-linear method of optimization offers a more intricate and meticulous procedure for the assessment of kinetic and isotherm parameters with an error evaluation to assess the ideal fit of the sorption parameters to the experimental findings [42].
To assess the appropriateness of a model calculation to the experimental findings, error function evaluation is generally considered necessary. Error functions are numerical equations employed to assess the variation between the hypothetically predicted data from the estimates of real experimental data [43]. The key usefulness of error functions is to measure the sorbent distribution and offer mathematical evaluation of the result; most critically is the authentication of the reliability of the experimental outcomes, which have led to the creation of sorption isotherm and kinetic [44].
The best fitting for the isotherm and kinetic models were authenticated using two statistical error functions. To assess the association between the model calculated data and the experimental data, the non-linear chi-square test (X2) and residual sum of squares (RSS) were implemented in this study using Originlab software (Eqs. 7 and 8). Using this error model, the various error values obtained for the isotherm/kinetic models forecast are further normalized [45].
N, qe(exp) and qe(cal) represent the number of experiments carried out, the numerical values of the experimental data and the calculated data by the model [46].
3 Results and Discussions
3.1 Optical and Structural Characterization
3.1.1 FTIR Analysis
FTIR studies were carried-out to ascertain the functional groups existing in the VA leaf extract, and the prepared MNPs before and after sorption of Rhd 6G dye using an array of wavenumber (400–4000 cm−1) (Fig. 3). The FTIR spectra in Fig. 3a show distinctive peaks of the VA leaf extract. The VA extract shows vibrational peaks at 3275, 2934, and 1051 cm−1 ascribed to the uniqueness of O–H stretching of inter-molecular bonded alcohol, C-H stretching alkanes, and C-N stretching vibration, respectively. The peaks at 1526–1597, 1402, 1261, and 866 cm−1 were ascribed to N–O stretching of the aliphatic-nitro group, C-H bending of the methylene group, C-O ester stretching of carboxylic acid and C-H deformation ring respectively [47,48,49,50,51]. After the synthesis of the MNPs, a new adsorption peak was noticed at 547.29 cm−1 and was attributed to the Fe–O vibration band, which is the distinguished spectra of Fe3O4 (Fig. 3b) [52, 53]. Before the sorption of Rhd 6G to MNPs, the observed spectra of the MNPs were 3372, 1611, 1352, and 1104 cm−1, which were ascribed to the uniqueness of O–H stretching of inter-molecular bonded alcohol, C = C stretching of aromatic C = C group, OH stretching of OH group (alcohol) and C-N stretching vibration. After the Rhd 6G dye sorption, a blue shift in the MNPs wavenumber (hypsochromic shift) was observed, suggesting the dye molecules’ sorption to the MNPs after treatment (Fig. 3b). The presence of O–H in the VA extract and the prepared MNPs improved the dye molecules’ sorption and could act as antimicrobial and antioxidant inhibitions [45, 48].

FTIR spectra of a VA leaf extract and b MNPs before and after adsorption of Rhd 6G
3.1.2 TEM Analysis of MNPs
The morphology of the prepared MNPs is shown in Fig. 4a. The TEM image of the MNPs shows that they are spherically shaped. Almost all of the particles show minor particle aggregation, which might have resulted from the VA thickening properties or OH− groups from the VA extract [54] The particle size distribution shows the particles are within the range of 2–13 nm with an average particle diameter of 4.81 ± 2.2 nm employing the ImageJ software and the Gaussian function in OriginLab software (Fig. 4b).

a TEM image of MNPs and b particle size distribution of MNPs
3.1.3 XRD Analysis
The XRD diffraction pattern of the prepared MNPs (Fig. 5) shows diffraction peaks at \(2\theta\)2 value of 30.28° (220), 35.57° (311), 43.17° (400), 53.93° (422), 57.19° (511), and 62.85° (440), corresponding to a faced centred cubic spinel crystalline structure of magnetite (Fe3O4) (JCPDS no. 19–0629) [55, 56]. To determine the MNPs’ regular crystalline size (DMNPs), the Debye–Scherrer equation was applied to a matching peak value of 35.57° (311) (Eq. 9).
k, λ, θ, and β signify the grain shape factor, the incident X-ray wavelength of Cu–Kα radiation, the Bragg’s angle, and the full-width half-maximum (FWHM) (radians), respectively. The determined particle size was 5.61 nm, which was consistent with the TEM result. The calculated interplanar spacing (d) and lattice parameters were 2.519 Å and 8.355 Å, which was determined using Eqs. 10 and 11. The determined lattice parameter was close to the Fe3O4 NPs standard [17, 25, 26, 28]. A slight shift and modification in the distinctive XRD peaks of the MNPs to higher intensity were noticed after the sorption of Rhd 6G dye. This indicates slight modification in the crystal structure of the MNPs without any physical change in its crystalline structure after Rhd 6G dye was sorbed. The shift in XRD peak after sorption was based on the size of the dye molecules sorbed. The size of dye molecules is lesser than the base metal, thus, the interstitial position was inhabited by the dye molecules leading to the lattice structure modification and smaller d-spacing between the atoms. Hence, an intensification in the XRD results in above-average angles. Also, when the dye molecules are larger than the base-metal, lower angles are achieved with enhanced d-spacing [20].
a and d represent the lattice parameter, and the interplanar spacing matching to Miller indices h, k, l [57].

XRD pattern a after sorption of Rhd 6G to MNPs and b before sorption of Rhd 6G dye to MNPs
3.1.4 UV–Vis Analysis of MNPs
To confirm the creation of the MNPs in an aqueous colloidal solution, a UV spectral assessment of the solution was carried out at a wavelength of 200-800 nm. The UV spectra of the VA extract-synthesized MNPs revealed absorption peaks at 227 and 298 nm. This is attributed to the existence of poly-phenolic compounds and other phytochemicals responsible for Fe3O4 fabrication and stabilization (Fig. 6a) [40]. The optical direct bandgap (Eg) of the MNPs was calculated using equation 12.
\(\alpha , A, h, and v,\) signify the sorption coefficient, a constant, Plank’s constant, and frequency, respectively [58]. Figure 6b shows the Tauc plot of \({\left(\alpha hv\right)}^{2}\) against hv and the Eg was approximated by extrapolating the linear plot to the x-axis and the estimated Eg of the MNPs was approximately 3.6 eV. The increase in the bandgap of the prepared MNPs was attributed to quantum confinement, where the confinement of electrons and holes leads to the energy bandgap increasing between the valence and conduction band with particle size decreasing [59, 60].

