
Abstract
To determine the diagnostic yield of Next-generation sequencing (NGS) in suspect Primary Immunodeficiencies Diseases (PIDs). This systematic review was conducted following PRISMA criteria. Searching Pubmed and Web of Science databases, the following keywords were used in the search: (“Next-generation sequencing”) OR “whole exome sequencing” OR “whole genome sequencing”) AND (“primary immunodeficiency disease” OR “PIDs”). We used STARD items to assess the risk of bias in the included studies. The meta-analysis included 29 studies with 5847 patients, revealing a pooled positive detection rate of 42% (95% CI 0.29–0.54, P < 0.001) for NGS in suspected PID cases. Subgroup analyses based on family history demonstrated a higher detection rate of 58% (95% CI 0.43–0.71) in patients with a family history compared to 33% (95% CI 0.21–0.46) in those without (P < 0.001). Stratification by disease types showed varied detection rates, with Severe Combined Immunodeficiency leading at 58% (P < 0.001). Among 253 PID-related genes, RAG1, ATM, BTK, and others constituted major contributors, with 34 genes not included in the 2022 IUIS gene list. The application of NGS in suspected PID patients can provide significant diagnostic results, especially in patients with a family history. Meanwhile, NGS performs excellently in accurately diagnosing disease types, and early identification of disease types can benefit patients in treatment.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Primary Immunodeficiency Diseases (PIDs), also recognized as Inborn Errors of Immunity (IEI), constitute a diverse set of monogenic disorders that impact the immune system. The clinical spectrum of PIDs encompasses various phenotypes, including infections, autoinflammation, autoimmunity, allergies, malignancies, and more [9]. Presently, over 430 genes associated with PIDs have been identified, predominantly inherited in a monogenic manner. The list of disease-causing genes continues to expand with ongoing discoveries. The International Union of Immunological Societies (IUIS) classifies IEI into 10 categories, addressing diseases with overlapping phenotypes (citation). These categories range from Combined Immunodeficiencies to Congenital Immune Deficiencies (citation). Given the broad clinical spectrum exhibited by PIDs, achieving an accurate diagnosis based solely on clinical manifestations poses a significant challenge. Hence, there is an urgent need in clinical practice for methods that are safe, rapid, and accurate for testing and diagnosing PIDs.
Before the advent of advanced technologies, Sanger sequencing was the primary method employed in clinical practice. However, its limitations became evident in the context of PIDs due to genetic pleiotropy and heterogeneity. Sanger sequencing, being time-consuming, labor-intensive, inefficient, and relatively expensive, faced challenges in identifying disease-causing genes [19, 23]. The emergence of next-generation high-throughput sequencing technologies marks a transformative era in PID diagnosis. Next-generation sequencing (NGS) has not only expanded the known gene list associated with PIDs but has also introduced a faster and more cost-effective means of evaluating the genome. Particularly in cases lacking clear candidate genes, NGS becomes indispensable. NGS encompasses various techniques, including Targeted Gene Panel (TGP) sequencing, Whole Exome Sequencing (WES), and Whole Genome Sequencing (WGS) [39].
TGP is a method focusing on a limited set of genes relevant to specific phenotypes or disease groups and is used for patients with well-defined clinical phenotypes [16, 31]. WES, targeting the exome region, can identify genetic variations associated with classical or atypical phenotypes, expanding the understanding of new phenotypes and genes. Diagnostic yields from WES vary widely (15–79%) depending on patient types and clinical phenotypes (citation). WGS, covering the entire genome, provides better resolution for detecting copy number variations (CNVs) and structural variations. However, it introduces the challenge of interpreting a substantial number of variants of uncertain significance, especially in non-coding regions [10, 20].
As technology advances and costs decrease, NGS plays an increasingly vital role in primary PID diagnosis. Nevertheless, literature reports indicate significant variation in the diagnostic rates of NGS in PIDs. Hence, there is a crucial need to comprehensively evaluate the technical performance and diagnostic efficacy of NGS in PID patients across diverse populations and diseases. This meta-analysis aims to fulfill this need through a comprehensive evaluation.
