
Abstract
The title compound, diethyl (6-methyl-2-pyridyl)aminoethylenemalonate (1), crystallises in the monoclinic space group P21/c (No. 14). The unit cell parameters are a = 10.5657(7) Å, b = 9.1784(5) Å, c = 14.5681(7) Å, β = 101.636(6)°, Z′ = 1 and Z = 4 at 150 K. The extended structure forms approximately orthogonal columns of stacked molecules. All bond lengths and angles are unremarkable. No disorder, twinning or co-crystallised solvent is present in the structure. An intramolecular hydrogen bond exists between the enamine nitrogen and carbonyl oxygen. Hirshfeld surface analysis reveals a short contact between a carbonyl oxygen and neighbouring aryl hydrogen, as well as a carbonyl–carbonyl interaction.
Graphical Abstract
The X-ray structure of diethyl (6-methyl-2-pyridyl)aminoethylenemalonate (1), an intermediate in the synthesis of ethyl nalidixate, is reported, and Hirshfeld surface analysis employed to identify intermolecular interactions within the structure.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Initially reported in 1962 [1], the title compound diethyl (6-methyl-2-pyridyl)aminoethylenemalonate (1) is commonly prepared as an intermediate in the synthesis of ethyl nalidixate in undergraduate medicinal chemistry laboratories. Ethyl nalidixate has since become ubiquitous in medicinal chemistry as a lead compound for the development of quinolone antibiotics [2]. Synthesis of 1 entails a condensation reaction between 2-aminopicoline and diethyl ethoxymethylenemalonate (Scheme 1). Despite its role as a key characterisation technique for inorganic and organometallic compounds, single crystal X-ray diffraction (SC-XRD) is much less routinely employed for the characterisation of neutral organic compounds, and the solid-state structure of 1 has not been previously reported.

Synthesis and molecular structure of 1
Hirshfeld surface analysis [3] has recently become prevalent for describing interactions within crystals, and provides a method to view molecules as ‘organic wholes’ [4]. It offers a tool for visualising interactions in crystal structures, allowing for comparison between polymorphs and chemically similar structures [5]. By mapping the distance from the nearest external nucleus (de) onto the Hirshfeld surface, represented as colour contours on the generated 3D visualisations, we can identify points of intermolecular interaction. Plotting de against di (the distance to the nearest internal nucleus) gives 2D-fingerprint plots which allow more detailed analysis of these interactions.
Experimental
NMR spectra were recorded on a JEOL Eclipse 400 MHz NMR spectrometer (399.60 MHz for 1H and 100.48 MHz for 13C). LC/MS was carried out using a Waters Acquity UPLC (Waters Corp, Milford, MA, USA), coupled to a Waters Xevo G2-XS Q-TOF mass spectrometer (Waters Corp, Milford, MA, USA). Melting point measurements are uncorrected. Yield and spectra were in close agreement with literature values [2].
Synthesis of 1
2-Aminopicoline (2.00 g, 18.49 mmol) and diethyl ethoxymethylenemalonate (4.00 g, 18.49 mmol) were combined and heated at 110 °C for 90 min with stirring. The precipitate formed on cooling was recrystallized from ethanol to afford 1 as a white powder. Yield: 4.11 g, 80%.
1H-NMR (400 MHz, CDCl3): δ 11.04 (1H, d br, J = 12.58 Hz, NH), 9.25 (1H, d, J = 13.04 Hz, C=C–H), 7.51 (1H, t, J = 7.78 Hz, Ar–H), 6.86 (1H, t, J = 7.55, Hz, Ar–H), 6.64 (1H, t, Ar–H), 4.27(4H, dq, J = 19.06 Hz, 7.08 Hz, OC–H2), 2.48 (3H, s, Ar–C–H3), 1.34 (3H, m, C–CH3).
13C-NMR (100 MHz, CDCl3): δ 14.28 (s), 14.40 (s), 24.29 (s), 60.14 (s), 60.48 (s), 94.99 (s), 108.61 (s), 119.08 (s), 138.68 (s), 149.89 (s), 150.08 (s), 165.69 (s).
LC/MS [M + H]+: Calc. for C14H19N2O4 = 279; found 279.
Melting point (ethanol): 104–106 °C.
SC-XRD quality crystals were obtained by slow evaporation of a solution of 1 in tetrahydrofuran over 3 days.
X-Ray Crystallography
Crystallographic measurements were performed at 150 K using an Oxford Xcaliubur Gemini diffractometer with a Sapphire 3 CCD plate (graphite-monochromated Cu Kα radiation, λ = 1.54184 Å). A crystal of suitable size and quality was selected, coated with Fomblin® Y oil, and mounted onto a nylon loop. The Numerical absorption correction was based on gaussian integration over a multifaceted crystal model [6]. Empirical absorption correction, using spherical harmonics, implemented in SCALE3. The ABSPACK scaling algorithm was applied for absorption correction [7]. Cell refinement, data collection and data reduction were performed using Rigaku CrysAlisPro 1.171.39.46 [8]. The crystal density was not measured.
Refinement
Using Olex2 [9], the structure was solved with the ShelXT structure solution program using intrinsic phasing and refined with the ShelXL refinement package using Least Squares minimisation [10, 11]. All hydrogen atoms were geometrically placed and refined using the riding model approximation. Methyl groups were refined as a rotating group. Structural and refinement parameters are given in Table 1. Bond lengths, angles and atomic displacement parameters are given in the supplementary information.
Structural Analysis
Bond parameters were measured using Mercury 3.10 [12,13,14]. Hirshfeld surface analysis was performed using CrystalExplorer [15]. The Hirshfeld surface was mapped using an isovalue of 0.5. Red contours indicate a contact less than the sum of the Van Der Waals radii of the respective elements. Blue and white contours indicate that the nearest external atom is at a distance greater than or equal to the sum of the Van Der Waals radii respectively from the atomic co-ordinate. As hydrogen atoms have been placed geometrically, e.s.d. are not included in quoted bond lengths.
Results and Discussion
Asymmetric Unit
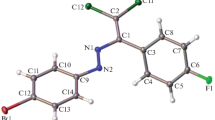
1 Crystallises in the monoclinic space group P21/c (No. 14). The asymmetric unit consists of one molecule of 1 (Fig. 1) and the unit cell consists of four molecules of 1.