a UV spectra of the prepared MNPs and b bandgap of the prepared MNPs
3.2 Influences of pH on Rhd 6G Dye Sorption
The pH is an important factor that regulates the sorption of dye molecules in a sorption medium to the sorbent surface active sites. The sorption medium’s initial pH is a critical element in defining the sorption mechanism and significantly and sensitively influences the sorption molecular particle ionization of the dyes and the sorbent surface charge [61, 62]. Hence, the uptake rate of pollutants may be improved or decreased based on the preliminary solution pH value. Consequently, hydrogen (H+) and hydroxyl (OH−) ions are sorbed effectively, thereby improving the sorption process as the functional groups of the sorption sites are fragmented, leading to the reaction of kinetics and equipoise attribute variations. Generally, cationic dyes’ uptake is impacted and sorption is reduced at the low solution pH value. While for anionic dyes, the uptake of dyes is improved with an increased solution pH value [63]. To ascertain the precise sorption mechanism, the pHPZC of the sorbent is assessed. For cationic and anionic dyes sorption on any solid surface of a material, sorption is favourable when the pH \(>\) pHPZC and pH \(<\) pHPZC [45]. The determined pHPZC of the MNPs was 9.1 (Fig. 7a). As noticed in Fig. 7b, the sorption of Rhd 6G to the MNPs improved as the solution pH value increased from pH 2–10, with optimum sequestration of Rhd 6G dye observed at pH 10 (\(\sim 91.4\%)\). The reduction in the % of Rhd 6G dye eliminated in acidic pH was attributed to the electrostatic repulsion between the protonated H+ sites on the sorbent and the cationic dye molecules in the acidic medium. With the subsequent increase in the solution pH (basic medium), there was a reported increase in the % of Rhd 6G dye confiscated onto the MNPs. This could be attributed to the electrostatic attraction taking place between the deprotonated sites on the MNPs sites and the cationic dye molecules in the basic medium. As reported in the study by Kumar et al. [64] and El-Nemr et al. [65] the sorption of Rhd 6G dye may have arisen from the ionic process (electrostatic interaction) and chemical interaction corresponding to the columbic force (attraction and repulsion), which significantly varies depending on the types of interactions between the sorbate molecules and sorbent binding sites. These two factors were accountable for the sorption of Rhd 6G dye to the MNPs [45]. Figure 7c depicts the electrostatic interaction between Rhd 6G dye and Fe3O4 NPs.

a The pHPZC of the MNPs, b the impact of solution pH on the sorption of Rhd 6G dye to the MNPs and c graphic representation of the electrostatic interaction between Fe3O4 and Rhd 6G dye (Conditions: initial concentration-100 mg.L−1, sorbent dosage-0.05 g, stirring speed-200 rpm and time of interaction-120 min)
3.3 Influence of Dosage on Rhd 6 g Dye Sorption
The impact of sorbent dosage is a critical factor that is measured as it enhances the treatment process’s effectiveness. Optimizing the sorbent dosage is crucial to estimate the least amount of sorbent dosage required to achieve ideal sorption [63, 66]. At a fixed Rhd 6G dye concentration (100 mg.L−1), the Rhd 6G dye removal was observed to increase from 86.8–92.6%, as the sorbent dosage was enhanced from 0.02–0.06 g (Fig. 8). The increase in the Rhd 6G dye % confiscated as the dosage of MNPs was enhanced was attributed to enhanced sorption surface and sorption sites available [52]. With the successive intensification in the dosage of MNPs from 0.06–0.07 g, no significant change in the % of Rhd 6G dye removed was observed. This was possibly due to the increased competition between the compounds released from the MNPs into the water-soluble solution and dye molecules, thereby hindering the sorption process. This could also be attributed to the decline in the diffusion of dye molecules owing to sorbent binding capacity or pore volume and the surface becoming saturated due to the aggregation of sorbent particles as the dosage increased from 0.06–0.07 mg [11, 46, 67, 68]. The sorption capacity declined from 217.05–65.98 mg.g−1 as the dosage of the MNPs was augmented from 0.02–0.07 g. This reduction in the sorption capacity was due to the drop in the active surface area or the inverse association between sorbent dosage and the qe (mg.g−1), according to Eq. 2 [69, 70]. Hence 0.06 g sorbent dosage was chosen as the optimum dose for subsequent experiments.

Impact of MNPs dosage on Rhd 6G dye sorption to MNPs (Conditions: initial concentration-100 mg.L−1, pH-10, stirring speed-200 rpm and time of interaction 120 min)
3.4 Impact of Initial Concentration on Rhd 6G Dye Sequestration
The initial concentration presents a crucial dynamic drive to enhance the bulk transfer challenge of pollutant molecules between the water-soluble solution and the solid phase. Hence, a more initial pollutant concentration improves the rate of sorption and requires an extended equilibrium time [63]. The result of the impact of initial dye concentration on the sorption capacity of MNPs is reported in Fig. 9. It was observed that the sorption capacity and the % of Rhd 6 g dye sequestered to MNPs improved and decreased with intensifying Rhd 6G dye concentrations (40–283 mg.g−1 and 97–85%). This was ascribed to the enhanced driving force due to the mass transfer that happens along with the variation from the solution phase to the solid phase or the capability of the dye molecules to be dispersed faster to the sorbent surface as the dye concentration improved. At low Rhd 6G dye concentrations, the accessible surface area or active sites were relatively high and hence the increased % removal with increased concentrations. But the ratio of accessible surface area or active site tends to reduce as the initial dye concentration is increased, and could be attributed to the active sites’ saturation at elevated dye concentrations, hence decreased % removal of Rhd 6 g dye molecules [46, 71, 72]. At higher initial dye concentration, there was an increased chance of dye molecules colliding with the sorption sites on the sorbent surface, thereby leading to increased sorption capacity. Furthermore, the driving force for the mass transfer of dye to the sorption sites on the sorbent surface was improved at elevated initial dye concentrations, which is favourable to decreasing the mass transfer resistance and improving the sorption capacity. The possibility of dye molecules in the unit volume was greater under improved concentration [73,74,75,76,77].