Method
Search strategy
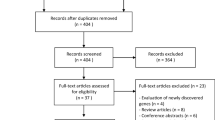
A comprehensive search for pertinent studies was systematically conducted in the Pubmed and Web of Science databases up to November 1st, 2023. The search strategy employed the following Boolean operators: ("next-generation sequencing" OR "whole exome sequencing" OR "whole genome sequencing") AND ("primary immunodeficiency disease" OR "PIDs"). A total of 958 studies were retrieved from Pubmed, and 836 studies were identified from Web of Science. No additional pertinent studies were discerned in the bibliographies of the encompassed studies. A transparent depiction of the search and literature screening process is presented in Fig. 1.

PRISMA flow diagram
Literature selection
In adherence to predefined inclusion and exclusion criteria, studies were meticulously selected for the meta-analysis. Inclusion criteria stipulated: (1) a study population exhibiting clinically evident manifestations of PIDs; (2) incorporation of genetic sequencing methodologies and provision of pertinent genetic testing information; (3) explicit reporting of both the total number of patients and the number of positive patients detected; and (4) documentation in the English language. Exclusion criteria comprised: (1) studies exclusively focused on the detection of copy number variations (CNVs); and (2) exclusion of duplicate publications, conference proceedings, case reports, reviews, and unpublished studies.
Data extraction and quality assessment
A rigorous evaluation of potentially relevant articles commenced with an independent screening of titles and abstracts by three authors. Subsequent data extraction, in concordance with predefined inclusion criteria, encompassed key parameters such as the first author, publication year, detection method, total number of patients, number of positive patients, and genetic locus information. The assessment of the risk of bias in the included studies adhered to the Standards for Reporting of Diagnostic Accuracy Studies (STARD) criteria. Any discrepancies in the extraction and assessment processes were resolved through meticulous discussion. In instances where consensus remained elusive, a third reviewer assumed the role of an arbitrator.
Analytical procedures
All statistical analyses were conducted utilizing the meta package in Stata 14. A meta-analysis of the gene testing detection rate for PIDs was undertaken utilizing a single-group rate approach. Effect size (ES) and a 95% confidence interval (CI) were computed employing a random-effects model. Subgroup analyses discerned the detection rate of gene testing for distinct populations and varied diseases. Heterogeneity was quantified employing the chi-square Q test and I2 test. A fixed-effects model was employed when I2 was less than 50% or the p value of the Q test exceeded 0.05. Conversely, in instances where I2 surpassed 50% or the p value of the Q test fell below 0.05, a random-effects model was applied.
Results
Characteristics of included studies
A total of 29 studies [1,2,3,4,5, 11, 14, 18, 21, 24, 27, 30, 32, 37, 38, 6,7,8, 12, 13, 15, 22, 25, 26, 28, 29, 36, 45, 46], encompassing 5847 patients, were included in this meta-analysis. Among these, 10 studies conducted familial analyses across various centers and countries. The studies employed diverse sequencing methodologies, including whole-exome sequencing (WES), Whole Genome Sequencing (WGS), targeted DNA sequencing, and clinical exome sequencing (CES). Detailed characteristics of the included studies are summarized in Table 1.
Primary outcome analysis
This study involved 5847 cases, with 1603 cases diagnosed as positive for NGS. Utilizing a random-effects model, the pooled positive detection rate was 42% (95% CI 0.29–0.54, P < 0.001) (Fig. 2). Additionally, we conducted subgroup analyses based on family history and different disease types. In patients with a family history, the detection rate could reach 58% (95% CI 0.43–0.71, P < 0.001), while in patients without a family history, the detection rate was 33% (95% CI 0.21–0.46, P < 0.001) (Fig. 3). We selected diseases mentioned in a larger number of articles as grouping criteria, including Severe Combined Immunodeficiency (SCID), Common Variable Immunodeficiency (CVID), Hyper IgE syndrome (HIES), and Combined Immunodeficiency (CID). The overall detection rate for these four diseases was 44% (95% CI 0.31–0.57, P < 0.001), with a detection rate of 58% (95% CI 0.43–0.74, P < 0.001) for SCID, 35% (95% CI 0.17–0.52, P < 0.001) for CVID, 35% (95% CI 0.20–0.50, P < 0.001) for HIES, and 24% (95% CI 0.03–0.44, P = 0.026) for CID (Fig. 4).