Representation of the solid-state structure of 1. Thermal ellipsoids are shown at 50% probability. Hydrogen atom labels are omitted for clarity. The O1–N2 hydrogen bond is shown as a tan dashed line
The molecular configuration of 1 is essentially planar in the solid state, with C4 and C14 having the largest deviation from the plane at 0.117 Å and 0.129 Å respectively. This planarity is supported by a hydrogen bonding interaction between O1 and N2, which forms a six-membered ring (O1–C9–C8–C7–N2–H2) and locks the conformation of the ester groups. The strongly downfield shift of the enamine proton in the 1H NMR spectrum (11.04 ppm) suggests that this hydrogen bonding interaction is maintained in solution. No disorder, twinning or co-crystallised solvent is present in the structure. All bond lengths and angles are unremarkable. The bond lengths and angles in the heterocyclic component of 1 are similar to pyridine [16].
Intermolecular Interactions
Molecules are stacked in a head-to-tail fashion (Fig. 2). The interplanar distance between molecules is 3.396 Å. Stacked molecules are fully co-planar, with an angle of 0.00° between them. These planes are offset by 2.84 Å. The resulting columns of stacked molecules are approximately orthogonal to one another with an angle of 86° between them.

a Parallel view of the asymmetric unit of 1 with stacked molecule above and below it. b Perpendicular view of the asymmetric unit of 1 with stacked molecule above and below it. The molecule planes and cell axes are shown. Atom labels and hydrogen atoms omitted for clarity
Database Survey
Eleven crystal structures possessing a 2-(2,2-dicarbalkoxyvinylamino)-pyridine substructure exist in the Cambridge Structural Database (CSD, Version 5.39, November 2017). The most comparable to 1 is 2-(N-(2,2-diformylethenyl)amino)pyridine (CCDC:297480, Fig. 3). Similarly to 1, a hydrogen bond interaction exists between the enamine N and a carbonyl O. The supramolecular structure is similar to 1, in that the molecule is planar, with a shorter intermolecular stacking distance (3.389 Å).

Structure of 2-(N-(2,2-diformylethenyl)amino)pyridine (CCDC:297480), showing the intramolecular hydrogen bond in grey
Hirshfeld Surface Analysis
The Hirshfeld analysis reveals a short contact between O3 and H5 of a neighbouring molecule of 2.402 Å, demonstrated by red contours on the Hirshfeld surface at these atoms (Fig. 4). A fairly short contact is also identified between C9 and C12 (3.444 Å). This is likely the result of a carbonyl–carbonyl stacking interaction. Such interactions have been found in other small molecules and the secondary structures of proteins, and are the result of n → π* and π → π* interactions [17]. There is also a short contract between H3 and C11 (2.817 Å).

