
Abstract
Electrophilic cyanation of the lithiated cymantrenes [(C5X4Li)Mn(CO)3] (X = H, Cl) yields the corresponding monocyanocymantrenes [(C5X4CN)Mn(CO)3] (1, 3). UV irradiation of 1 in the presence of PPh3 leads to the formation of [(C5H4CN)Mn(CO)2PPh3] (2). The molecular and crystal structures of 1, 2 and 3 were determined. The cyano groups take part in intermolecular C-X⋯N (X = H, Cl) interactions for all compounds.
Graphical Abstract

The crystal structures of the cyanocymantrenes [(C5X4CN)Mn(CO)2L] X = H, Cl, L= CO, PPh3) show numerous C-X⋯Y (X= H, Cl; Y= N, O, C, H) and for L= PPh3 also C-H⋯π interactions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nitriles, particularly aromatic nitriles, are amongst the most-studied organic functional groups. On one hand, they have multiple industrial applications, as in pharmaceuticals or agrochemicals [1, 2]. On the other hand, they can easily be transformed into other important functional groups [3], which again have widespread applications. Last, but not least, nitriles are useful as ligands in coordination chemistry, as they form thermodynamically stable, yet kinetically rather labile complexes [4]. Thus it is not surprising that numerous methods for their preparation exist [5,6,7,8,9,10]. This statement should also hold for ferrocenyl and other metallocenyl nitriles, which are a sub-group of aromatic nitriles. In comparison, however, their chemistry, particularly of the metallocenes apart from ferrocene, seems to be underdeveloped. Although they are known for a while [11], only very few studies devoted to applications have been reported [12]. So far, the main interest seems to have been in their use as complex ligands [13,14,15]. This is also true, when it comes to crystal structure determinations. A search in the CSD (accessed on October 29th, 2021) using “(C5H4CN⋯TR)” as query mask yields 63 hits, of which 60 contain derivatives of cyanoferrocene, and 46 of these have the cyanoferrocene as a ligand to another metal like Cu, Ag, Pd or Pt; changing the search mask to “(C5H3ZCN⋯TR)” yields 12 more hits, all of which contain ferrocene derivatives. The only “non-ferrocene” structures are [(C5H4CN)Co(C4Ph4)] (CYCBCO10) [16], [(C5H4CN)Cr(CO)2(NO)] (KAFCAV) [17] and [(C5H4CN)Ru(C5H5)] (SUFGIL) [18]. It seems quite astonishing, that although it has been stated that “perhaps the most highly studied half-sandwich transition metal compound is..MnCp(CO)3” [19] and “the metal carbonyl complex …most confluent with ferrocene is …MnCp(CO)3” [20] (particularly, when it comes to pharmaceutical studies), there were virtually no structural studies on cyanocymantrenes until very recently [21]. We felt that this class of compounds deserves more attention, and here we report our synthetic and crystallographic studies on three different monocyanocymantrenes [(C5X4CN)Mn(CO)2L] (X = H, Cl; L = CO, PPh3).
Experimental
Starting Materials, Reagents and Instrumentation
[(C5H5)Mn(CO)3] was obtained commercially and was used as received. [(C5X4Br)Mn(CO)3] (X = Cl, Br) were prepared from Mn(CO)5Br and C5X4N2 as described in the literature [22, 23]. The n-BuLi solutions and dimethyl malonodinitrile CMe2(CN)2 were obtained commercially and used as received. Phenyl cyanate PhOCN and ethanedinitrile (CN)2 were prepared as described in the literature [24, 25].
NMR spectra (1H, 13C, 31P) were measured in CDCl3 with a Jeol Eclipse 400 + instrument and were processed and evaluated with the MestReNova program. 1H-NMR spectra were referenced to the residual CHCl3 signal at δ = 7.26 ppm and 13C-NMR spectra to the CDCl3 signal at δ = 77.16 ppm. IR spectra for 1 and 2 were measured on a Bruker IFS 66v/S instrument, while 3 was measured on a Perkin-Elmer 841 instrument.