Impact of initial dye concentrations on Rhd 6 g dye adsorption (Conditions: sorbent dosage-0.06 g, pH-10, time of interaction-120 min, and stirring speed-200 rpm)
3.5 Effect of Time and Kinetic Studies
A critical parameter for the treatment of system design is the sorption rate [11]. The sorption kinetics varies with the interface of the solid–liquid interaction and the process of diffusion [45]. The sorption kinetics studies of Rhd 6G dye to MNPs were explored at two different preliminary concentrations (50 and 100 mg.L−1), and the results are presented in Fig. 10. There was rapid sorption of Rhd 6G dye to the MNPs as time progressed (20 min), with sorption equilibrium obtained in 40 min for both concentrations. The time needed to achieve sorption equilibrium is noted to be contrary relative to the preliminary dye concentration. Hence, the solution with a lesser concentration will attain equilibrium than that with a comparatively elevated concentration, owing to the presence of sturdy sorption driving forces in solutions with a lesser concentration. The decreased sorption after 40 min was suggestive of an absence of accessible active sites for the sorption of dye molecules after achieving equilibrium (Fig. 10a) [45, 78]. To study the rate of sorption and understand the mechanism of sorption involved in the sorption process of Rhd 6G dye to the MNPs, the PFOM, pseudo-second-order model (PSOM) and intra-particles diffusion (INT) models were explored using their non-linear and linear forms as given by Eqs. 13–17, and their fitting results are depicted in Fig. 10b-e. The determined parameters of the various linear and non-linear kinetic models are also depicted in Tables 1 and 2.
k1, k2, KINT and C signify the rate constants of PFOM and PSOM models, INT rate constant (mg.g−1.min0.5) and the boundary layer thickness respectively [79]. Comparing the R2 of the PFOM, PSOM and INT, it was observed that the PSOM model (R2 = 0.9999 and 0.9999) best depicted the sorption of Rhd 6G dye (50 and 100 mg.L−1) to the MNPs using the linear fitting (LFG) in comparison to the PFOM and INT models (Table 1).

a Impact of time on the adsorption of Rhd 6G dye, b PFOM, c PSOM, d INT models and e NL simulations using the various models
The kinetic model that best described the sorption process was determined by the highest values of the coefficient of regression (R2) and lowest values of X2 and RSS. A high value of X2 and RSS was suggestive of a high bias between the calculated and experimental model values [43, 43]. The ideal model for the sorption of Rhd 6 g dye was assessed using the NLFG of the models, and the ideal model was one with the highest values of R2 and the lowest values of X2 and RSS values. The parameters determined from the non-linear fitting (NLFG) of all models showed that the PFOM model best described the sorption process of dye molecules of Rhd 6G dye, which was closely trailed by the PSOM model and the INT model, with the calculated parameters of R2, X2 and RSS determined to be 0.9999/0.9999, 0.0178/0.0090 and 0.0711/0.0359 (PFOM), 0.9583/0.9567, 1.8883/6.8242 and 1884.5328/6810.5555 (PSOM) and 0.5201/0.5032, 21.7748/78.3399 and 21,704.6532/78183.2168 (INT) for 50 and 100 mg.L−1 concentrations of Rhd 6G dye respectively (Table 2). The values of qe (mg.g−1) of the PFOM and PSOM models for the LFG and NLFG were relatively close to the experimental values. The plots of the INT model of the sorption process of Rhd 6G dye were not rate controlling and diffused through the liquid film of the MNPs, as the straight line did not pass through the origin of the plot, and the R2 values were relatively poor. Hence, the sorption of Rhd 6G dye to MNPs was centred on physisorption, which further collaborates with the findings from the study of Hashem et al., [45].
3.6 Isotherm Studies
The sorption isotherm signifies the sorbent interaction with the sorbate at a fixed temperature and the viability of various sorption processes to sorb certain sorbate. It also shows the sorption molecules’ distribution between the solid and liquid phases when the equilibrium state is achieved [45]. Sorption isotherm guarantees that various basic physicochemical data assess the sorption processes application. The sorption isotherm is considered by particular coefficients whose values specify the surface features and sorbent affinity and determine the sorptive capacity of a mass [69, 71]. In this study, the Langmuir (LNR), FRH and Temkin (TKN) models were employed to quantify the sorption process taking place using MNPs for the removal of Rhd 6G dye.
The LNR model envisages the presence of monolayer coverage of the sorbate molecules over a consistent sorbent surface with no critical interface among the sorbed species. This model adopts that the surface of the sorbent comprises of only a binding site, hence the sorption energy is continuous. The non-linear and linear forms of this model are given by Eqs. 18 and 19.
KL and qm refer to the LNR constant associated with the sorption capacity (mg.g−1) and the maximum sorption capacity (qm) (L.g−1) [69].
The FRH model adopts a diverse sorption surface with active sites having various energy measures grounded on multi-layer sorption. The model’s non-linear and linear forms are described by Eqs. 20 and 21.
kf and 1/n refer to the sorption capacity in ((mg.g−1)(L/mg)1/n) and the sorption intensity, which indicates the relative distribution of energy and sorbate site diversity [80].
While the TKN model presumes that the sorption heat of all molecules in the sorbed layers reduces linearly with contact from the sorbent/sorbate interaction [6]. The model’s NLFG and LFG are presented by Eqs. 22 and 23.
B (RT/b) and KT relate to the TKN constant, which is associated with the sorption heat (J/mol) and TNK constant (L/mg) [81,82,83].
To understand the mechanism of sorption involved in the Rhd 6G dye sorption to MNPs, the LNR, FRH and TNK models were explored using their linear and non-linear forms, and their fitting results are depicted in Fig. 11. Tables 3 and 4 show the determined parameters of the LFG and NLFG of the various isotherm models, with their equivalent R2, X2 and RSS describing the best perfect model that describes the sorption of Rhd 6G dye to MNPs. It was observed from the calculated parameters of the LFG and NLFG of all models (highest and lowest values of R2 and X2 and RSS) that the sorption of Rhd 6G dye was best described by the FRH model (0.9724, 0.9844, 156.8812 and 470.6434) in comparison to the LNR and TKN models. The obtained optimum sorption capacity from the LNR model was 400/454 mg.g−1, and the dimensionless constant (RL) was determined to be 0.0591–0.3345/0.0828–0.4193 (LFG/NLFG), which was in the array of 0 and 1, which was suggestive of a positive sorption process. There was a considerable difference between the calculated and experimental sorption capacity, which could be due to the absence of attraction or repulsion between the sorbent and sorbate in the case of the LNR assumption, hence causing a high sorption capacity. On the other hand, the experimental sorption capacity was impacted by the electrostatic interaction and possibly repulsion between the sorbate-sorbate and sorbent-sorbent, hence restraining the interaction between the dye molecules and MNPs and causing a reduction in the sorption capacity [36]. The n values from both fittings were 1.7970 and 1.7429, which were greater than 1, suggesting the suitability of the MNPs for the sorption of Rhd 6G dye. The sorption of Rhd 6G dye to MNPs shows sorption with multilayer sorption on a distinct surface with various active sites and energy distribution using the MNPs. The sorption mechanism is attributed to the electrostatic interaction between the cationic Rhd 6G dye and negatively charged sites on the sorbents as well as hydrogen bonding attraction between the molecules of Rhd 6G dye and the surface of the MNPs or the surface-to-dye molecules pore diffusion [84]. Table 5 compares the sorption capacity of MNPs with other magnetic sorbents employed for the sequestration of Rhd 6G dye. The table shows that the sorption capacity of the MNPs was comparative with that of other sorbents employed for Rhd 6G dyes confiscation.