The overall physical examination rate of primary immunodeficiency diseases

The detection rate of primary immunodeficiency diseases in different populations

The detection rate of different types of primary immunodeficiency diseases
Genetic landscape of PID-related genes
A total of 253 PID-related genes were examined, with notable pathogenic mutations mainly involving RAG1, ATM, BTK, LRBA, DOCK8, STAT3, IL2RG, JAK3, RAG2, and WAS, each accounting for more than 3% of the cases, with RAG1 accounting for 6% (Fig. 5). Notably, 34 genes were not included in the updated 2022 IUIS gene list, and additional information on these genes can be found in Table 2.

Gene types
Quality assessment of included studies
Quality assessment utilizing a modified version of the Standards for Reporting of Diagnostic Accuracy (STARD) specific to this project indicated high-quality studies (Fig. 6). As the analysis is a single-group rate analysis with descriptive results, publication bias assessment was deemed unnecessary.

Quality assessment of included studies
Discussion
Primary Immunodeficiency Diseases (PIDs) represent a class of inherent immunodeficiency disorders, attributed to genetic mutations impacting distinct facets of the immune system. This diverse group manifests through recurrent infections, autoimmunity, autoinflammation, hypersensitivity reactions, and malignancies [9]. PIDs exhibit genetic heterogeneity, with different mutations in the same gene yielding varied clinical and immune phenotypes, while distinct gene mutations may produce similar clinical outcomes [42]. The broad clinical spectrum, coupled with genetic heterogeneity and pleiotropy, renders the diagnosis and treatment of PIDs challenging. A precise molecular diagnosis is crucial for accurate recognition and tailored intervention in clinical practice.
Our study reveals a positive detection rate of 42% (95% CI 0.29–0.54, P < 0.001) for Next-Generation Sequencing (NGS) in patients displaying clinical symptoms associated with PIDs. Previous literature reports a variable detection rate for PIDs, ranging from 15 to 80%. In our analysis of 5847 cases, the observed 42% detection rate falls within an intermediate range. Three key factors contribute to this result: Firstly, the expansive array of clinical manifestations related to PIDs has led to a surge in suspected cases. Secondly, the limited nucleotide coverage of NGS diminishes the potential for detecting pathogenic mutations. Additionally, our study excludes cases diagnosed through Copy Number Variation (CNV) detection [17, 35, 44], a method reported in the literature to offer a substantial number of additional genetic diagnoses [35]. Due to the complexity of these factors, determining an average diagnostic rate poses challenges. Enhancing the accuracy of this rate involves the meticulous collection of comprehensive clinical data.
Our subgroup analysis, differentiating patients based on the presence or absence of a family history and various disease types, yielded intriguing insights. Notably, the detection rate among patients with a family history stands at an impressive 58% (95% CI 0.43–0.71, P < 0.001), while their counterparts without a family history exhibit a lower detection rate of 33% (95% CI 0.21–0.46, P < 0.001). This stark contrast underscores the substantial impact of familial factors on the detection rate of Primary Immunodeficiency Diseases (PIDs). Supporting this finding, existing literature posits that a majority of PIDs follow an autosomal recessive (AR) inheritance pattern. Identifying individuals with a singular clinical and immune phenotype within consanguineous families emerges as a pivotal strategy for uncovering novel pathogenic genes[2]. Our study, incorporating data from 10 relevant articles [2, 4, 5, 13, 15, 18, 32, 38, 25, 26] involving patients with a family history, aligns with this genetic landscape. Among these, 2 cases inherited PIDs as autosomal recessive (AR) traits, 1 as a combination of AR and X-linked (XL) traits, 6 as a blend of AR, autosomal dominant (AD), and XL traits, and 1 did not specify the genetic features. This consistency with prior research underscores the crucial role of families with consanguinity in unraveling intricate phenotypes associated with PIDs. Exploring consanguineous relationships further reveals their potential to unveil new pathogenic genes, delineate genetic patterns of known genes, identify novel clinical phenotypes, and elucidate novel manifestations linked to established PID-causing genes. Within this context, Next-Generation Sequencing (NGS) emerges as a promising tool for eugenics in families with consanguinity. Recommendations for NGS testing in families, especially post-pregnancy following a PID diagnosis, offer valuable insights into gauging the likelihood of disease inheritance in subsequent generations.