Synthesis
[(C5H4CN)Mn(CO)3] (1). A solution of [(C5H5)Mn(CO)3] (0.30 g, 1.47 mmol) in THF (10 mL) was treated with 2.5 m n-BuLi solution (0.71 mL, 1.76 mmol) at − 78 °C with stirring for 30 min. After addition of CMe2(CN)2 (0.17 g, 1.76 mmol) stirring was continued for 16 h, while the temperature was gradually raised to ambient. The solution was evaporated to dryness. The residue was taken up in the minimum amount of Et2O and filtered through a silica plug. After evaporation the crude product was chromatographed on silica using petroleum ether/Et2O 8:2 as eluent. The main fraction yielded 1 as a yellow solid (0.29 g, 1.25 mmol, 85%). The spectroscopic data agreed with the literature.
1H-NMR (CDCl3, 400 MHz): δ = 5.31 (t, J = 2.3 Hz, 2H), 4.84 (t, J = 2.2 Hz, 2H). IR (KBr; cm−1): ν (CN, CO) = 2236, 2022, 1920.
[(C5H4CN)Mn(CO)2(PPh3)] (2). A solution of 1 (0.20 g, 0.87 mmol) and PPh3 (0.26 g, 1.00 mmol) in THF (120 mL) was irradiated for 7 h with a Hanau Heraeus TQ150 high-pressure mercury UV lamp. The solvent was evaporated in vacuo, the obtained residue was taken up with the minimum amount of Et2O and filtered through a silica plug. The fitrate was evaporated and the residue was chromatographed on silica using petroleum ether/Et2O 85:15 as eluent. The main fraction yielded 2 as a yellow solid (0.26 g, 0.56 mmol, 64%). The spectroscopic data agreed with the literature.
1H-NMR (CDCl3, 400 MHz): δ = 7.54–7.36 (m, 15H, PPh3), 4.81 (m, 2H, C5H4), 4.27 (m, 2H, C5H4).–13C{1H}-NMR (CDCl3, 101 MHz): δ = 230.3 (d, J = 26.6 Hz), 136.7 (d, J = 42.3 Hz), 132.9 (d, J = 10.4 Hz), 130.1, 128.6 (d, J = 9.8 Hz), 116.7, 87.6, 83.8, 64.1. –31P{1H}-NMR (CDCl3, 162 MHz): δ = 88.4. – IR (KBr; cm−1): ν (CN, CO) = 2237, 1937, 1869.
[(C5Cl4CN)Mn(CO)3] (3). A solution of [(C5Cl4Br)Mn(CO)3] (0.21 g, 0.50 mmol) in Et2O (15 mL) was treated at − 78 °C with 1.6 m n-BuLi solution (0.31 mL, 0.50 mmol) with stirring for 10 min. Then freshly prepared gaseous (CN)2 was condensed into this solution with continuous stirring. The colour of the solution changed to a deep violet. After 2 h, the solvent was evaporated in vacuo at − 78 °C. The residue was extracted with pentane (two 25 mL portions). Evaporation of the solution yielded a yellow solid, which changed its colour to dark green upon standing for several days. IR spectroscopy identified the product as impure 3 (impurities show up in the ν (CH) region and in the region 1500–600 cm−1). CAUTION: Ethanedinitrile is a highly toxic gas! Working in a well-ventilated hood is absolutely necessary!
IR (KBr, cm−1): ν (CN, CO) = 2245w; 2043vs, 1976vs,b. [Lit. 2246vw, 2055, 1995, 1991 [26]]
Upon standing for several months (vide infra) very few yellow crystals formed. The amount of formed crystals did not allow for the measurement of a 13C-NMR spectrum; however, an X-ray diffraction experiment was possible.
IR (KBr, cm−1): ν (CN, CO) = 2246w, 2051vs, 1985vs
Crystallization and Data Collection
The pre-purified compounds 1 and 2 were dissolved at r.t. in the minimum amount of an 85:15 mixture of petroleum ether/diethyl ether and transferred in an open vial to a refrigerator operating at + 5 °C. After standing for several days and slow evaporation of the solvent, yellow crystals were obtained.
Upon standing for 6 months, a few yellow crystals formed amidst the green solid obtained in the synthesis of 3.
