
Abstract
Background
As the application of next generation sequencing (NGS) is moving to earlier stages in the diagnostic pipeline for primary immunodeficiencies (PIDs), re-evaluation of its effectiveness is required. The aim of this study is to systematically review the diagnostic yield of NGS in PIDs.
Methods
PubMed and Embase databases were searched for relevant studies. Studies were eligible when describing the use of NGS in patients that had previously been diagnosed with PID on clinical and/or laboratory findings. Relevant data on study characteristics, technological performance and diagnostic yield were extracted.
Results
Fourteen studies were eligible for data extraction. Six studies described patient populations from specific PID subcategories. The remaining studies included patients with unsorted PIDs. The studies were based on populations from Italy, Iran, Turkey, Thailand, the Netherlands, Norway, Saudi Arabia, Sweden, the UK, and the USA. Eight studies used an array-based targeted gene panel, four used WES in combination with a PID filter, and two used both techniques. The mean reported reading depth ranged from 98 to 1337 times. Five studies described the sensitivity of the applied techniques, ranging from 83 to 100%, whereas specificity ranged from 45 to 99.9%. The percentage of patients who were genetically diagnosed ranged from 15 to 79%. Several studies described clinical implications of the genetic findings.
Discussion
NGS has the ability to contribute significantly to the identification of molecular mechanisms in PID patients. The diagnostic yield highly depends on population and on the technical circumstances under which NGS is employed. Further research is needed to determine the exact diagnostic yield and clinical implications of NGS in patients with PID.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Primary immunodeficiencies (PIDs) are a diverse group of congenital diseases affecting different parts of the immune system. Patients usually present with a varying degree of recurrent, unusual or severe infections, autoimmunity, autoinflammation, allergy and/or malignancies [1,2,3]. Identification and clinical diagnosis of the exact type of PID have important consequences in terms of prognosis, treatment, and genetic counseling [4,5,6,7]. However, phenotypic and genotypic heterogeneity, causing atypical presentations and overlap of symptoms between diseases, impedes reaching a definitive molecular diagnosis [8,9,10,11]. The introduction of NGS-based sequencing techniques, facilitating testing of panels of disease-related genes, can overcome these diagnostic difficulties [12]. Currently, over 360 genes involved in immunodeficiencies have been identified and are classified on a yearly basis by the International Union of Immunological Societies (IUIS) [13].
Several DNA sequencing techniques are currently being used to detect disease-causing mutations. Until 2010, in the clinical situation, routine genetic analyses were primarily performed by means of Sanger sequencing [14]. The introduction of next generation sequencing (NGS) has, next to its contribution to the expansion of the list of genes known to cause PIDs, provided a much quicker and now cheaper way to evaluate large portions of the genome [9, 15, 16]. Especially in situations where there is no obvious candidate gene, NGS is preferred to Sanger sequencing [14]. However, array-based targeted gene panels and whole exome sequencing (WES) may have the disadvantage of insufficient coverage of specific regions of the genome thereby creating the possibility of missing mutations. Whole genome sequencing (WGS) provides a more complete picture, with improved identification of CNV’s and other genomic rearrangements, but complicates analysis due to the generation of even larger amounts of data [17]. Moreover, due to the frequent use of short read technologies, limitations such as GC bias, difficulties with mapping to repetitive elements, trouble discriminating paralogous sequences and identification of large indels complicate its use. In some cases, Sanger sequencing therefore remains essential in the confirmation of mutations identified by NGS [14].
Due to technical advances and the continuously decreasing costs of NGS [18], its place in the diagnostic pipeline of PID requires re-evaluation as it is moving to the earlier stages. In order to do so, this review aims to describe the technological performance and diagnostic yield of NGS in PID patients.
Methods
Search Strategy
We performed a systematic review in order to analyze the current literary framework describing the use of NGS in PIDs. We searched the Pubmed and Embase databases for relevant studies in June 2018 using the terms primary immunodeficiency and PID combined with next generation sequencing and related techniques including whole exome sequencing and whole genome sequencing. The full search string is presented in Appendix 1, Table 4. We retrieved 219 studies from Pubmed and 297 from Embase. No further relevant studies were found in the reference lists of the included studies.
