
Abstract
Reaction of 4,6-dinitroresorcinol (1) and the nitrogen base 1,4-diazabicyclo[2·2·2]octane (2) affords the 1:2 salt and proton-transfer compound 1,4-diazabicyclo[2·2·2]octane-1,4-diium bis(5-hydroxy-2,4-dinitrophenolate) (3). Compound 3 crystallizes in the triclinic crystal system (space group P-1) with a = 8.3242(5) Å, b = 11.9915(7) Å, c = 12.4595(7) Å, α = 116.282(2)°, β = 100.576(3)°, γ = 101.051(2)°, 1042.30(11) Å3 and Z = 2. The dication 2-\({\text{H}}_{2}^{2+}\) forms charge assisted donating bifurcated N+−H⋅⋅⋅O− hydrogen bonds to the phenolate moieties of two monoanions of 1. The latter exhibit an intramolecular O−H⋅⋅⋅O hydrogen bond between the hydroxy group and the nitro group in ortho position. The crystal structure of 3 features pseudo B-centering of the lattice, which relates the two crystallographically distinct monoanions of 1 by a pseudo translation. The possible B-centring is broken by the ethylene groups of 2-H22+, which are related in neighbouring molecules by centres of symmetry.
Graphical Abstract

The 1:2 proton-transfer compound 1,4-diazabicyclo[2.2.2]octane-1,4-diium bis(5-hydroxy-2,4-dinitrophenolate) features pseudo B-centering of the lattice.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
4,6-Dinitroresorcinol (systematic name: 4,6-dinitrobenzene-1,3-diol, 1, Fig. 1) is a useful precursor to 4,6-diaminoresorcinol for the production of the synthetic polymer material poly(p-phenylene-2,6-benzobisoxazole) (PBO) [1], which is marketed under the tradename Zylon®. Crystallographic data for 1 were published for the first time in 1952, albeit without atom positions [2]. A full crystal structure determination of the compound was reported by Kolev et al. in 2000 [3]. In the crystal, the molecules feature two intramolecular O−H⋅⋅⋅O hydrogen bonds, each formed between a hydroxy group and the nitro group in ortho position, as previously observed in the gas-phase [4].
The electron-withdrawing effect of the nitro groups in ortho and para position of each hydroxy group in 1 significantly increase the acidity (pKa1 = 3.71; pKa2 = 7.49 [5]) in comparison with resorcin (pKa1 = 9.48; pKa2 = 12.08 [6]), but reports on structurally characterized salts of 1 are surprisingly scarce. To the best of our knowledge and based on a search of the Cambridge Structural Database (CSD; version 5.43 with November 2022 updates [7]), the sole example is the crystal structure of the monocesium salt [8], whereas structurally characterized organic salts of 1 appear to be unknown. For salt formation, the nitrogen base 1,4-diazabicyclo[2·2·2]octane (2, Fig. 1) attracted our interest. Compound 2 is a versatile reagent, which has found many applications in organic chemistry [9]. It exhibits two protonation sites (2-\({\text{H}}_{2}^{2+}\): pKa1 = 3.2; pKa2 = 9.0 [10]), which are readily accessible owing to the bicyclic structure of the molecule.
The molecular structure of 2 lacks a centre of symmetry. Its capability to form non-centrosymmetric crystalline salts with achiral anions has been demonstrated, for example, in the dihydrate of the 1:2 proton transfer compound with chloranilic acid (CSD refcode: BOXJOQ) [11] or in the 1:1 salt of 2,4-dinitrobenzoic acid (CSD refcode: CERRUN) [12]. Such materials are potentially interesting for applications in non-linear optics (NLO). In this context, we investigated salt formation of 2 with 1. Herein, we describe the crystal structure of the 1:2 salt and proton-transfer compound of 1 and 2, namely 1,4-diazabicyclo[2·2·2]octane-1,4-diium bis(5-hydroxy-2,4-dinitrophenolate) (3), the title compound (Fig. 1). As far as we are able to ascertain, compound 3 represents the first structurally characterized organic salt of 1.