Hirshfeld surface of 1. a View down a axis, b view down b axis, c view down c axis
The fingerprint plots (Fig. 5) show that the predominant intermolecular interaction is H–H interactions; these account for 60% of the Hirshfeld surface (Fig. 5c). These interactions are presumed to be primarily the result of attractive dispersion forces [18]. C–C interactions, primarily the result of stacking interactions, also contribute to the Hirshfeld surface (Fig. 5b).

Hirshfeld fingerprint plots for a all intermolecular interactions. b C–C interactions. c H–H interactions. d O(inside)–All(outside) interactions. e N(inside)–All(outside). f All(inside)–O(outside). g All(inside)–N(outside)
Conclusions
Here, we have presented the solid-state structural analysis of a common organic small molecule. Intramolecular H-bonding is present in the structure, and Hirshfeld surface analysis has been used to reveal intermolecular interactions that would otherwise have been difficult to elucidate.
References
Lesher GY, Froelich EJ, Gruett MD, Bailey JH, Brundage RP (1962) Communication to the editor: 1,8-naphthyridine derivatives. A new class of chemotherapeutic agents. J Med Chem 70:1063–1065
Leslie R, Leeb E, Smith RB (2012) Synthesis of ethyl nalidixate: a medicinal chemistry experiment. J Chem Educ 89:144–146. https://doi.org/10.1021/ed200148h
Spackman MA, Byrom PG (1997) A novel definition of a molecule in a crystal. Chem Phys Lett 267:215–220. https://doi.org/10.1016/S0009-2614(97)00100-0
Weber E, Aoyama Y, Caira M, Desiraju G, Glusker J et al (1998) Design of organic solids. Springer, Berlin, Heidelberg
Spackman MA, Jayatilaka D (2009) Hirshfeld surface analysis. Cryst Eng Comm 11:19–32. https://doi.org/10.1039/b818330a
Clark RC, Reid JS (1995) The analytical calculation of absorption in multifaceted crystals. Acta Crystallogr Sect A 51:887–897. https://doi.org/10.1107/S0108767395007367
Oxford Diffraction (2005) SCALE3 ABSPACK—An oxford diffraction program (Oxford Diffraction)
Rigaku Oxford Diffraction (2018) CrysAlisPro 1.171.39.46 (Rigaku Oxford Diffraction)
Dolomanov OV, Bourhis LJ, Gildea RJ et al (2009) OLEX2: a complete structure solution, refinement and analysis program. J Appl Crystallogr 42:339–341. https://doi.org/10.1107/S0021889808042726
Sheldrick GM (2015) SHELXT integrated space-group and crystal-structure determination. Acta Crystallogr Sect A 71:3–8. https://doi.org/10.1107/S2053273314026370
Sheldrick GM (2015) Crystal structure refinement with SHELXL. Acta Crystallogr Sect C 71:3–8. https://doi.org/10.1107/S2053229614024218
Macrae CF, Bruno IJ, Chisholm JA et al (2008) Mercury CSD 2.0—new features for the visualization and investigation of crystal structures. J Appl Crystallogr 41:466–470. https://doi.org/10.1107/S0021889807067908
Macrae CF, Edgington PR, McCabe P et al (2006) Mercury: visualization and analysis of crystal structures. J Appl Crystallogr 39:453–457. https://doi.org/10.1107/S002188980600731X
Bruno IJ, Cole JC, Edgington PR et al (2002) New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallogr Sect B 58:389–397. https://doi.org/10.1107/S0108768102003324
Wolff SK, Grimwood DJ, McKinnon JJ, et al (2012) CrystalExplorer (Version 3.1)
Mootz D, Wussow HG (1981) Crystal structures of pyridine and pyridine trihydrate. J Chem Phys 75:1517–1522. https://doi.org/10.1063/1.442204
Rahim A, Saha P, Jha KK et al (2017) Reciprocal carbonyl-carbonyl interactions in small molecules and proteins. Nat Commun 8:1–12. https://doi.org/10.1038/s41467-017-00081-x
Rösel S, Quanz H, Logemann C et al (2017) London dispersion enables the shortest intermolecular hydrocarbon H···H contact. J Am Chem Soc 139:7428–7431. https://doi.org/10.1021/jacs.7b01879
Acknowledgements
We would like to thank Dr. Toby Blundell and Dr. Benjamin Lake for helpful discussions on crystallography.
Funding
Funding was provided by Nottingham Trent University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Coughlin, O., De Bruyn, N., Kilgour, D.P.A. et al. Crystal Structure and Hirshfeld Surface Analysis of Diethyl (6-methyl-2-pyridyl)aminoethylenemalonate. J Chem Crystallogr 50, 212–218 (2020). https://doi.org/10.1007/s10870-019-00789-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-019-00789-2