Eligibility
We aimed to study the use of NGS in a clinical setting. To this end, we selected all papers that described the use of NGS in patients that had previously been clinically diagnosed with a primary immunodeficiency or were highly suspected of having one according to clinical parameters as described by the authors. Exclusion criteria included prior knowledge of a genetic mutation, and the use of NGS for diagnosing other disease categories than PID according to the IUIS 2017 guidelines. We disregarded studies describing diseases with both PID-non-PID-related causes. We further rejected studies that included less than n = 10 patients, and studies with results that were not written in English. Case series of patients within the same family were also excluded due to a high probability of all patients having the same causative mutation.
Selection of Studies
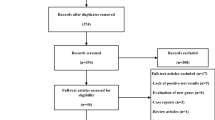
After duplicate removal, all 404 remaining studies were first screened by title and abstract by a first author (HY), followed by a second author (KE). After agreeing on conflicts, 367 studies were excluded. A significant amount consisted of conference abstracts that had not yet been excluded in earlier stages. The 37 studies that remained were assessed by both authors by analysis of the full text. Fourteen studies were determined to be eligible for data extraction. The selection of studies is summarized in Fig. 1.

PRISMA flow diagram
Data Extraction
The following background information was extracted from the eligible papers: year of publication, country, population, number of patients included, and previous genetic analysis technique. Technical variables included the sequencing technique, sequencing platform, number of genes included and the rationale behind it, coverage of base pairs, reading depth, sensitivity, specificity, and additional analyses performed by the researchers in order to ensure completeness. Finally, information regarding the diagnostic yield, rationale behind evaluation of mutations, analyses, and clinical implications were collected.
Critical Appraisal
Most appraisal tools for diagnostic studies were designed for studies that compare a reference standard with an index test, which was not the focus of this article. Therefore, it was decided to use a modified version of the 2015 STARD criteria [19]. Using this list, the included studies were critically assessed for possible bias and completeness of reporting. In short, articles were scored on fourteen items that mainly reflected population background, rationale behind and quality of the analysis and general completeness. If four or more of the included items were found to be missing, completeness of reporting was considered inadequate.
Results
Study Characteristics
The fourteen studies eligible for data extraction are described in Table 1. Eight of these used NGS in a mixed PID population; the other six studies described patients from specific subcategories of PID. The number of included patients in the studies describing unsorted populations ranged from 15 to 278 (median = 41). The six papers focusing on specific PID subcategories included 19–696 patients (median = 38). Eight studies were based on Western patient populations. Others were based on populations from Iran, Turkey, Thailand, and Saudi Arabia. Several studies did not describe clinical characteristics such as gender, specific symptoms, and severity of the phenotypes in the affected patients in detail. Of the studies that did specify these data, seven described patients who received genetic testing prior to NGS, whereas patients from several other studies (Yu et al. and Maffucci et al.) had not [20, 21].
Technical Performance
Of the 14 eligible studies, eight used an array-based targeted gene panel, four used WES combined with a PID filter, and two studies used both techniques. The technical evaluation of these techniques is described in Table 2. Most authors used one of three sequencing platforms: SOLiD (n = 1), ion system (n = 4), or illumina (n = 8), and one study did not specify the used platform. The number of genes included in the analysis ranged from 12 to 365 in the specific PID group and from 60 to 571 in the mixed population group. Not all authors clarified their choice for the selection of the included genes. Reading depth and/or coverage were presented in all papers, except in one study [22]. Complete and in-depth comparison of efficacy of detecting mutations between studies could not be performed due to incomplete presentation of sufficient technical parameters. The studies that did mention these parameters reported a minimum of 88% of base pairs covered at least once. A total of five studies performed additional copy number variant (CNV) analyses on their samples (Table 3), which increased the diagnostic yield by on average 4.2%.
The mean reading depth ranged from 80 to 1337. Five studies described the sensitivity of their technique and reported an overall sensitivity of 83–100%. Nijman et al. made a distinction between SNVs and indels [23], whereas Al-Mousa et al. analyzed SNVs and short indels on the one hand and CNVs on the other [24]. Both found higher sensitivity rates for SNVs, or SNVs and indels respectively. Specificity ranged from 45 to 99.9% with the lowest percentage found for the analysis of CNVs in the study by Al-Mousa et al.