a LNR, b FRH, c TKN models and d NL simulation of the various models
3.7 Thermodynamics
Temperature variation has a key impact on the rate of sorption as it improves the sorbent molecules’ diffusion rate via the exterior layer and the sorbent’s inner pores, in recognition of the reduction in the solution viscosity. Also, the tendency of sorbent deagglomeration for a certain sorbate is increased due to temperature variations [63]. To ascertain the viability, spontaneity and heat transformation of the sorption process, thermodynamic factors like change in the Gibbs free energy (\(\Delta \mathrm{G}^\circ\)), enthalpy (\(\Delta \mathrm{H}^\circ\)) and entropy (\(\Delta \mathrm{S}^\circ\)) were used [69]. For substantial sorption to take place, the ∆G of the sorption process must be negative and it is defined by Eq. 24.
R, T and KD signify the gas constant (8.314*10–3 kJ.mol−1 K−1), the absolute temperature and the equilibrium constant which is given by \({K}_{D}=\frac{{q}_{e}}{{C}_{e}}\).
The \(\Delta \mathrm{H}\)° and \(\Delta \mathrm{S}\)° were assessed using the following association (Eqs. 25 and 26). Their values are estimated from the intercept and slope of the plot lnKD versus 1/T as observed in Fig. 12 [71].

Van’t Hoff plot for the sorption of Rhd 6G dye to MNPs
According to Table 6, the negative values of ∆G \(^\circ\) show that the sorption of Rhd 6G dye to the MNPs was spontaneous and reasonable thermodynamically. The decrease in the value of ∆G° from -3.88 to -4.14 as the temperature increased shows that the sorption process was favourable at elevated temperatures. While the positive changes in the \(\Delta \mathrm{H}^\circ\) (1.58 kJ/mol) and \(\Delta \mathrm{S}^\circ\) (13.03 kJ/mol.K) states show that the sorption of Rhd 6G dye to MNPs was endothermic and growing randomness at the interface between the MNPs and the water-soluble solution. \(\Delta \mathrm{H}^\circ\) value smaller than 40 kJ/mol indicated a physisorption process and endothermic reaction [70, 91]. Hence, the magnitude of \(\Delta \mathrm{H}^\circ\) shows that the sorption type was physical sorption, as the equilibrium was generally swiftly achieved and was effortlessly reversible owing to the energy requisite being low [69].
3.8 Adsorption–Desorption Studies
The desorption research is performed to restore the expended sorbent to its prior feature. In large-scale practice, the restoration process plays a key part in the viable treatment process thereby reducing the volume of resulting waste [92]. The desorption process was carried out to desorb Rhd 6 g dye molecules from the exhausted sorbent using 0.1 M HCl and NaOH. After four cycles of the adsorption–desorption process (Fig. 13), the study shows that HCl was effective in desorbing the dye molecules from the sorbent surface. By utilizing acidic eluent as a desorbing facilitator, the sorbent surface gets more H+ ions from the solution, thereby leading to a cationic exchange occurring more instinctively, and resulting in the release of Rhd 6 g dye from the sorbent surface. The adsorption–desorption process shows a reduced elimination of Rhd 6 g dye as the cycle is repeated, with a drastic reduction in the % of Rhd 6 g dye eliminated noticed at the fourth cycle. This was ascribed to the loss of NPs after each adsorption–desorption cycle or the possibility of HCl acid destroying the structure of MNPs when used as an eluent for the desorption process [93].

Adsorption–desorption cycle using MNPs for Rhd 6 g confiscation
4 Conclusion
In treating industrial effluents, the sorption process is a well-thought-out promising technique using especially MNPs created with the support of plant extracts. In this study, VA leaf extract was employed as a reducing and stabilising agent in the fabrication of MNPs. This was then used to confiscate Rhd 6G dye molecules owing to the metabolites found in VA leaf extract. The Rhd 6G sequestration to the MNPs hugely depended on the solution pH, with optimum confiscation of the dye molecules at pH 11. The sorption process of Rhd 6G dye to MNPs was ideally described by employing the FRH and PFOM models, with the optimum sorption capacity established to be 454 mg.g−1. The thermodynamic process was spontaneous, endothermic, and with increased randomness for Rhd 6G confiscation. The sorption mechanism was attributed to the electrostatic interaction between the cationic dye and negatively charged surface sites on the MNPs, as well as the hydrogen bonding between the dye molecules and the MNPs’ surface sites. Also, the Rhd 6 g dye molecules effectively desorbed from the spent sorbent using 0.1 M HCl for four adsorption–desorption cycles. Hence, the results from this study show that MNPs synthesized with the support of VA leaf extract (bitter leaf) via the green synthesis route were effective for confiscating Rhd 6G dye and can be harnessed and employed for the treatment of industrial wastewater containing dye.
Data Availability
Data sharing does not apply to this article.
References
A. Alswieleh, Efficient removal of dyes from aqueous solution by adsorption on L-Arginine-modified mesoporous silica nanoparticles. Processes 10(6), 1079 (2022)
M. Kanani, N. Kanani, N. Batooie, A. Bozorgian, A. Barghi and S. Rezania, "Removal of rhodamine 6G dye using one-pot synthesis of magnetic manganese graphene oxide: Optimization by response surface methodology," Environmental Nanotechnology, Monitoring & Management, p. 100709, 2022.