Crystals of 1 and 2 were measured on a Bruker D8 Venture diffractometer, while a crystal of 3 was examined on a Syntex R3/Siemens P4 4-circle diffractometer using ω-2θ scans. The latter crystals showed substantial decomposition during the measurement, as calculated from the intensity decay of the 3 check reflections (59%). The intensities of 1 and 2 were corrected for absorption effects using the sadabs-2016/2 option of the diffractometer software [27]. For the absorption correction of 3 the “Numerical/cylindrical” option of Wingx [28] was applied. The structures of 1 and 2 were solved with Shelxt [29,30,31], while the structure of 3 was solved with Shelxs86. Refinements of all structures were performed with Shelxl 2018/3. Table 1 presents general experimental details of the structure determinations.
Special Remarks on the Structure Refinements
One low angle reflection in the refinement of structure 1 and seven low angle reflections in structure 2 had to be omitted because of likely interference with the beam stop. All hydrogen positions were geometrically positioned and refined according to the “riding model”. No further restraints or constraints were applied.
The data collection of the crystals of 3 was performed quite a while ago with a four-circle diffractometer. This meant that reflections with θ > 25° were not collected due to very low intensity; furthermore, due to the decay of the crystal in the X-ray beam only little more than one half of the Ewald sphere was measured. Consequently, the data: parameter ratio amounts only to 11.4 and is thus much poorer than with the other two compounds. Still we think, it is good enough for this dataset to be included in this discussion. There were no reflections omitted and no restraints applied. The unit cell contains two symmetry independent molecules, which show slight differences in the relative orientations of the Mn(CO)3 tripod with respect to the cyclopentadienyl ring. A Platon molfit diagram can be seen in Fig. S1.
Results and Discussion
Synthesis
Cyanocymantrene [(C5H4CN)Mn(CO)3] (1) was first reported in 1967 as the product of the reaction of [Mn(CO)5Cl] with K(C5H4CN) in 58% yield [32, 33]. Later on, alternative synthetic procedures using thermolysis of [(C6H5N3)Mn(CO)3]+ (22% yield) [34] or dehydration of the intermediate oxime [(C5H4CHNOH)Mn(CO)3] in an overall yield of 51% [35]. We used the strategy of electrophilic cyanation of lithiated cymantrene [(C5H4Li)Mn(CO)3] with dimethylmalononitrile, which we had employed recently for the synthesis of [(C5H4CN)Mn(CO)2PPh3] (2) [21]. 1 was obtained in a yield of 85%.
The Dicarbonyl-triphenylphosphine complex [(C5H4CN)Mn(CO)2PPh3] (2), had been prepared by us before—as just mentioned-by lithiation of [(C5H4Br)Mn(CO)2PPh3] followed by electrophilic capture with CMe2(CN)2, in 45% yield. Alternatively, 2 can also be obtained via irradiation of a THF solution of 1 in the presence of PPh3, in a yield of 64%.
[(C5Cl4CN)Mn(CO)3] (3) had been obtained by us in low yield by dehydration of [(C5Cl4CONH2)Mn(CO)3] using POCl3 [26], in a multistep synthesis starting from the lithiated [(C5Cl4Li)Mn(CO)3]. A more “direct” procedure would be electrophilic cyanation of this lithiated intermediate. While there are numerous reagents available for this purpose, we decided to use cyanogen (CN)2, which had been introduced for “direct cyanation of aromatics” back in 1980 [36] and had already been used successfully for the cyanation of Li(C5Me5) [37]. In the present case the formation of intensely coloured solutions and the instability of the isolated solid product hinted to the formation of some radicals. Although a few crystals of 3 could be obtained for a crystal structure determination, we discarded this synthetic pathway.