Clinical Performance
All studies explored the diagnostic yield of their NGS analyses, the results of which can be found in Table 3. The percentage of patients who were genetically diagnosed ranged from 15 to 79%. The diagnostic yield of NGS in mixed PID groups ranged from 15 to 46% (median = 25%). Within the specific PID subcategories, these values ranged from 30 to 79% (median = 42%), but were less evenly distributed. All studies described the pipeline used to establish pathogenicity of detected variants, except one. We evaluated information regarding several steps in the process. The results of variants on the amino acid sequence, such as missense-, nonsense-, or splice-site-altering, were collected. Other variables include the reference database to which the variants were compared, variant analyses, and further studies providing lines of evidence regarding the pathogenicity of a specific variant, such as parental cosegregation analysis, functional assays, and genotype-phenotype linkage. Cutoff values for comparison with healthy population databases differed between studies. Nijman et al. and Mukda et al. reported all variants present in < 5% of a healthy cohort [23, 25]. The other studies used a lower cutoff value of < 1%. Several studies mentioned the clinical impact of their analyses on patients. Stray-Pedersen et al. described significant changes in management in up to 25% of cases [26]. Rae et al. found an even higher number of 37% [17]. Four studies found a number of patients that were clinically reclassified according to their molecular diagnosis.
Critical Appraisal
The results of the critical appraisal can be found in Appendix 2, Table 5. The reporting of variables was found to be incomplete for seven studies according to the aforementioned criteria. All articles explained the aim of their research, confirmed variants by Sanger sequencing, and proposed a set of possible implications of the research. The included studies scored relatively low on patient eligibility criteria, clinical background of their included patients, and analysis of sensitivity.
Discussion
In this review, we analyzed test characteristics and performance of next generation sequencing techniques in patients with clinically defined, but genetically undiagnosed PIDs. After a systematic search, we collected fourteen studies describing the diagnostic yield of NGS in PID patients. A broad range in diagnostic yields was found (15–79%). This was explained by methodological differences (e.g., number of PID-related genes evaluated) and by different a priori risks for monogenetic causes of PID between the study populations. Overall, NGS-based evaluations performed well in a clinical setting.
Several studies described the clinical impact of their diagnoses. It was previously described that a genetic diagnosis is important for understanding the molecular mechanism of disease, for initiation of targeted therapy, for family counseling and reproductive advice, and because it can end the so called “diagnostic Odyssey” [22]. We found eight papers describing these clinical implications; they were frequent and ranged from changes in therapeutic approach to screening for malignancies [27].
A number of observations can be made regarding the patient populations in this review. First, as primary immunodeficiencies are rare disorders, there were few large cohorts describing specific PID phenotypes [2], with the exception of the studies by Abolhassani et al. reporting large cohorts in patients with PID subcategories [22, 27]. Stray-Pedersen et al. found varying diagnostic yields between disease subgroups in their cohort, ranging from 13% for autoinflammatory disorders to 100% for patients with SCID [26]. This illustrates that NGS may be more useful for certain PID sub-populations than others, depending on factors such as complexity of the underlying genetic mechanisms, parental consanguinity rate, and environmental factors.
Second, eight out of 14 studies were performed in Western countries, which could make the results less representative for other regions. As the prevalence of PIDs varies greatly due to differences in carrier rates of mutations, parental consanguinity rates, and varying patterns of expression, the descent of the included patients and the location where the research was performed may have significantly influenced results [28]. Especially the two studies by Abolhassani et al. illustrate this, reporting overall diagnostic yields of 79 and 68% in highly consanguineous patient populations. Even in a subcategory of patients with antibody deficiencies, a diagnostic yield of 68% was reported [22, 27].
Third, six studies mention that a subset of patients had received extensive genetic testing (for example, by Sanger sequencing) prior to NGS-based evaluation. This may have influenced the yield of NGS-based testing in a negative way as obvious well-known candidate mutations were likely to have been identified by prior Sanger-based testing. This difference is illustrated by Al-Mousa et al. where a higher diagnostic yield was found for new cases without any previous genetic work-up in comparison with cases without prior genetic evaluation [24].