Our investigation underscores distinctive detection rates across various primary immunodeficiency diseases (PIDs). Severe Combined Immunodeficiency (SCID) emerges with the highest detection rate, followed by Common Variable Immunodeficiency (CVID), Hyper-IgE Syndrome (HIES), and Combined Immunodeficiency (CID). Clinical manifestations of Severe Combined Immunodeficiency (SCID) denote a complex spectrum of disorders characterized by impaired T lymphocyte development, impacting the quantity and functionality of B cells and NK cells [34]. SCID stands out as a profoundly severe subset within the broader PID landscape, marked by early-onset dermatitis, dermal complications, persistent enteritis, pneumonia, oral candidiasis, and other distinctive manifestations [43]. Notably, more than 50% of SCID patients have been reported to harbor mutations in the RAG1 or RAG2 genes [33, 41]. In the absence of immune reconstitution, the survival prognosis for SCID patients beyond 6–12 months is exceedingly poor. Nonetheless, this patient cohort commonly demonstrates a favorable response to allogeneic hematopoietic stem cell transplantation (HSCT) [41]. Hence, early disease recognition and expeditious intervention hold the potential to substantially augment patient survival rates. Similarly, diseases such as Common Variable Immunodeficiency (CVID), Hyper-IgE Syndrome (HIES), and Combined Immunodeficiency (CID) constitute prevalent categories within the PID spectrum. The extensive range of diseases within PIDs manifests shared clinical manifestations, necessitating timely and precise identification of disease types. Next-generation sequencing (NGS) emerges as a critical tool for optimizing treatment effectiveness and providing early benefits to patients. However, it is crucial to acknowledge that the literature data incorporated into our study, while informative, is not derived from large-scale studies. The potential introduction of slight errors in the results emphasizes the need for more extensive clinical data to validate our conclusions thoroughly. Large-scale studies will enhance the robustness of our findings, contributing to a more comprehensive understanding of the disease-specific landscape within PIDs.
A critical facet of our study involves scrutinizing primary immunodeficiency diseases (PIDs)-related genes updated by the International Union of Immunological Societies (IUIS) in 2022 [40]. Our analysis identified 34 genes absent from the IUIS list, prompting a deeper investigation. Consulting the Online Mendelian Inheritance in Man (OMIM) database revealed six genes with significant relevance to PIDs' clinical manifestations. CD40L's potential involvement in Immunodeficiency and hyper-IgM, RECQL4's associations with Baller-Gerold syndrome, RAPADILINO syndrome, and Rothmund-Thomson syndrome, and IL7RA's connection to severe combined immunodeficiency exemplify the intricate genetic landscape. Additionally, SP1NK5, ITGA2B, and CR2 exhibit diverse implications, linking to Netherton syndrome, bleeding disorders, and common variable immunodeficiency, respectively. Despite the potential of these genes as targets for clinical testing, their validation is hindered by limited clinical data, underscoring the need for comprehensive validation efforts.
This article still has certain limitations. Firstly, the meta-analysis included a total of 29 articles, and some of the sequencing results in these articles were not validated using Sanger sequencing. Secondly, we have excluded positive cases determined through CNVs detection when entering the data, which may provide accurate information. Thirdly, some of the included literature had a small number of patients tested (< 20), which could affect the pooled results and lead to minor errors. More clinical sequencing data is required to validate the relevant conclusions.
Conclusion
The application of NGS in suspected PID patients can provide significant diagnostic results, especially in patients with a family history. Meanwhile, NGS performs excellently in accurately diagnosing disease types, and early identification of disease types can benefit patients in treatment.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
References
Abolhassani H, Chou J, Bainter W, Platt CD, Tavassoli M, Momen T, et al. Clinical, immunologic, and genetic spectrum of 696 patients with combined immunodeficiency. J Allergy Clin Immunol. 2018;141(4):1450–8. https://doi.org/10.1016/j.jaci.2017.06.049.
Al-Herz W, Chou J, Delmonte OM, Massaad MJ, Bainter W, Castagnoli R, et al. Comprehensive genetic results for primary immunodeficiency disorders in a highly consanguineous population. Front Immunol. 2019. https://doi.org/10.3389/fimmu.2018.03146.
Al-Mousa H, Abouelhoda M, Monies DM, Al-Tassan N, Al-Ghonaium A, Al-Saud B, et al. Unbiased targeted next-generation sequencing molecular approach for primary immunodeficiency diseases. J Allergy Clin Immunol. 2016;137(6):1780–7. https://doi.org/10.1016/j.jaci.2015.12.1310.