C. Osagie, A. Othmani, S. Ghosh, A. Malloum, Z. Esfahani, S. Ahmadi, Dyes adsorption from aqueous media through the nanotechnology: A review. J. Market. Res. 14, 2195–2218 (2021)
P. Abdullah, L. Wen, H. Awang, S. Azmin, Rhodamine 6G removal from aqueous solution with coconut shell-derived nanomagnetic adsorbent composite (Cs-nmac): Isotherm and kinetic studies. Pertanika Journal of Science and Technology 29(3), 1535–1556 (2021)
Y. Chao, J. Pang, Y. Bai, P. Wu, J. Luo, J. He, Y. Jin, X. Li, J. Xiong, H. Li, W. Zhu, Graphene-like BN@ SiO2 nanocomposites as efficient sorbents for solid-phase extraction of Rhodamine B and Rhodamine 6G from food samples. Food Chem. 320, 126666 (2020)
Y. Wang, X. Wang, W. Ma, R. Lu, W. Zhou, H. Gao, Recent developments in rhodamine-based chemosensors: A review of the years 2018–2022. Chemosensors 10(10), 399 (2022)
U. Aigbe, K. Ukhurebor, R. Onyancha, O. Osibote, H. Darmokoesoemo and H. Kusuma, "Fly ash-based adsorbent for adsorption of heavy metals and dyes from aqueous solution: A review," Journal of Materials Research and Technology, 2021.
S. Phoemphoonthanyakit, P. Seeharaj, P. Damrongsak and K. Locharoenrat, "Effect of adsorption characteristics of rhodamine 6G dye solution in Fe3O4 magnetic nanoparticles on fluorescence quantum yield," Journal of Spectroscopy, vol. 2019, 2019.
A. Chandrashekar, S. Vargheese, J. Vijayan, J. Gopi and T. Prabhu, "Highly efficient removal of Rhodamine B dye using nanocomposites made from cotton seed oil-based polyurethane and silylated nanocellulose," Journal of Polymers and the Environment, pp. 1–13, 2022.
S. Low, M. Tan, N. Chin, Effect of ultrasound pre-treatment on adsorbent in dye adsorption compared with ultrasound simultaneous adsorption. Ultrason. Sonochem. 48, 64–70 (2018)
T. Kamilya, S. Mondal, R. Saha, Effect of magnetic field on the removal of copper from aqueous solution using activated carbon derived from rice husk. Environ. Sci. Pollut. Res. 29(14), 20017–20034 (2022)
N. Awoke, D. Pandey, A. Habtemariam, Synthesis of tin (IV) oxide nanoparticles using plant leaf extracts of Vernonia amygdalina and Mentha spicata. Regenerative Engineering and Translational Medicine 8(3), 407–412 (2022)
K. Pal, S. Chakroborty, N. Nath, Limitations of nanomaterials insights in green chemistry sustainable route: Review on novel applications. Green Processing and Synthesis 11(1), 951–964 (2022)
G. Weldegebrieal, A. Sibhatu, Photocatalytic activity of biosynthesized α-Fe2O3 nanoparticles for the degradation of methylene blue and methyl orange dyes. Optik 241, 167226 (2021)
S. Syarifah, C. Imawan, W. Handayani and D. Djuhana, "Biosynthesis of ferric oxide nanoparticles using Pometia pinnata JR Frost. & G. Forst. leaves water extract," in AIP Conference Proceedings, AIP Publishing, 2018.
M. Gupta, R. Tomar, S. Kaushik, R. Mishra, D. Sharma, Effective antimicrobial activity of green ZnO nano particles of Catharanthus roseus. Front. Microbiol. 9, 2030 (2018)
A. Dash, M. Ahmed, R. Selvaraj, Mesoporous magnetite nanoparticles synthesis using the Peltophorum pterocarpum pod extract, their antibacterial efficacy against pathogens and ability to remove a pollutant dye. J. Mol. Struct. 1178, 268–273 (2019)
A. Samrot, P. Senthilkumar, S. Rashmitha, P. Veera, C. Sahithya, Azadirachta indica influenced biosynthesis of super-paramagnetic iron-oxide nanoparticles and their applications in tannery water treatment and X-ray imaging. Journal of Nanostructure in Chemistry 8(3), 343–351 (2018)
M. Harja, N. Lupu, H. Chiriac, D. Herea, G. Buema, Studies on the removal of congo red dye by an adsorbent based on fly-ash@ Fe3O4 mixture. Magnetochemistry 8(10), 125 (2022)
S. Shukla, R. Khan, A. Daverey, Synthesis and characterization of magnetic nanoparticles, and their applications in wastewater treatment: A review. Environ. Technol. Innov. 24, 101924 (2021)
S. Karunakaran, S. Ramanujam, B. Gurunathan, Green synthesised iron and iron-based nanoparticle in environmental and biomedical application: A review. IET Nanobiotechnol. 12(8), 1003–1008 (2018)
P. Khandel, R. Yadaw, D. Soni, L. Kanwar, S. Shahi, Biogenesis of metal nanoparticles and their pharmacological applications: present status and application prospects. Journal of Nanostructure in Chemistry 8(3), 217–254 (2018)
C. Sangode, S. Mahant, P. Tidke, M. Umekar, R. Lohiya, Green synthesized of novel iron nanoparticles as promising antimicrobial agent: A review. GSC Biological and Pharmaceutical Sciences 15(2), 117–127 (2021)
C. Hano, B. Abbasi, Plant-based green synthesis of nanoparticles: Production, characterization and applications. Biomolecules 12(1), 31 (2021)
K. Sirdeshpande, A. Sridhar, K. Cholkar, R. Selvaraj, Structural characterization of mesoporous magnetite nanoparticles synthesized using the leaf extract of Calliandra haematocephala and their photocatalytic degradation of malachite green dye. Appl. Nanosci. 8(4), 675–683 (2018)
R. Selvaraj, G. Murugesan, G. Rangasamy, R. Bhole, N. Dave, S. Pai, K. Balakrishna, R. Vinayagam, T. Varadavenkatesan, As (III) removal using superparamagnetic magnetite nanoparticles synthesized using Ulva prolifera−optimization, isotherm, kinetic and equilibrium studies. Chemosphere 308, 136271 (2022)
N. Elizondo-Villarreal, L. Verástegui-Domínguez, R. Rodríguez-Batista, E. Gándara-Martínez, A. Alcorta-García, D. Martínez-Delgado, E. Rodríguez-Castellanos, F. Vázquez-Rodríguez, C. Gómez-Rodríguez, Green synthesis of magnetic nanoparticles of iron oxide using aqueous extracts of lemon peel waste and its application in anti-corrosive coatings. Materials 15(23), 8328 (2022)
R. Vinayagam, C. Zhou, S. Pai, T. Varadavenkatesan, M. Narasimhan, S. Narayanasamy, R. Selvaraj, Structural characterization of green synthesized magnetic mesoporous Fe3O4NPs@ME. Mater. Chem. Phys. 262, 124323 (2021)
C. Das, S. Sen, T. Singh, T. Ghosh, S. Paul, T. Kim, S. Jeon, D. Maiti, J. Im and G. Biswas, "Green synthesis, characterization and application of natural product coated magnetite nanoparticles for wastewater treatment," Nanomaterials, 10(8), p.1615., vol. 10, no. 8, p. 1615, 2020.