Apart from the type of patients undergoing testing, the overall performance of NGS depends on several other technical variables, including sequencing method, number of genes analyzed, and interpretation pipelines. Due to the limited number of studies included in this review, statistical analyses to identify factors that accounted for differences in yield over time could not be performed. Most studies used array-based PID panels, and no clear difference in diagnostic yield was found between studies using this approach and those using WES. No studies using WGS were included in this review, although we identified one case series using WGS that identified the genetic cause of disease in 6/6 patients, indicating the diagnostic potential of WGS [29]. Regarding the role of the number of PID-related genes evaluated per study, we found that most studies included a set of genes referenced in the IUIS guidelines. A range of 12–365 PID-related genes in the panels for the specific PID populations was found and 60–571 in the panels for unsorted PID populations. The heterogeneity in the number of genes used in the different studies illustrates that currently only limited consensus exists as to which genes should be investigated in patients with suspected PIDs. Due to the frequent discovery of new PID-related genes, any standardized gene set in a PID panel will have to undergo regular updates to include new genes; this may be especially challenging for PIDs caused by a variety of genes such as CVID [30, 31]. In the Netherlands, in an attempt to provide uniform testing, all genetic laboratories have adopted the same nationwide PID gene panel which undergoes three monthly updates by consensus meetings.
Another technical factor that influences the diagnostic yield of NGS is coverage of nucleotides. Low coverage decreases the likelihood of NGS to retrieve pathogenic mutations, and may be caused by a variety of reasons including insufficient reads of a specific region and mapping problems. For example, Nijman et al. reported a set of nine genes that could not be adequately sequenced by NGS, an issue also reported in several other studies [23, 32, 33]. This may be due to the presence of pseudogenes (for example IKBKG and NCF1) or to high CG-content [34]. These regions cannot be reliably analyzed using a NGS approach based on short reads, and should be targeted using alternative techniques. Long read technologies with low SNV error rates will solve these limitations, but are still under development. The third reason for low coverage may be the presence of CNV’s, which may be missed by NGS and are identified more reliably by WGS-based techniques [4, 35]. The fact that CNV analyses can provide a considerable percentage of additional genetic diagnoses (on average 4.2% in the three studies that provided these data) indicates that CNV analyses can be a very valuable part of the diagnostic pipeline [26].
Finally, the clinical performance of NGS has been shown to depend on method of subsequent interpretation pipelines. We noted that different reference sets of DNA variants were used in the pathogenicity analysis between the studies. In the studies by Nijman et al. and Mukda et al., the cutoff values for frequency of SNPs in healthy control population was < 5% whereas most other studies used < 1% or even lower cutoff values [23, 25]. Moreover, most studies did not include intronic or synonymous variants in their analyses. Substantial heterogeneity was also found between papers in the means used to evaluate the pathogenicity of the DNA variants. Stray-Pedersen and Rae followed the American College of Medical Genetics and Genomics guidelines recommended for Mendelian disorders, while others gave limited description how they classified DNA variants [17, 26]. Software pathogenicity prediction tools were employed most often, sometimes in combination with functional assays, familial cosegregation analysis, or other additional analyses. As the final diagnostic yield greatly depends on the type and quality of these procedures, some variants may have been falsely marked as pathogenic, whereas other disease-causing variants may have been missed. The heterogeneity between the articles on this topic illustrates the need for more standardized procedures to evaluate the disease-causing potential of mutations.
The heterogeneity between the included studies is one of the most important limitations of this review. An average diagnostic yield is difficult to establish as a broad variety of factors has to be taken into account. For instance, we noted large differences in yields between the studies performed in populations with low consanguinity rates versus high consanguinity rates. A second limitation is that case studies and case series < n = 10 cases were excluded. Many of these reports did in fact identify causative genetic mutations in the majority of their patients. Pooling of these patients in future studies could provide additional information. Studies that included patients with VEO-IBD phenotypes were also excluded. Suzuki et al., for instance, used WES in order to identify the molecular mechanism of disease for pediatric IBD in 35 patients. Fifty-five genes were investigated, providing 14% of all patients with a genetic diagnosis [36]. Interestingly, all diagnosed patients were found to have PID-associated mutations. Future research could investigate the efficacy of targeted NGS PID-panels within this and other specific PID-related patient groups.