Chemical diagrams of 1–3
Experimental Section
Synthesis and Crystallisation
Reagent grade 1,4-diazabicyclo[2·2·2]octane (2) was purchased and used as received, and 4,6-dinitroresorcinol (1) was prepared as described previously [3]. Compound 2 (224 mg, 2 mmol) dissolved in 40 mL of methanol was mixed with 1 (400 mg, 2 mmol) dissolved in 60 mL of methanol under continuous stirring. The mixture was stirred for four hours and then left undisturbed at ambient conditions. Orange-yellow crystals of 3 suitable for single-crystal X-ray diffraction analysis were obtained after two weeks.
X-ray Crystallography
Diffraction data were collected on a Bruker AXS X8 Proteum diffractometer with a FR591 rotating anode X-ray source. The data were processed with the SAINT software [13]. The crystal structure was solved with SHELXS-97 [14] and initial independent atom model refinement (IAM) was carried out with SHELXL-2019/3 [15]. The final structure refinement was carried out by Hirshfeld atom refinement with non-spherical atomic form factors using NoSpherA2 [16, 17] partitioning in Olex2 [18] based on electron density from iterative single-determinant SCF single-point DFT calculations using ORCA (version 5.0) [19] with a B3LYP functional [20, 21] and a def2-TZVPP basis set [22]. The hydrogen atom positions were refined with isotropic atomic displacement parameters. For carbon-bound hydrogen atoms, Uiso(H) was constrained to 1.2 Ueq(C). Structure pictures were drawn with Diamond [23] and Mercury [24]. Root mean square (r.m.s.) deviations of atomic positions (hydrogen atoms excluded) were calculated with Mercury. Table 1 lists crystal data and refinement details for 3.
Results and Discussion
Reaction of equimolar amounts of the base 1,4-diazabicyclo[2·2·2]octane and 4,6-dinitroresorcinol in methanol afforded thin orange-yellow plate-shaped crystals of the proton-transfer compound 3, as revealed by X-ray crystallography. The compound crystallizes in the centrosymmetric space group P-1. The asymmetric unit comprises one 2-\({\text{H}}_{2}^{2+}\) cation and two 5-hydroxy-2,4-dinitrophenolate anions (Fig. 2). The transfer of a phenolic acidic proton of 1 to both nitrogen atoms of 2 was evident from the Fobs−Fcalc(IAM) difference map (Fig. 3). Moreover, the average C9−O1 bond length is shorter than the average C11−O2 bond length by 0.09 Å, consistent with deprotonation of O1. The C−N−C bond angles of 2 are increased by 1.7° upon protonation compared with the crystal structure of the free base (polymorph II, CSD refcode: TETDAM08) [25]. With the above-mentioned pKa values for 1 and 2, we calculate ΔpKa = pKa [protonated base] − pKa [acid] = 3.2 − 3.71 = −0.51 for the second protonation step of 2. This suggests that predicting the location of the proton based on aqueous pKa values alone would be difficult [26]. It is assumed that the environment in the crystal has a bearing on the position of the acidic hydrogen atom.

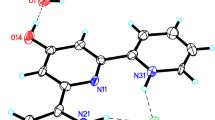
Asymmetric unit of 3. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms are represented by small spheres of arbitrary radius. Dashed lines illustrate hydrogen bonds. The numbers after the underscore indicate the two crystallographically unique 5-hydroxy-2,4-dinitrophenolate anions

F obs−Fcalc(IAM) difference map in a plane through the nitrogen atoms and one of the ethylene bridges in the 2-H22+ cation in 3. Contours are drawn from −0.30 eÅ−3 (red dotted line) through zero (blue dashed line) to 0.60 eÅ−3 in 0.10 eÅ−3 intervals. The hydrogen atoms bonded to N1 and N4 were omitted from the structure factor calculations and thus show up as positive difference electron density close to the parent nitrogen atoms (Color figure online)
The 2-\({\text{H}}_{2}^{2+}\) ion in 3 exhibits approximate molecular D3h point group symmetry (r.m.s. deviation: 0.041 Å). The two protonated tertiary amino groups form charge-assisted N+−H⋅⋅⋅O− hydrogen bonds with the two crystallographically distinct 5-hydroxy-2,4-dinitrophenolate anions. These hydrogen bonds are best described as donating asymmetrically bifurcated (three-centred). The graph set descriptor for the pattern is \({\text{R}}_{1}^{2}\left(6\right)\) [27]. Such a hydrogen bond pattern was, for example, also encountered in the bis(2,4,6-trinitrophenolate) salt of 2 (CSD refcode: QUYPOQ) [28]. Table 2 lists hydrogen bond parameters in 3. These suggest that the