M. Sivakami, K. Renuka Devi and R. Renuka, "Phytomediated synthesis of magnetic nanoparticles by Murraya koenigii leaves extract and its biomedical applications," Applied Physics A, vol. 128, no. 4, pp. 1–13, 2022.
K. Parajuli, A. Sah, H. Paudyal, Green synthesis of magnetite nanoparticles using aqueous leaves extracts of Azadirachta indica and its application for the removal of As (V) from water. Green and Sustainable Chemistry 10(4), 117–132 (2020)
S. Mohammadi, B. Lashkari, A. Khosravan, S. Fouladi, Synthesis of a new magnetic adsorbent using green tea leaf extract and its application in phenol removal by RSM method. J. Mater. Sci.: Mater. Electron. 33(14), 11212–11226 (2022)
A. Ramesh, K. Pal, A. Kodandaram, B. Manjula, D. Ravishankar, H. Gowtham, M. Murali, A. Rahdar, G. Kyzas, Antioxidant and photocatalytic properties of zinc oxide nanoparticles phyto-fabricated using the aqueous leaf extract of Sida acuta. Green Processing and Synthesis 11(1), 857–867 (2022)
E. Daniel, M. Ebeagwu, P. Okafor, W. Emori, I. Udoh, Exploring the efficacy of phytoconstituents from vernonia amygdalina on mild steel protection in acid environment: Combined experimental and theoretical study. Journal of Bio-and Tribo-Corrosion 7(3), 1–15 (2021)
I. Oyeyemi, A. Akinlabi, A. Adewumi, A. Aleshinloye, O. Oyeyemi, Vernonia amygdalina: A folkloric herb with anthelminthic properties. Beni-suef University Journal of Basic and Applied Sciences 7(1), 43–49 (2018)
D. Zewde, B. Geremew, Removal of Congo red using Vernonia amygdalina leaf powder: optimization, isotherms, kinetics, and thermodynamics studies. Environmental Pollutants and Bioavailability 34(1), 88–101 (2022)
A. Sebastian, A. Nangia, M. Prasad, Cadmium and sodium adsorption properties of magnetite nanoparticles synthesized from Hevea brasiliensis Muell. Arg. bark: Relevance in amelioration of metal stress in rice. J. Hazard. Mater. 371, 261–272 (2019)
A. Habtemariam, Biosynthesis of magnetite (Fe3O4) nanostructures using Vernonia amygdalina leaves extract. Lett. Appl. Nanobiosci 10, 2777–2783 (2021)
J. Joseph, K. Khor, E. Moses, V. Lim, M. Aziz, N. Samad, In vitro anticancer effects of Vernonia amygdalina leaf extract and green-synthesised silver nanoparticles. Int. J. Nanomed. 16, 3599–3612 (2021)
P. Dhar, P. Saha, M. Hasan, M. Amin, M. Haque, Green synthesis of magnetite nanoparticles using Lathyrus sativus peel extract and evaluation of their catalytic activity. Cleaner Engineering and Technology 3, 100117 (2021)
A. Prasad, T. Santhi, S. Manonmani, Recent developments in preparation of activated carbons by microwave: Study of residual errors. Arab. J. Chem. 8(3), 343–354 (2015)
S. Dubey, D. Gusain, Y. Sharma, Kinetic and isotherm parameter determination for the removal of chromium from aqueous solutions by nanoalumina, a nanoadsorbent. J. Mol. Liq. 219, 1–8 (2016)
E. Jasper, V. Ajibola, J. Onwuka, Nonlinear regression analysis of the sorption of crystal violet and methylene blue from aqueous solutions onto an agro-waste derived activated carbon. Appl Water Sci 10(6), 1–11 (2020)
H. Hami, "An overview of using error function in adsorption isotherm modeling," Muthanna Journal of Pure Science (MJPS), vol. 8, no. 1, 2021.
A. Hashem, C. Aniagor, O. Morsy, A. Abou-Okeil and A. Aly, "Apricot seed shell: an agro-waste biosorbent for acid blue193 dye adsorption," Biomass Conversion and Biorefinery, pp. 1–14, 2022.
J. Jabar, M. Adebayo, I. Owokotomo, Y. Odusote, M. Yılmaz, Synthesis of high surface area mesoporous ZnCl2–activated cocoa (Theobroma cacao L) leaves biochar derived via pyrolysis for crystal violet dye removal. Heliyon 8(10), e101873 (2022)
R. Bashir, Y. Mukhtar, I. Chimbekujwo, D. Aisha, S. Fatima, S. Salamatu, Phytochemical screening and fourier transform infrared spectroscopy (FT-IR) analysis of Vernonia amygdalina Del. (Bitter leaf) methanol leaf extract. FUTY Journal of the Environment 14(2), 35–41 (2020)
O. Alara, N. Abdurahman, S. Mudalip, O. Olalere, Characterization and effect of extraction solvents on the yield and total phenolic content from Vernonia amygdalina leaves. Journal of Food Measurement and Characterization 12(1), 311–316 (2018)
O. Alara, N. Abdurahman, C. Ukaegbu, N. Kabbashi, Extraction and characterization of bioactive compounds in Vernonia amygdalina leaf ethanolic extract comparing Soxhlet and microwave-assisted extraction techniques. Journal of Taibah University for Science 13(1), 414–422 (2019)
A. Widyaningtyas, Y. Yulizar and D. Apriandanu, "Ag2O nanoparticles fabrication by Vernonia amygdalina Del. leaf extract: synthesis, characterization, and its photocatalytic activities," in IOP Conference Series: Materials Science and Engineering, 2019.
Y. Ch’ng, Y. Loh, C. Tan, M. Ahmad, M. Asmawi, W. Wan Omar and M. Yam, "Vasorelaxant properties of Vernonia amygdalina ethanol extract and its possible mechanism," Pharmaceutical Biology, vol. 55, no. 1, pp. 2083–2094, 2017.