Molecular Structure of [(C5H4CN)Mn(CO)3], 1
Compound 1 crystallizes in the triclinic space group P-1 with one molecule in the asymmetric unit. As can be seen in Fig. 1, the cyano group is in relative “trans” position to one Mn–CO group, while the other carbonyl groups eclipse one C–H bond each. The bond from Mn1 to the cyano group bearing cyclopentadienyl carbon C1 is the shortest; however, there is a relatively small spread (ca. 6σ) in the Mn–Ccp distances (Table 2). The C–C bonds in the ring show a distortion towards a “butadiene-yl” structure (two short and three longer bonds). The cyano group is significantly bent away to the distal side of the ring plane (distance 0.141(3) Å). The length of the C–N bond is comparable to the values found for the above-mentioned Ru (1.148 Å), Cr (1.132 Å) and Co (1.16/1.19 Å) cyanocyclopentadienyl complexes, as well as the bending towards the distal side of the ring. However, the ring distortion is not observed in these three complexes.

Top view of compound 1. 30% probability ellipsoids
Molecular Structure of [(C5H4CN)Mn(CO)2(PPh3)], 2
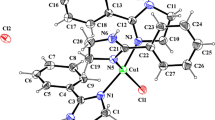
Compound 2 crystallizes in the orthorhombic space group Pbca with one molecule in the asymmetric unit. Figure 2 shows an ortep3 top view of the structure. While the cyano group is still in a “transoid” position with respect to the PPh3 ligand (torsion C1–Ct–Mn1–P1 153.1°), the Mn–P bond prefers an eclipsed position with the C3–H3 bond. The distance of the manganese atom from the cyclopentadienyl centroid is longer (> 13σ) than in 1, and there is also a much wider spread (> 25σ) in the individual Mn–Ccp distances, with the bond from Mn1 to the cyano bearing carbon being the shortest again. The C–C bonds of the cyclopentadienyl ring show no alternation, with the shortest bond occurring between the two “meta” carbons. The cyano group is only slightly bent away from the ring plane (distance 0.044(2) Å).

Top view of compound 2, 30% probability ellipsoids
It is also interesting to compare 2 with the recently published structures of [{C5H3(CN)2}Mn(CO)2PPh3] and [{C5H2(CN)3}Mn(CO)2PPh3] [26]. In these compounds the C–N bonds are in the range 1.138(6)–1.147(6) Å with C–C–N angles in-between 174.3(4) ° and 179.4 (2)°, with the longer bonds and larger angles with the dicyano compound. The distances of the Mn atoms from the cyclopentadienyl ring centroids are reported to be around 1.772(2) Å. In both compounds the Mn → P vector eclipses a cyclopentadienyl C–H bond. The Mn–P bond in 2 measures 2.2473(5) Å, while in the dicyano compound 2.2549(5) Å and the tricyano compound an average distance of 2.264(1) Å is found. As can be seen from these data the major effect of increasing cyano substitution is an increase of the Mn–P bond lengths.
Molecular Structure of [(C5Cl4CN)Mn(CO)3], 3
Compound 3 crystallizes in the triclinic space group P-1 with two molecules in the asymmetric unit (Fig. 3).

Top view of the molecular structure of compound 3 (left: Mol.A, right: Mol.B). 30% probability ellipsoids
As can be seen from Figs. 3 and S1 of the Supporting Information and Table 2, there are only slight but distinct differences between both molecules. The major difference occurs in the relative orientations of the cyclopentadienyl ring and the Mn(CO)3 tripod. While in molecule A the cyano group is nearly perfectly in a “trans” position to the Mn1–C7–O2 group, which thus bisects the C3–C4 bond of the ring, the Mn2–C17–O12 group in molecule B, while still “transoid” with respect to the cyano group, prefers a more eclipsed orientation with respect to the C14–Cl7 bond. The C–N bond is in both molecules shorter than in compounds 1 and 2. The distances from manganese to the ring centroids are the same as in compound 1 and shorter than in 2, which leads to the conclusion that for this parameter the presence of a phosphine ligand is more important than the substituents on the cyclopentadienyl ring. The C–C bonds within the cyclopentadienyl rings show a similar distortion towards a “diene-yl” form (two shorter and three longer bonds) as compound 1, although this has to be regarded with caution due to the relatively high standard deviations in 3. All ring substituents in both molecules are bent away from the metal atoms, with the cyano nitrogens being particularly displaced from the ring planes (0.249(18) Å in Mol. A, and 0.144(14) Å in Mol. B).