Aluri J, Desai M, Gupta M, Dalvi A, Terance A, Rosenzweig SD, et al. Clinical, immunological, and molecular findings in 57 patients with severe combined immunodeficiency (SCID) from India. Front Immunol. 2019. https://doi.org/10.3389/fimmu.2019.00023.
Arts P, Simons A, AlZahrani MS, Yilmaz E, AlIdrissi E, van Aerde KJ, et al. Exome sequencing in routine diagnostics: a generic test for 254 patients with primary immunodeficiencies. Genome Med. 2019. https://doi.org/10.1186/s13073-019-0649-3.
Arunachalam AK, Maddali M, Aboobacker FN, Korula A, George B, Mathews V, et al. Primary immunodeficiencies in India: molecular diagnosis and the role of next-generation sequencing. J Clin Immunol. 2020;41(2):393–413. https://doi.org/10.1007/s10875-020-00923-2.
Asgardoon MH, Azizi G, Yazdani R, Sohani M, Pashangzadeh S, Kalantari A, et al. Monogenic primary immunodeficiency disorder associated with common variable immunodeficiency and autoimmunity. Int Arch Allergy Immunol. 2020;181(9):706–14. https://doi.org/10.1159/000508817.
Borghesi A, Trück J, Asgari S, Sancho-Shimizu V, Agyeman PKA, Bellos E, et al. Whole-exome sequencing for the identification of rare variants in primary immunodeficiency genes in children with sepsis: a prospective. Popul Based Cohort Study Clin Infect Dis. 2020;71(10):e614–23. https://doi.org/10.1093/cid/ciaa290.
Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human inborn errors of immunity: 2019 update of the IUIS phenotypical classification. J Clin Immunol. 2020;40(1):66–81. https://doi.org/10.1007/s10875-020-00758-x.
Chinn IK, Chan AY, Chen K, Chou J, Dorsey MJ, Hajjar J, et al. Diagnostic interpretation of genetic studies in patients with primary immunodeficiency diseases: a working group report of the primary immunodeficiency diseases committee of the American academy of allergy, asthma and immunology. J Allergy Clin Immunol. 2020;145(1):46–69. https://doi.org/10.1016/j.jaci.2019.09.009.
Cifaldi C, Brigida I, Barzaghi F, Zoccolillo M, Ferradini V, Petricone D, et al. Targeted NGS platforms for genetic screening and gene discovery in primary immunodeficiencies. Front Immunol. 2019. https://doi.org/10.3389/fimmu.2019.00316.
Elsink K, Huibers MMH, Hollink IHIM, Simons A, Zonneveld-Huijssoon E, van der Veken LT, et al. Implementation of early next-generation sequencing for inborn errors of immunity: a prospective observational cohort study of diagnostic yield and clinical implications in dutch genome diagnostic centers. Front Immunol. 2021. https://doi.org/10.3389/fimmu.2021.780134.
Engelbrecht C, Urban M, Schoeman M, Paarwater B, van Coller A, Abraham DR, et al. Clinical utility of whole exome sequencing and targeted panels for the identification of inborn errors of immunity in a resource-constrained setting. Front Immunol. 2021. https://doi.org/10.3389/fimmu.2021.665621.
Erman B, Bilic I, Hirschmugl T, Salzer E, Boztug H, Sanal Ö, et al. Investigation of genetic defects in severe combined immunodeficiency patients from turkey by targeted sequencing. Scand J Immunol. 2017;85(3):227–34. https://doi.org/10.1111/sji.12523.
Erman B, Çipe F. Genetic screening of the patients with primary immunodeficiency by whole-exome sequencing. Pediatr Allergy Immunol Pulmonol. 2020;33(1):19–24. https://doi.org/10.1089/ped.2019.1097.
Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15(7):565–74. https://doi.org/10.1038/gim.2013.73.
Heimall JR, Hagin D, Hajjar J, Henrickson SE, Hernandez-Trujillo HS, Tan Y, et al. Use of genetic testing for primary immunodeficiency patients. J Clin Immunol. 2018;38(3):320–9. https://doi.org/10.1007/s10875-018-0489-8.