S. Hussein-Al-Ali, S. Abudoleh, Q. Abdallah Abualassal, Z. Abudayeh, Y. Aldalahmeh and M. Hussein, "Arginine and chitosan modified magnetic nanoparticles in ciprofloxacin delivery: A comparative, characterization and in vitro release study," Materials Express, vol. 12, no. 2, pp. 241–254, 2022.
M. Manzo, H. Ahmed, S. Nasrazadani, Study on emission spectral lines of hematite and magnetite for purity’s differentiation. AIP Adv. 10(10), 105327 (2020)
R. Selvaraj, S. Pai, G. Murugesan, S. Pandey, R. Bhole, D. Gonsalves, T. Varadavenkatesan, R. Vinayagam, Green synthesis of magnetic α–Fe2O3 nanospheres using Bridelia retusa leaf extract for Fenton-like degradation of crystal violet dye. Appl. Nanosci. 11(8), 2227–2234 (2021)
N. Kobylinska, D. Klymchuk, A. Shakhovsky, O. Khainakova, Y. Ratushnyak, V. Duplij, N. Matvieieva, Biosynthesis of magnetite and cobalt ferrite nanoparticles using extracts of “hairy” roots: preparation, characterization, estimation for environmental remediation and biological application. RSC Adv. 11(43), 26974–26987 (2021)
G. Sulaiman, A. Tawfeeq, A. Naji, Biosynthesis, characterization of magnetic iron oxide nanoparticles and evaluations of the cytotoxicity and DNA damage of human breast carcinoma cell lines. Artificial Cells, Nanomedicine, and Biotechnology 46(6), 1215–1229 (2018)
S. Venkateswarlu, B. Kumar, C. Prasad, P. Venkateswarlu, N. Jyothi, Bio-inspired green synthesis of Fe3O4 spherical magnetic nanoparticles using Syzygium cumini seed extract. Physica B 449, 67–71 (2014)
J. Emegha, K. Ukhurebor, U. Aigbe, J. Damisa and A. Babalola, "Synthesis and characterization of copper zinc iron sulphide (CZFS) thin films," Heliyon, p. e10331, 2022.
M. Singh, M. Goyal, K. Devlal, Size and shape effects on the band gap of semiconductor compound nanomaterials. Journal of Taibah University for Science 12(4), 470–475 (2018)
D. Idisi, E. Benecha, S. Moloi, S. Ray, Effects of gold nanoparticles (Au-NPs) on the electrical properties of reduced graphene oxide: An experimental and DFT study. J. Mater. Res. 37(5), 1037–1046 (2022)
S. Ethaib, S. Al-Qutaifia, N. Al-Ansari, S. Zubaidi, Function of nanomaterials in removing heavy metals for water and wastewater remediation: A review. Environments 9(10), 123 (2022)
B. Vojnović, M. Cetina, P. Franjković, A. Sutlović, Influence of Initial pH value on the adsorption of reactive black 5 dye on powdered activated carbon: Kinetics, mechanisms, and thermodynamics. Molecules 27(4), 1349 (2022)
A. Badawi, M. Abd Elkodous and G. Ali, "Recent advances in dye and metal ion removal using efficient adsorbents and novel nano-based materials: An overview," RSC Advances, vol. 11, no. 58, pp. 36528–36553, 2021.
N. Kumar, H. Mittal, V. Parashar, S. Ray and J. Ngila, "Efficient removal of rhodamine 6G dye from aqueous solution using nickel sulphide incorporated polyacrylamide grafted gum karaya bionanocomposite hydrogel," RSC Advances, vol. 6, no. 26, 2016.
M. El-Nemr, U. Aigbe, K. Ukhurebor, R. Onyancha, A. El Nemr, S. Ragab, O. Osibote and M. Hassaan, "Adsorption of Cr6+ ion using activated Pisum sativum peels-triethylenetetramine," Environmental Science and Pollution Research, pp. 1–25, 2022.
T. Eldeeb, U. Aigbe, K. Ukhurebor, R. Onyancha, M. El-Nemr, M. Hassaan, O. Osibote, S. Ragab, B. Okundaye, V. Balogun and A. El Nemr, "Biosorption of acid brown 14 dye to mandarin-CO-TETA derived from mandarin peels," Biomass Conversion and Biorefinery, pp. 1–21, 2022.
A. da Silva, N. Serrão, G. de Gusmão Celestino, M. Takeno, N. Antunes, S. Iglauer, L. Manzato, F. de Freitas and P. Maia, "Removal of rhodamine 6G from synthetic effluents using Clitoria fairchildiana pods as low-cost biosorbent," Environmental Science and Pollution Research, vol. 27, no. 3, pp. 2868–2880, 2020.
A. Inyinbor, F. Adekola, G. Olatunji, Kinetics, isotherms and thermodynamic modeling of liquid phase adsorption of Rhodamine B dye onto Raphia hookerie fruit epicarp. Water Resources and Industry 15, 14–27 (2016)
H. Senturk, D. Ozdes, C. Duran, Biosorption of Rhodamine 6G from aqueous solutions onto almond shell (Prunus dulcis) as a low cost biosorbent. Desalination 252(1–3), 81–87 (2010)
R. Vinayagam, G. Murugesan, T. Varadavenkatesan, R. Bhole, L. Goveas, A. Samanth, M. Ahmed and R. Selvaraj, "Algal biomass-derived nano-activated carbon for the rapid removal of tetracycline by adsorption: Experimentation and adaptive neuro-fuzzy inference system modeling," Bioresource Technology Reports, p. 101291, 2022.
M. Keshavarz, R. Foroutan, F. Papari, L. Bulgariu, H. Esmaeili, Synthesis of CaO/Fe2O3 nanocomposite as an efficient nanoadsorbent for the treatment of wastewater containing Cr (III). Sep. Sci. Technol. 56(8), 1328–1341 (2021)
L. Maknun and F. Fajaroh, "Removal methylene blue from aqueous solution using silica aerogel prepared from bagasse ash," in IOP Conference Series: Earth and Environmental Science, 2019.
J. Di, Z. Ruan, S. Zhang, Y. Dong, S. Fu, H. Li, G. Jiang, Adsorption behaviors and mechanisms of Cu2+, Zn2+ and Pb2+ by magnetically modified lignite. Sci. Rep. 12(1), 1394 (2022)
F. Gorzin and M. Bahri Rasht Abadi, "Adsorption of Cr (VI) from aqueous solution by adsorbent prepared from paper mill sludge: Kinetics and thermodynamics studies," Adsorption Science & Technology, vol. 36, no. 1–2, pp. 149–169, 2018.