Intermolecular Interactions
When looking at weak interactions in the crystal of compound 1, there are two kinds of “non-classical” hydrogen bonds present. First, there are two C–H⋯O interactions (Fig. 4, left).

“Non-classical” hydrogen bonds in 1: symmetry operators left: for O1: − x,2−y,1−z, for O2: x−1,y,z; right: for N1: 1−x,2−y,− z
Two inversion-related molecules are joined pairwise via H2⋯O1 (d = 2.68 Å, dC–O = 3.588(3) Å) interactions, and these “pairs” are connected in a direction via two H4⋯O2 (d = 2.64 Å, dC–O = 3.333(42) Å) interactions. Second, there are two C–H⋯N interactions (d = 2.59 Å, dC–N = 3.367(4) Å) (Fig. 4, right), that join pairwise another pair of inversion-related molecules.
In comparison to compound 1, one might expect for 2 many more C–H⋯X interactions due to the presence of 15 phenyl CH groups. Indeed, Fig. 5 shows that this is the case.

“Non-classical” hydrogen bonds in 2: symmetry operators left: for O1: 1−x,1−y,1−z; for O2: x−½, y, ½ − z; right: for N1: 1−x, 1−y, − z, (towards H5) and: ½−x,1−y, z−½ (towards H125)
Again, there are two C–H⋯O interactions (Fig. 5, left): Two inversion related molecules are joined pairwise via H123⋯O1 (d = 2.45 Å, dC–O = 3.266(2) Å), and these dimers are doubly joined via the a glide using H106⋯O2 interactions (d = 2.66 Å, dC–O = 3.278(2) Å). Second, there are two independent C–H⋯N interactions (Fig. 5, right). One of them (H5⋯N1) joins two inversion related molecules (different from the pair in Fig. 5 left; translational shift along c) (d = 2.67 Å, dC–N = 3.388(2) Å), while the other joins a glide- related molecules via H125⋯N1 (d = 2.57 Å, dC–N = 3.449(2) Å). Besides these C–H⋯O and C–H⋯N interactions there are also C–H⋯C interactions in the crystal of 2. What might be best described as “edge-to-face phenyl interaction” is characterized by two short contacts between H104 and C124/C125 (d = 2.77 and 2.89 Å) and a distance between H104 and the C121–C126 ring centroid of 2.81 Å (see Fig. S2 of the Supporting Information). What might look like a face-to-face π-interaction in Fig. 5 (left picture), is too offset (centroid…centroid distance 4.267 Å) to be regarded as an important interaction.
Since compound 3 does not contain any H atoms, hydrogen bonds have no stabilization effect on the crystal structure. However, as a packing diagram shows (Fig. 6), there are some stabilizing intermolecular interactions involving the chlorine atoms.

Packing diagram of 3, watched along a, showing the stabilizing Cl⋯N, Cl⋯O and Cl⋯Cl contacts
There are two Cl⋯N interactions: N1⋯Cl6 (d = 3.096(9) Å; symm. op. x, y−1, z−1) and N2⋯Cl1 (d = 3.100(9) Å, symm. op. x, y−1, z); one Cl⋯O interaction between O2 and Cl7 (d = 3.109(7) Å, symm. op. 1−x, 1−y, 2−z) and one rather strong Cl⋯Cl interaction between Cl2 and Cl5 (d = 3.484(4) Å, symm. op. x, y−1, z). Figure 6 shows also the formation of double strands along the c axis. The double strands are not interconnected in b direction.