Kamae C, Imai K, Kato T, Okano T, Honma K, Nakagawa N, et al. Clinical and immunological characterization of ICF syndrome in Japan. J Clin Immunol. 2018;38(8):927–37. https://doi.org/10.1007/s10875-018-0559-y.
Kircher M, Kelso J. High-throughput DNA sequencing–concepts and limitations. BioEssays. 2010;32(6):524–36. https://doi.org/10.1002/bies.200900181.
Lee K, Abraham RS. Next-generation sequencing for inborn errors of immunity. Hum Immunol. 2021;82(11):871–82. https://doi.org/10.1016/j.humimm.2021.02.011.
Maffucci P, Filion CA, Boisson B, Itan Y, Shang L, Casanova J-L, et al. Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front Immunol. 2016. https://doi.org/10.3389/fimmu.2016.00220.
Mørup SB, Nazaryan-Petersen L, Gabrielaite M, Reekie J, Marquart HV, Hartling HJ, et al. Added value of reanalysis of whole exome- and whole genome sequencing data from patients suspected of primary immune deficiency using an extended gene panel and structural variation calling. Front Immunol. 2022. https://doi.org/10.3389/fimmu.2022.906328.
Mu W, Lu HM, Chen J, Li S, Elliott AM. Sanger confirmation is required to achieve optimal sensitivity and specificity in next-generation sequencing panel testing. J Mol Diagn. 2016;18(6):923–32. https://doi.org/10.1016/j.jmoldx.2016.07.006.
Nijman IJ, van Montfrans JM, Hoogstraat M, Boes ML, van de Corput L, Renner ED, et al. Targeted next-generation sequencing: a novel diagnostic tool for primary immunodeficiencies. J Allergy Clin Immunol. 2014;133(2):529-534.e521. https://doi.org/10.1016/j.jaci.2013.08.032.
Okano T, Imai K, Naruto T, Okada S, Yamashita M, Yeh T-W, et al. Whole-exome sequencing-based approach for germline mutations in patients with inborn errors of immunity. J Clin Immunol. 2020;40(5):729–40. https://doi.org/10.1007/s10875-020-00798-3.
Platt CD, Zaman F, Bainter W, Stafstrom K, Almutairi A, Reigle M, et al. Efficacy and economics of targeted panel versus whole-exome sequencing in 878 patients with suspected primary immunodeficiency. J Allergy Clin Immunol. 2021;147(2):723–6. https://doi.org/10.1016/j.jaci.2020.08.022.
Rae W, Ward D, Mattocks C, Pengelly RJ, Eren E, Patel SV, et al. Clinical efficacy of a next-generation sequencing gene panel for primary immunodeficiency diagnostics. Clin Genet. 2018;93(3):647–55. https://doi.org/10.1111/cge.13163.
Rawat A, Sharma M, Vignesh P, Jindal AK, Suri D, Das J, et al. Utility of targeted next generation sequencing for inborn errors of immunity at a tertiary care centre in North India. Sci Rep. 2022. https://doi.org/10.1038/s41598-022-14522-1.
Ripen AM, Chear CT, Baharin MF, Nallusamy R, Chan KC, Kassim A, et al. A single-center pilot study in Malaysia on the clinical utility of whole-exome sequencing for inborn errors of immunity. Clin Exp Immunol. 2021;206(2):119–28. https://doi.org/10.1111/cei.13626.
Rudilla F, Franco-Jarava C, Martínez-Gallo M, Garcia-Prat M, Martín-Nalda A, Rivière J, et al. Expanding the clinical and genetic spectra of primary immunodeficiency-related disorders with clinical exome sequencing: expected and unexpected findings. Front Immunol. 2019;10:234. https://doi.org/10.3389/fimmu.2019.02325.
Seleman M, Hoyos-Bachiloglu R, Geha RS, Chou J. Uses of next-generation sequencing technologies for the diagnosis of primary immunodeficiencies. Front Immunol. 2017;8:847. https://doi.org/10.3389/fimmu.2017.00847.
Shahbazi Z, Yazdani R, Shahkarami S, Shahbazi S, Hamid M, Sadeghi-Shabestari M, et al. Genetic mutations and immunological features of severe combined immunodeficiency patients in Iran. Immunol Lett. 2019;216:70–8. https://doi.org/10.1016/j.imlet.2019.10.001.