NGS is a relatively new technique, and has given rise to several questions outside the direct scope of this review. First, the more genes sequenced, the more variants will be detected of which the clinical significance is unknown. When misinterpreted, these variants of unknown significance (VUS) may hamper proper treatment and may cause unnecessary distress to patients. A second issue (especially in the application of unfiltered WES or WGS) is the possibility of discovering incidental findings: pathogenic mutations in genes related to other illnesses [17]. This is an important consideration as unfiltered WES and WGS are likely to become increasingly more relevant in the future [4]. Due to its comprehensive nature, WES and WGS without PID filter have the ability to provide patients with an alternative genetic diagnosis than PID. Careful counseling of patients on this topic is indispensable. Currently, however, the large amounts of data generated by WES and WGS complicate its use as a routine first-line investigation, reason why currently the output of WES and WGS is usually interpreted first with the application of a PID filter [34]. Third, even though NGS can be cheaper than Sanger sequencing in certain groups of patients, it remains expensive. Further research should focus on the most appropriate place to use NGS in the diagnostic pipeline in order to ensure the highest level of cost-effectiveness. Last, even though the costs of NGS are decreasing, the application of Sanger sequencing remains important in several situations. For example, in patients with a high suspicion of only one or two disease-related genes, Sanger sequencing can be more effective than NGS due to its high sensitivity and specificity and usually relatively short time to diagnosis. Finally, NGS may be a quicker and more comprehensive alternative, but it can fail to detect certain mutations. For this reason, Sanger sequencing is also required for the sequencing of parts of genes that are poorly covered by NGS. Sanger sequencing can also be used for confirmation of pathogenic mutations identified by NGS, but this is only necessary when results are inconclusive.
In conclusion, this systematic review shows that NGS has the ability to contribute significantly to the identification of molecular mechanisms in PID patients, thereby altering clinical management. This highlights the potential value of NGS in clinical practice. The diagnostic yields presented in this review highly depended on their context such as clinical background and technological performance of the diagnostic method. Therefore, further research should be performed in order to determine the efficacy and associated costs of NGS in patients with PIDs. Moreover, a more standardized means of analysis should be conceptualized in order to correctly identify the causative genetic defect in PID patients.
Abbreviations
- ALPS:
-
autoimmune lymphoproliferative syndrome
- BTK:
-
Bruton’s tyrosine kinase
- CID:
-
Combined immunodeficiency
- CMC:
-
Chronic mucocutaneous candidiasis
- CNV:
-
Copy number variant
- CVID:
-
Common variable immunodeficiency
- GC:
-
Guanine-cytosine
- HLH:
-
Hemophagocytic lymphohistiocytosis
- (VEO)-IBD:
-
(Very early-onset) inflammatory bowel disease
- MHC:
-
Major histocompatibility complex
- NGS:
-
Next generation sequencing
- PID:
-
Primary immunodeficiency
- SCID:
-
Severe combined immunodeficiency
- SNP:
-
Single-nucleotide polymorphism
- SNV:
-
Single-nucleotide variant
- VUS:
-
Variant of unknown significance
- WES:
-
Whole exome sequencing
- WGS:
-
Whole genome sequencing
- XLP:
-
X-linked lymphoproliferative disease
References
van der Spek J, Groenwold RHH, van der Burg M, van Montfrans JM. TREC based newborn screening for severe combined immunodeficiency disease: a systematic review. J Clin Immunol. 2015;35:416–30. https://doi.org/10.1007/s10875-015-0152-6.
Gray PEA, Namasivayam M, Ziegler JB. Recurrent infection in children: when and how to investigate for primary immunodeficiency? J Paediatr Child Health. 2012;48:202–9. https://doi.org/10.1111/j.1440-1754.2011.02080.x.
Shillitoe B, Bangs C, Guzman D, Gennery AR, Longhurst HJ, Slatter M, et al. The United Kingdom primary immune deficiency (UKPID) registry 2012 to 2017. Clin Exp Immunol. 2018;192:284–91. https://doi.org/10.1111/cei.13125.
Heimall JR, Hagin D, Hajjar J, Henrickson SE, Hernandez-Trujillo HS, Tan Y, et al. Use of genetic testing for primary immunodeficiency patients. J Clin Immunol. 2018;38:320–9. https://doi.org/10.1007/s10875-018-0489-8.
Gallo V, Dotta L, Giardino G, Cirillo E, Lougaris V, D’Assante R, et al. Diagnostics of primary immunodeficiencies through next-generation sequencing. Front Immunol. 2016;7. https://doi.org/10.3389/fimmu.2016.00466.
Oliveira JB, Fleisher TA. Laboratory evaluation of primary immunodeficiencies. J Allergy Clin Immunol. 2010;7. https://doi.org/10.3389/fimmu.2016.00466.
Alkan G, Keles S, Reisli İ. Evaluation of clinical and immunological characteristics of children with common variable immunodeficiency. Int J Pediatr. 2018;2018:1–8. https://doi.org/10.1155/2018/3527480.