N1−H1⋅⋅⋅O1_1 and N4−H4⋅⋅⋅O1_2 interactions involving the phenolate oxygen atoms are, as expected, stronger than the N1−H1⋅⋅⋅O6_1 and N4−H4⋅⋅⋅O6_2 interactions with the nitro oxygen atoms. The nitro group in ortho position to the phenolate oxygen atom is markedly tilted out of the plane of the benzene ring, as revealed by the C9−C14−N2−O6 torsion angles of 19.2(2)° for anion 1 and −10.4(2)° for anion 2. In contrast, the nitro group in ortho position to the hydroxy group is virtually coplanar with the benzene ring in both unique anions and serves as acceptor for an intramolecular O−H⋅⋅⋅O hydrogen formed by the hydroxy group, resulting in a six-membered ring. The graph set notation for this ring is S(6). The hydrogen bond pattern encountered in 3 agrees with Etter’s hydrogen bond rules for organic crystals [29], especially rule 2 stating that six-membered ring intramolecular hydrogen bonds form in preference to intermolecular hydrogen bonds. In accord with this rule, the parent neutral 1 features two such six-membered ring intramolecular hydrogen bonds between hydroxy and nitro groups with the latter being virtually coplanar with the benzene ring [3, 30].
In the chosen asymmetric unit, as shown in Fig. 2, the two crystallographically unique 5-hydroxy-2,4-dinitrophenolate anions are approximately related by local inversion symmetry, which is not fulfilled by the non-centrosymmetric cation. In the crystal, neutral hydrogen-bonded assemblies forming 3 (coinciding with the chosen asymmetric unit) are stacked along the [101] direction via π⋅⋅⋅π stacking of the 5-hydroxy-2,4-dinitrophenolate anions (Fig. 4). Details of the of the π⋅⋅⋅π interactions in the crystal between the rings defined by and C9_1-C14_1 (plane 1) and C9_2-C14_2 (plane 2) are given in Table 3. The sheets so formed are layered in alternating fashion so that no voids remain and are associated via weak van der Waals forces. The crystal structure features pseudo symmetry, namely pseudo B-centring of the lattice with 83% fit, as calculated with ADDSYM in PLATON [31]. This is reflected in the mean intensity of the lattice exceptions for B-centering in the diffraction data, which is half those of possible A-, C-, I- or F-centering, although this difference is less significant when the errors of the intensities are taken into account. As shown in Fig. 5, two crystallographically distinct 5-hydroxy-2,4-dinitrophenolate anions are related by a pseudo translation by the vector 0.5c + 0.5a. The additional translational symmetry is, however, not fulfilled by the 2-\({\text{H}}_{2}^{2+}\) ions. These are related by crystallographic inversion symmetry, as can be seen in Fig. 5.

Packing diagram of 3 a viewed along the a axis direction and b along the [101] direction,, showing the π⋅⋅⋅π stacking of the 5-hydroxy-2,4-dinitrophenolate anions on neighbouring neutral hydrogen-bonded assemblies forming 3 (one such unit is highlighted as a space filling model) Colour scheme: carbon, grey; hydrogen, white; nitrogen, blue; oxygen, red. Hydrogen atoms are omitted for clarity (Color figure online)

Packing diagram of 3. The orange and the green highlightings of the crystallographically unique 5-hydroxy-2,4-dinitrophenolate anions illustrate their respective relation by pseudo symmetry (see text). Neighbouring 2-\({\text{H}}_{2}^{2+}\) cations are related by translation centres of symmetry. Colour scheme: carbon, grey; nitrogen, blue; oxygen, red. Hydrogen atoms are omitted for clarity (Color figure online)
Conclusions
We have structurally characterized the 1:2 proton-transfer compound 3, resulting from reaction of equimolar amounts of 2 and 1, by X-ray crystallography. Cations and anions are linked through charge-assisted intermolecular N+−H⋅⋅⋅O− hydrogen bonds, and the anions exhibit intramolecular O−H⋅⋅⋅O hydrogen bonds. In the crystal, neighbouring neutral hydrogen-bonded assemblies forming 3 are held together by a combination of π⋅⋅⋅π stacking in a zip fastener fashion and van der Waals forces between adjacent layers. Since the second protonation can hardly be predicted from aqueous pKa values alone and we have no evidence that the structure is disordered, we conclude that the crystal environment has a bearing on the proton-transfer. The crystal structure of 3 features pseudo B-centering of the lattice. The centrosymmmetry of the crystal structure renders 3 unsuitable for NLO applications.