H. DOLAS, "Activated carbon synthesis and methylene blue adsorption from pepper stem using microwave assisted impregnation method: Isotherm and kinetics," Journal of King Saud University-Science, p. 102559, 2023.
A. Bayu, T. Abeto Amibo and S. Beyan, "Adsorptive capacity of calcinated hen eggshell blended with silica gel for removal of lead II ions from aqueous media: kinetics and equilibrium studies," Journal of Environmental and Public Health, vol. 2022, 2022.
R. Kayiwa, H. Kasedde, M. Lubwama, J. Kirabira, Active pharmaceutical ingredients sequestrated from water using novel mesoporous activated carbon optimally prepared from cassava peels. Water 14(21), 3371 (2022)
U. Jinendra, D. Bilehal, B. Nagabhushana, A. Kumar, Adsorptive removal of Rhodamine B dye from aqueous solution by using graphene–based nickel nanocomposite. Heliyon 7(4), e06851 (2021)
M. Motitswe, K. Badmus, L. Khotseng, Development of adsorptive materials for selective removal of toxic metals in wastewater: A review. Catalysts 12(9), 1057 (2022)
K. Shen, M. Gondal, Removal of hazardous Rhodamine dye from water by adsorption onto exhausted coffee ground. J. Saudi Chem. Soc. 21, S120–S127 (2017)
K. Yu, X. Lee, H. Ong, W. Chen, J. Chang, C. Lin, P. Show, T. Ling, Adsorptive removal of cationic methylene blue and anionic Congo red dyes using wet-torrefied microalgal biochar: Equilibrium, kinetic and mechanism modeling. Environ. Pollut. 272, 115986 (2021)
N. Ayawei, A. Ebelegi and D. Wankasi, "Modelling and interpretation of adsorption isotherms," Journal of Chemistry, 2017.
A. Benmessaoud, D. Nibou, E. Mekatel, S. Amokrane, A comparative study of the linear and non-linear methods for determination of the optimum equilibrium isotherm for adsorption of Pb2+ ions onto Algerian treated clay. Journal of Chemistry and Chemical Engineering (IJCCE) 39(4), 153–171 (2020)
T. Eldeeb, U. Aigbe, K. Ukhurebor, R. Onyancha, M. El-Nemr, M. Hassaan, S. Ragab, O. Osibote and A. El Nemr, "Adsorption of methylene blue (MB) dye on ozone, purified and sonicated sawdust biochars," Biomass Conversion and Biorefinery, pp. 1–23, 2022.
E. Ranjbari, M. Hadjmohammadi, F. Kiekens, K. De Wael, Mixed hemi/Ad-micelle sodium dodecyl sulfate-coated magnetic iron oxide nanoparticles for the efficient removal and trace determination of rhodamine-B and rhodamine-6G. Anal. Chem. 87(15), 7894–7901 (2015)
Y. Chang, C. Ren, Q. Yang, Z. Zhang, L. Dong, X. Chen and D. Xue, "Preparation and characterization of hexadecyl functionalized magnetic silica nanoparticles and its application in Rhodamine 6G removal," Applied Surface Science, vol. 257, 2011.
T. Suwunwong, P. Patho, P. Choto, K. Phoungthong, Enhancement the rhodamine 6G adsorption property on Fe3O4-composited biochar derived from rice husk. Materials Research Express 7(2), 025511 (2020)
F. Gomri, G. Finqueneisel, T. Zimny, S. Korili, A. Gil, M. Boutahala, Adsorption of Rhodamine 6G and humic acids on composite bentonite–alginate in single and binary systems. Appl Water Sci 8(6), 1–10 (2018)
M. Ajmal, M. Siddiq, N. Aktas, N. Sahiner, Magnetic Co–Fe bimetallic nanoparticle containing modifiable microgels for the removal of heavy metal ions, organic dyes and herbicides from aqueous media. RSC Adv. 5(54), 43873–43884 (2015)
A. Gollakota, V. Munagapati, K. Shadangi, G. Reddy, J. Wen, C. Shu, Encapsulating toxic Rhodamine 6G dye, and Cr (VI) metal ions from liquid phase using AlPO4-5 molecular sieves. Preparation, characterization, and adsorption parameters. J. Mol. Liq. 336, 116549 (2021)
A. Hidayat, D. Sulistiono, I. Murwani, B. Endrawati, H. Fansuri, L. Zulfa, R. Ediati, Linear and nonlinear isotherm, kinetic and thermodynamic behavior of methyl orange adsorption using modulated Al2O3@ UiO-66 via acetic acid. J. Environ. Chem. Eng. 9(6), 106675 (2021)
S. Sonal, P. Prakash, B. Mishra, G. Nayak, Synthesis, characterization and sorption studies of a zirconium (iv) impregnated highly functionalized mesoporous activated carbons. RSC Adv. 10(23), 13783–13798 (2020)
L. Dong, C. Shan, Y. Liu, H. Sun, B. Yao, G. Gong, X. Jin, S. Wang, Characterization and mechanistic study of heavy metal adsorption by facile synthesized magnetic xanthate-modified chitosan/polyacrylic acid hydrogels. Int. J. Environ. Res. Public Health 19(17), 11123 (2022)
Acknowledgements
The authors are grateful to the Cape Peninsula University of Technology for funding this project and to the Centre for Nanomaterials and Science Research, University of Johannesburg, Johannesburg, South Africa; Department of Chemical Sciences (Formerly Applied Chemistry) for the use of their facility and equipment.
Funding
Open access funding provided by Cape Peninsula University of Technology. This research work was funded by the Cape Peninsula University of Technology, South Africa.
Author information
Authors and Affiliations
Contributions
Dr UO Aigbe conducted the experiment, calculation, theoretical models and writing of the original manuscript. Mr Rodyne Maluleke and Dr Thabang C Lebepe assisted with the laboratory work and characterization of the prepared nanoparticles. Prof SO Oluwafemi and Dr OA Osibote supervised the experimental work, reviewed the original manuscript and provided financial support for the experiment work.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Aigbe, U.O., Maluleke, R., Lebepe, T.C. et al. Rhodamine 6G Dye Adsorption Using Magnetic Nanoparticles Synthesized With the Support of Vernonia Amygdalina Leaf Extract (Bitter Leaf). J Inorg Organomet Polym 33, 4012–4031 (2023). https://doi.org/10.1007/s10904-023-02639-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-023-02639-3