An alternative way of looking at intermolecular interactions is by performing a Hirshfeld surface analysis [38]. For this purpose, we employed the freely available program CrystalExplorer [39]. Figure S3 of the Supporting Information shows the calculated Hirshfeld surfaces of the three compounds. The red “spots” on the surfaces close to the CN, CO and CH groups show the presence of close interactions with distances smaller than the sum of van der Waals radii between atoms inside and outside the surface. While there are many aspects of intermolecular interactions that can be visualized with this program, we want to concentrate on the “Fingerprints” (Figs. S4, 7) and “Interaction Energies” (Figs. S5, S6, 8). “Fingerprint plots” visualize the number of close contacts between nuclei inside a Hirshfeld surface and those outside, either in general or separated according to specific atom pairs [40]. Figure S3 shows the general Fingerprints, summing up all close interactions. Figure 7 shows the different H⋯X (compounds 1 and 2) and Cl⋯X (compound 3), respectively, contacts, with the percentages referring to the absolute number of close contacts. Simple addition of the numbers shows, that for compound 1 the contacts involving hydrogen make up for roughly two thirds of all contacts (67.8%), while in compound 2 they are responsible for nearly all of them (94.1%). In compound 3, close contacts involving chlorine make up for nearly three quarters of all close contacts (72.8%). While H⋯H contacts are virtually not existent for compound 1, they are the largest single contributor in compound 2. It can also be concluded, that C–H⋯π interactions determine the π stacking interactions and not the π–π interaction between the phenyl rings.

“Fingerprint plots” of compounds 1 (first row), 2 (second row) and 3 (third row)

Interaction energies (HF/3-21G) for the crystal of compound 1
Another interesting aspect of intermolecular interactions are the involved interaction energies [41,42,43]. Figure 8 shows the calculated energies (as a combination of the electrostatic, polarization, dispersion and repulsion terms, using HF/3-21G) between a central molecule of compound 1 and its 13 closest neighbours. The corresponding visualizations for compounds 2 and 3 are shown in Fig. S5/6 in the Supporting Information. As can be seen from these figures, Etot ranges from slightly destabilizing (+ 1.0 kJ/mol in compound 1) to strongly stabilizing (− 39.0 kJ/mol in compound 1, − 38.7 kJ/mol in compound 2). While electrostatic terms are important in some cases, in most cases the interplay between dispersion and repulsion terms determines the total outcome. As might be expected the largest influence of dispersion terms (up to − 57 kJ/mol) occurs in compound 2 due to its many phenyl- phenyl interactions.
Conclusions
Cyanocymantrenes 1 and 3 have been obtained by electrophilic cyanation of the lithiated cymantrenes [(C5X4Li)Mn(CO)3] with either dimethylmalononitrile or ethanedinitrile, with the former one being the reagent of choice. While an analogous preparation of 2 had been reported before, irradiation of 1 in the presence of PPh3 yielded the desired compound in higher yield. The C–N bond in 3 is significantly shorter than in the other two compounds. However, otherwise the molecular bond parameters of the two tricarbonyl complexes are much more similar to each other than to the dicarbonyl complex 2. As we had observed before, the replacement of one CO by PPh3 has a larger effect on the molecular parameters than the substitution of hydrogen by chlorine within the cyclopentadienyl moiety. For the intermolecular interactions the formation of weak C-H⋯X (X = N, O, C, H) and C–Cl⋯X “bonds” are most important for all three compounds; in the PPh3 complex 2 they are responsible for nearly 95% of all interactions.
Data Availability
CCDC 2123987–2123989 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. The authors declare that all other data supporting the findings of this study are available within the article and its supplementary information files.