Data Availability
CCDC 2304129 contain the supplementary crystallographic data for this paper. The data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
References
Durairaj RB (2005) Resorcinol–chemistry, technology and applications. Springer, Heidelberg
McCrone WC, Corvin I (1952) Crystallographic Data.63. 4,6-Dinitroresorcinol. Anal Chem 24:2008–2009. https://doi.org/10.1021/ac60072a046
Kolev T, Berkei M, Hirsch C, Preut H, Bleckmann P, Radomirska V (2000) Crystal structure of 4,6-dinitroresorcinol, C6H4N2O6. Z Kristallogr NCS 215:483–484. https://doi.org/10.1515/ncrs-2000-0412
Borisenko KB, Zauer K, Hargittai I (1995) Intramolecular hydrogen bonding and molecular geometry of 4,6-dinitroresorcinol from gas-phase electron diffraction. J Phys Chem 99:13808–13813. https://doi.org/10.1021/j100038a010
Milliaresi EE, Ruchkin VE (1974) Use of an empirical method for assigning bands in Uv spectra of mono- and diionized forms of 4,6-dinitroresorcinol and its monomethyl ether. Ukr Khim Zh (Russ Ed) 40:65
Rappoport Z (1984) CRC Handbook of tables for Organic compound identification, 3rd edn. CRC Press/Taylor and Francis, Boca Raton, Florida, USA
Groom CR, Bruno IJ, Lightfoot MP, Ward SC (2016) The Cambridge Structural Database. Acta Crystallogr B 72:171–179. https://doi.org/10.1107/S2052520616003954
Kolev T, Kleb DC, Yancheva D, Schürmann M, Preut Η, Bleckmann P (2001) Crystal structure of cesium 4,6-dinitroresorcinolate,CsC6H3O2(NO3)1. Z Kristallogr NCS 216:63–64. https://doi.org/10.1524/ncrs.2001.216.14.63
Chakraborty N, Mitra AK (2023) The versatility of DABCO as a reagent in organic synthesis: a review. Org Biomol Chem 21:6830–6880. https://doi.org/10.1039/d3ob00921a
Guzonas DA, Irish DE (1988) A Raman and infrared spectroscopic study of triethylenediamine (DABCO) and its protonated forms. Can J Chem 66:1249–1257. https://doi.org/10.1139/v88-203
Sosa-Rivadeneyra MV, Vasquez-Ríos MG, Vargas-Olvera EC, Mendoza ME, Varela-Caselis JL, Meza-León RL et al (2020) Crystal structures of organic salts of chloranilic acid and 2,2′-bi(3-hydroxy-1,4-naphthoquinone) acting as proton donors to 4,4′-Bipyridine and 1,4-Diazabicyclo[2.2.2]octane: 3D networks with bifurcated N+-H···O–/O or N+-H···O/Cl synthons. J Mol Struct 1205:127609. https://doi.org/10.1016/j.molstruc.2019.127609
Rosli MM, Fun H-K, Lee BS, Chantrapromma S (2006) 4-Aza-1-azoniabicyclo[2.2.2]octane 2,4-dinitrobenzoate. Acta Crystallogr Sect E 62(10):o4575–o7. https://doi.org/10.1107/S1600536806037585
SAINT V8.40B, Bruker AXS Inc, Madison, Wisconsin, USA, 2019
Sheldrick GM (2008) A short history of SHELX. Acta Crystallogr A 64:112–122. https://doi.org/10.1107/S0108767307043930
Sheldrick GM (2015) Crystal structure refinement with SHELXL. Acta Crystallogr C 71:3–8. https://doi.org/10.1107/S2053229614024218
Kleemiss F, Dolomanov OV, Bodensteiner M, Peyerimhoff N, Midgley L, Bourhis LJ et al (2021) Accurate crystal structures and chemical properties from NoSpherA2. Chem Sci 12:1675–1692. https://doi.org/10.1039/D0SC05526C
Midgley L, Bourhis LJ, Dolomanov OV, Grabowsky S, Kleemiss F, Puschmann H et al (2021) Vanishing of the atomic form factor derivatives in non-spherical structural refinement - a key approximation scrutinized in the case of Hirshfeld atom refinement. Acta Crystallogr A 77:519–533. https://doi.org/10.1107/S2053273321009086
Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H (2009) OLEX2: a complete structure solution, refinement and analysis program. J Appl Crystallogr 42:339–341. https://doi.org/10.1107/S0021889808042726
Neese F, Wennmohs F, Becker U, Riplinger C (2020) The ORCA quantum chemistry program package. J Chem Phys 152:224108. https://doi.org/10.1063/5.0004608
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652. https://doi.org/10.1063/1.464913
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789. https://doi.org/10.1103/PhysRevB.37.785
Weigend F, Ahlrichs R (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to rn: design and assessment of accuracy. Phys Chem Chem Phys 7:3297–3305. https://doi.org/10.1039/B508541A
Brandenburg K (2018) Diamond. 3.2k4 ed. Crystal Impact GbR, Bonn, Germany
Macrae CF, Sovago I, Cottrell SJ, Galek PTA, McCabe P, Pidcock E et al (2020) Mercury 4.0: from visualization to analysis, design and prediction. J Appl Crystallogr 53:226–235. https://doi.org/10.1107/S1600576719014092
Churakov A (2017) CCDC 1580071: Experimental crystal structure determination. https://doi.org/10.5517/ccdc.csd.cc1q1610
Cruz-Cabeza AJ (2012) Acid–base crystalline complexes and the pKa rule. CrystEngComm 14:6362–6365. https://doi.org/10.1039/C2CE26055G
Bernstein J, Davis RE, Shimoni L, Chang NL (1995) Patterns in Hydrogen Bonding: functionality and graph set analysis in crystals. Angew Chem Int Ed 34:1555–1573. https://doi.org/10.1002/anie.199515551
SiMa W (2010) 1,4-Diazoniabicyclo[2.2.2]octane bis(2,4,6-trinitrophenolate). Acta Crystallogr Sect E 66:o1584. https://doi.org/10.1107/S1600536810021021
Etter MC (1990) Encoding and decoding hydrogen-bond patterns of organic compounds. Acc Chem Res 23:120–126.https://doi.org/10.1021/ar00172a005
Zhang X (2019) CCDC 1511234: Experimental Crystal Structure Determination. https://doi.org/10.5517/ccdc.csd.cc1mqkhd
Spek AL (2009) Structure validation in chemical crystallography. Acta Crystallogr D 65:148–155. https://doi.org/10.1107/S090744490804362X
Acknowledgements
We would like to thank Professor Christian W. Lehmann for providing access to the X-ray diffraction facility at the Max-Planck-Institut für Kohlenforschung. R.W.S. and T.M.K. are grateful to the late Professor William S. Sheldrick for his support of this research, and T.M.K. would like to thank the late Professor Rüdiger Wortmann.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Conceptualization, RWS and TMK; methodology, RWS and RG; validation, RWS and RG; formal analysis, RWS and RG; investigation, RG and TMK; resources, RG and TMK; data curation, RWS and RG; writing—original draft preparation, RWS; writing—review and editing, RG and TK; visualization, RWS and RG; supervision, TMK; project administration, RWS and TMK.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Dedicated to Professor Carl Krüger on the occasion of his 90th birthday.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Seidel, R.W., Goddard, R. & Kolev, T.M. The Pseudo Symmetric Crystal Structure of 1,4-Diazabicyclo[2·2·2]octane-1,4-diium bis(5-hydroxy-2,4-dinitrophenolate). J Chem Crystallogr 54, 125–131 (2024). https://doi.org/10.1007/s10870-023-01004-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-023-01004-z