Shearer WT, Dunn E, Notarangelo LD, Dvorak CC, Puck JM, Logan BR, et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: the Primary Immune Deficiency Treatment Consortium experience. J Allergy Clin Immunol. 2014;133(4):1092–8. https://doi.org/10.1016/j.jaci.2013.09.044.
Sponzilli I, Notarangelo LD. Severe combined immunodeficiency (SCID): from molecular basis to clinical management. Acta Biomed. 2011;82(1):5–13.
Stray-Pedersen A, Sorte HS, Samarakoon P, Gambin T, Chinn IK, Coban Akdemir ZH, et al. Primary immunodeficiency diseases: genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol. 2017;139(1):232–45. https://doi.org/10.1016/j.jaci.2016.05.042.
Sun J, Yang L, Lu Y, Wang H, Peng X, Dong X, et al. Screening for primary immunodeficiency diseases by next-generation sequencing in early life. Clin Trans Immunol. 2020;9(5):234. https://doi.org/10.1002/cti2.1138.
Suzuki T, Sasahara Y, Kikuchi A, Kakuta H, Kashiwabara T, Ishige T, et al. Targeted sequencing and immunological analysis reveal the involvement of primary immunodeficiency genes in pediatric IBD: a Japanese multicenter study. J Clin Immunol. 2016;37(1):67–79. https://doi.org/10.1007/s10875-016-0339-5.
Tafakori Delbari M, Cheraghi T, Yazdani R, Fekrvand S, Delavari S, Azizi G, et al. Clinical manifestations, immunological characteristics and genetic analysis of patients with hyper-immunoglobulin M Syndrome in Iran. Int Arch Allergy Immunol. 2019;180(1):52–63. https://doi.org/10.1159/000500197.
Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol. 2020;40(1):24–64. https://doi.org/10.1007/s10875-019-00737-x.
Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. J Clin Immunol. 2022;42(7):1473–507. https://doi.org/10.1007/s10875-022-01289-3.
Tasher D, Dalal I. The genetic basis of severe combined immunodeficiency and its variants. Appl Clin Genet. 2012;5:67–80. https://doi.org/10.2147/tacg.S18693.
Villa A, Sobacchi C, Notarangelo LD, Bozzi F, Abinun M, Abrahamsen TG, et al. V(D)J recombination defects in lymphocytes due to RAG mutations: severe immunodeficiency with a spectrum of clinical presentations. Blood. 2001;97(1):81–8. https://doi.org/10.1182/blood.v97.1.81.
Wadbudhe AM, Meshram RJ, Tidke SC. Severe combined immunodeficiency (SCID) and its new treatment modalities. Cureus. 2023;15(10): e47759. https://doi.org/10.7759/cureus.47759.
Whitford W, Lehnert K, Snell RG, Jacobsen JC. Evaluation of the performance of copy number variant prediction tools for the detection of deletions from whole genome sequencing data. J Biomed Inform. 2019;94: 103174. https://doi.org/10.1016/j.jbi.2019.103174.
Yu H, Zhang VW, Stray-Pedersen A, Hanson IC, Forbes LR, de la Morena MT, et al. Rapid molecular diagnostics of severe primary immunodeficiency determined by using targeted next-generation sequencing. J Allergy Clin Immunol. 2016;138(4):1142-1151.e1142. https://doi.org/10.1016/j.jaci.2016.05.035.
Zhu T, Gong X, Bei F, Ma L, Sun J, Wang J, et al. Primary immunodeficiency-related genes in neonatal intensive care unit patients with various genetic immune abnormalities: a multicentre study in China. Clin Transl Immunol. 2021. https://doi.org/10.1002/cti2.1266.
Funding
This work was supported by Science and Technology Plan Project of Guangzhou (No. 202201010985) and Plan on enhancing scientific research in GMU (No. 0803030040).
Author information
Authors and Affiliations
Contributions
YYC, DRL and JWY contributed to the study design, while JLX, MX and QQ contributed to the data collection. Statistical analyses and interpretation of results were performed by YYC and DRL, whereas WLY drafted the manuscript and edited the language. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Y., Li, D., Yin, J. et al. Diagnostic yield of next-generation sequencing in suspect primary immunodeficiencies diseases: a systematic review and meta-analysis. Clin Exp Med 24, 131 (2024). https://doi.org/10.1007/s10238-024-01392-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10238-024-01392-2