References
Fleming FF, Yao L, Ravikumar PC, Funk L, Shook BC (2010) J Med Chem 53:7902–7917
Kleemann A, Engel J, Kutscher B, Reichert D (2001) Pharmaceutical substances: synthesis, patents, applications, 4th edn. Georg Thieme Verlag, Stuttgart, p 241
Fatiadi J (1983) In: Patai S, Rappoport Z (eds) Preparation and synthetic applications of cyano compounds. Wiley-VCH, New York
Rach SF, Kühn FE (2009) Chem Rev 109:2061–2080
Wang L, Shao Y, Cheng J (2021) Org Biomol Chem 19:8646–8655
Cheng H, Guo P, Ma J, Hu X (2021) Catal Sci Technol 11:3308–3325
Ahmad MS, Puldindi IN, Li C (2020) New J Chem 44:17177–17197
Neetha M, Afsina CMA, Aneeja T, Anilkumar G (2020) RSC Adv 10:33683–33699
Nauth AM, Opatz T (2019) Org Biomol Chem 17:11–23
Cui J, Song J, Liu Q, Liu H, Dong Y (2018) Chem Asian J 13:482–495
Aghoramurthy K (1956) J Sci Ind Res 15B:11–13
Quintana C, Klahn AH, Artigas V, Fuentealba M, Arancibia R (2015) Inorg. Chem Commun 55:48–50
Vosahlo P, Harmach P, Cisarova I, Stepnicka P (2021) J Organomet Chem 953:122065
Strehler F, Hildebrandt A, Korb M, Rüffer T, Lang H (2014) Organometallics 33:4279–4289
Wilkinson LA, Yue TTC, Massey E, White AJP, Long NJ (2019) Dalton Trans 48:72–78
Villa AC, Coghi L, Manfredott AG, Guastini C (1974) Acta Cryst Sect B 30:2101
Rogers RD, Shakir R, Atwood JL, Macomber DW, Wang YP, Rausch MD (1988) J Crystallogr Spectrosc Res 18:767
Strehler F, Korb M, Lang H (2015) Acta Cryst Sect E 71:398
Laws DR, Chong D, Nash K, Rheingold AL, Geiger WE (2008) J Amer Chem Soc 130:9859–9870
Wu K, Pudasaini B, Park JY, Top S, Jaouen G, Baik M, Geiger WE (2020) Organometallics 39:679–687
Klein-Heßling C, Blockhaus T, Sünkel K (2021) Inorg Chim Acta 528:120619
Reimer KJ (1975) ShaverA. Inorg Chem 14:2707–2716
Herrmann WA, Huber M (1977) J Organomet Chem 140:55–61
Murray RE, Zweifel G (1980) Synthesis 150–151
Brauer G (1978) Handbuch der Präparativen Anorganischen Chemie, 3. Aufl. Bd. 2, S. 630, Ferdinand Enke Verlag, Stuttgart
Sünkel K, Steiner D (1989) J Organomet Chem 368:67–76
Krause L, Herbst-Irmer R, Sheldrick GM, Stalke D (2015) J Appl Cryst 48:3–10
Farrugia LJ (2012) J Appl Cryst 45:849–854
Sheldrick GM (2015) Acta Cryst A 71:3–8
Sheldrick GM (2015) Acta Cryst C 71:3–8
Sheldrick GM (1985) J Mol Struct 130:9–16
Nesmeyanov AN, Kolobova NE, Anisimov KN, Makarov YN (1967) Bull Acad Sci USSR Chem Sci 16:928
Christopher RE, Venanzi LM (1973) Inorg Chim Acta 7:219–225
Munro GAM, Pauson PL (1978) J Organomet Chem 160:177–181
Kolobova NE, Valueva ZP, Solodova MY (1980) Bull Acad Sci USSR Chem Sci 29:1701–1705
So YH, Miller LL (1980) J Am Chem Soc 102:7119–7120
Jutzi P, Schwartzen KH, Mix A (1990) Chem Ber 123:837–840
Spackman MA, Jayatilaka D (2009) CrystEngComm 11:19–32
Spackman PR, Turner MJ, McKinnon JJ, Wolff SK, Grimwood DJ, Jayatilaka D, Spackman MAJ (2021) Appl Cryst 54:1006–1011
Spackman MA, McKinnon JJ (2002) CrystEngComm 4:378–392
Mackenzie CF, Spackman PR, Jayatilaka D, Spackman MA (2017) IUCrJ 4:575–587
Turner MJ, Thomas SP, Shi MW, Jayatilaka D, Spackman MA (2015) Chem Commun 51:3735–3738
Spackman MA (2015) Cryst Growth Des 15:5624–5628
Acknowledgements
We thank Dr. P. Mayer for performing the X-ray measurements. A special “thanks” to Peter Spackman for his invaluable support in getting started with “CrystalExplorer”.
Funding
Open Access funding enabled and organized by Projekt DEAL. The authors did not receive support from any organization for the submitted work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to declare that are relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sünkel, K., Reimann, D. & Klein-Heßling, C. Synthesis, Molecular and Crystal Structures of Some Monocyanocymantrenes. J Chem Crystallogr 52, 315–323 (2022). https://doi.org/10.1007/s10870-022-00929-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-022-00929-1