Stoddard JL, Niemela JE, Fleisher TA, Rosenzweig SD. Targeted NGS: a cost-effective approach to molecular diagnosis of PIDs. Front Immunol. 2014;5. https://doi.org/10.3389/fimmu.2014.00531.
Seleman M, Hoyos-Bachiloglu R, Geha RS, Chou J. Uses of next-generation sequencing technologies for the diagnosis of primary Immunodeficiencies. Front Immunol. 2017;8. https://doi.org/10.3389/fimmu.2017.00847.
Notarangelo LD, Sorensen R. Is it necessary to identify molecular defects in primary immunodeficiency disease? J Allergy Clin Immunol. 2008;122:1069–73. https://doi.org/10.1016/j.jaci.2008.08.038.
Raje N, Soden S, Swanson D, Ciaccio CE, Kingsmore SF, Dinwiddie DL. Utility of next generation sequencing in clinical primary immunodeficiencies. Curr Allergy Asthma Rep. 2014;14:468. https://doi.org/10.1007/s11882-014-0468-y.
Bisgin A, Boga I, Yilmaz M, Bingol G, Altintas D. The utility of next-generation sequencing for primary immunodeficiency disorders: experience from a clinical diagnostic laboratory. Biomed Res Int. 2018;2018:1–5. https://doi.org/10.1155/2018/9647253.
Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International Union of Immunological Societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol. 2018;38(1):96–128.
Picard C, Fischer A. Contribution of high-throughput DNA sequencing to the study of primary immunodeficiencies. Eur J Immunol. 2014;44:2854–61. https://doi.org/10.1002/eji.201444669.
Engelhardt KR, Xu Y, Grainger A, Germani Batacchi MGC, Swan DJ, Willet JDP, et al. Identification of heterozygous single- and multi-exon deletions in IL7R by whole exome sequencing. J Clin Immunol. 2017;37:42–50. https://doi.org/10.1007/s10875-016-0343-9.
van Schouwenburg PA, Davenport EE, Kienzler A-K, Marwah I, Wright B, Lucas M, et al. Application of whole genome and RNA sequencing to investigate the genomic landscape of common variable immunodeficiency disorders. Clin Immunol. 2015;160:301–14. https://doi.org/10.1016/j.clim.2015.05.020.
Rae W, Ward D, Mattocks C, Pengelly RJ, Eren E, Patel SV, et al. Clinical efficacy of a next-generation sequencing gene panel for primary immunodeficiency diagnostics. Clin Genet. 2018;93:647–55. https://doi.org/10.1111/cge.13163.
Fang M, Abolhassani H, Lim CK, Zhang J, Hammarström L. Next generation sequencing data analysis in primary immunodeficiency disorders - future directions. J Clin Immunol. 2016;36:68–75. https://doi.org/10.1007/s10875-016-0260-y.
Bossuyt PM, Reitsma JB, Bruns DE, Gatsonis CA, Glasziou PP, Irwig LM, et al. Towards complete and accurate reporting of studies of diagnostic accuracy: the STARD initiative. standards for reporting of diagnostic accuracy. Clin Chem. 2003 Jan;49(1):1–6. Available from http://www.ncbi.nlm.nih.gov/pubmed/12507953.
Yu H, Zhang VW, Stray-Pedersen A, Hanson IC, Forbes LR, de la Morena MT, et al. Rapid molecular diagnostics of severe primary immunodeficiency determined by using targeted next-generation sequencing. J Allergy Clin Immunol. 2016;138:1142–1151.e2. https://doi.org/10.1016/j.jaci.2016.05.035.
Maffucci P, Filion CA, Boisson B, Itan Y, Shang L, Casanova J-L, et al. Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front Immunol. 2016;7. https://doi.org/10.3389/fimmu.2016.00220.
Abolhassani H, Aghamohammadi A, Fang M, Rezaei N, Jiang C, Liu X, et al. Clinical implications of systematic phenotyping and exome sequencing in patients with primary antibody deficiency. Genet Med [Internet. 2019;21(1):243–51 Available from: http://www.ncbi.nlm.nih.gov/pubmed/29921932.
Nijman IJ, van Montfrans JM, Hoogstraat M, Boes ML, van de Corput L, Renner ED, et al. Targeted next-generation sequencing: a novel diagnostic tool for primary immunodeficiencies. J Allergy Clin Immunol. 2014;133:529–534.e1. https://doi.org/10.1016/j.jaci.2013.08.032.
Al-Mousa H, Abouelhoda M, Monies DM, Al-Tassan N, Al-Ghonaium A, Al-Saud B, et al. Unbiased targeted next-generation sequencing molecular approach for primary immunodeficiency diseases. J Allergy Clin Immunol. 2016;137:1780–7. https://doi.org/10.1016/j.jaci.2015.12.1310.
Mukda E, Trachoo O, Pasomsub E, Tiyasirichokchai R, Iemwimangsa N, Sosothikul D, et al. Exome sequencing for simultaneous mutation screening in children with hemophagocytic lymphohistiocytosis. Int J Hematol. 2017;106:282–90. https://doi.org/10.1007/s12185-017-2223-3.
Stray-Pedersen A, Sorte HS, Samarakoon P, Gambin T, Chinn IK, Coban Akdemir ZH, et al. Primary immunodeficiency diseases: genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol. 2017;139:232–45. https://doi.org/10.1016/j.jaci.2016.05.042.
Abolhassani H, Chou J, Bainter W, Platt CD, Tavassoli M, Momen T, et al. Clinical, immunologic, and genetic spectrum of 696 patients with combined immunodeficiency. J Allergy Clin Immunol. 2018;141:1450–8. https://doi.org/10.1016/j.jaci.2017.06.049.
Al-Herz W, Aldhekri H, Barbouche M-R, Rezaei N. Consanguinity and primary immunodeficiencies. Hum Hered. 2014;77:138–43. https://doi.org/10.1159/000357710.
Mousallem T, Urban TJ, McSweeney KM, Kleinstein SE, Zhu M, Adeli M, et al. Clinical application of whole-genome sequencing in patients with primary immunodeficiency. J Allergy Clin Immunol. 2015;136:476–479.e6. https://doi.org/10.1016/j.jaci.2015.02.040.
Kienzler A-K, Hargreaves CE, Patel SY. The role of genomics in common variable immunodeficiency disorders. Clin Exp Immunol. 2017;188:326–32. https://doi.org/10.1111/cei.12947.
Ameratunga R, Lehnert K, Woon S-T, Gillis D, Bryant VL, Slade CA, et al. Review: diagnosing common variable immunodeficiency disorder in the era of genome sequencing. Clin Rev Allergy Immunol. 2018;54:261–8. https://doi.org/10.1007/s12016-017-8645-0.
Moens LN, Falk-Sörqvist E, Asplund AC, Bernatowska E, Smith CIE, Nilsson M. Diagnostics of primary immunodeficiency diseases: a sequencing capture approach. PLoS One. 2014;9:e114901. https://doi.org/10.1371/journal.pone.0114901.
Erman B, Bilic I, Hirschmugl T, Salzer E, Boztug H, Sanal Ö, et al. Investigation of genetic defects in severe combined immunodeficiency patients from Turkey by targeted sequencing. Scand J Immunol. 2017;85:227–34. https://doi.org/10.1111/sji.12523.
Meyts I, Bosch B, Bolze A, Boisson B, Itan Y, Belkadi A, et al. Exome and genome sequencing for inborn errors of immunity. J Allergy Clin Immunol. 2016;138:957–69. https://doi.org/10.1016/j.jaci.2016.08.003.
Whitford W, Lehnert K, Snell RG, Jacobsen JC. Evaluation of the performance of copy number variant prediction tools for the detection of deletions from whole genome sequencing data. J Biomed Inform [Internet. 2019;94:103174. https://doi.org/10.1016/j.jbi.2019.103174.
Suzuki T, Sasahara Y, Kikuchi A, Kakuta H, Kashiwabara T, Ishige T, et al. Targeted sequencing and immunological analysis reveal the involvement of primary immunodeficiency genes in pediatric IBD: a Japanese multicenter study. J Clin Immunol. 2017;37:67–79. https://doi.org/10.1007/s10875-016-0339-5.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
J.M. van Montfrans served on an advisory board for Shire.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Appendices
Appendix 1
Appendix 2
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yska, H.A.F., Elsink, K., Kuijpers, T.W. et al. Diagnostic Yield of Next Generation Sequencing in Genetically Undiagnosed Patients with Primary Immunodeficiencies: a Systematic Review. J Clin Immunol 39, 577–591 (2019). https://doi.org/10.1007/s10875-019-00656-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-019-00656-x