
Abstract
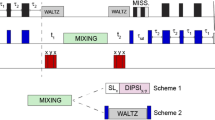
Carbohydrates influence many essential biological events such as apoptosis, differentiation, tumor metastasis, cancer, neurobiology, immunology, development, host-pathogen interactions, diabetes, signal transduction, protein folding, and many other contexts. We now report on the structure determination of pregnane glycosides isolated from the aerial parts of Ceropegia fusca Bolle (Asclepiadaceae). The observation of cicatrizant, vulnerary and cytostatic activities in some humans and animals of Ceropegia fusca Bolle, a species endemic to the Canary Islands, encouraged us to begin a pharmacological study to determine their exact therapeutic properties. High resolution 1H-NMR spectra of pregnane glycosides very often display well-resolved signals that can be used as starting points in several selective NMR experiments to study scalar (J coupling), and dipolar (NOE) interactions. ROESY is especially suited for molecules such that ωτc ~ 1, where τc are the motional correlation times and ω is the angular frequency. In these cases the NOE is nearly zero, while the rotating-frame Overhauser effect spectroscopy (ROESY) is always positive and increases monotonically for increasing values of τc. The ROESY shows dipolar interactions cross peaks even in medium-sized molecules which are helpful in unambiguous assignment of all the interglycosidic linkages. Selective excitation was carried out using a double pulsed-field gradient spin-echo sequence (DPFGSE) in which 180° Gaussian pulses are sandwiched between sine shaped z-gradients. Scalar interactions were studied by homonuclear DPFGSE-COSY and DPFGSE-TOCSY experiments, while DPFGSE-ROESY was used to monitor the spatial environment of the selectively excited proton. Dipolar interactions between nuclei close in space can be detected by the 1D GROESY experiment, which is a one-dimensional counterpart of the 2D ROESY method. The C-12 and C-17 configurations were determined by ROESY experiments.
Similar content being viewed by others


Introduction
Chemical methods, such as acid hydrolysis followed by TLC, GC or HPLC analysis are frequently used to identify glycoside monosaccharides. A range of ionization mechanisms can occur in water (Smith et al. 1999). Water is useless as a solvent because of two kinds of leveling effect. When an acid is stronger than hydronium cations, it reacts with water and is converted essentially completely to hydronium cations. The second kind of leveling effect is found with acids weaker than water, since the amount of hydronium cations produced by the acid is less than that produced by the autoprotolysis of the water and thus cannot be measured. Acid or base strength depends on the acidity or basicity of the solvent since it has an influence on activity coefficients, but other characteristics of the solvent are also important, such as the dielectric constant, and it is important because it is a measure of the ion-solvating ability of the solvent. In low dielectric constant solvents (ε < 10) the amount of free ions may even be negligible, then ions aggregate, such that ion pairs and larger aggregates are present. In such solvents molecular ionogens such as HCl, HNO3 or HCOOH behave like other protic molecules (Chabanel 1990; Skvortsov et al. 2009; Wang et al. 2009). Solvents with high dielectric constants, like water (ε > 10) completely solvate each ion.
Studies of aqueous solutions of carbohydrates are complicated because they are present in solution in several forms (Junquera et al. 2002). By means of modern NMR techniques the problems derived from free sugar mutarotation can be avoided (specific optical rotation of aqueous sugar solution changes over time).
Pregnane glycosides are of considerable interest because of their antineoplasic effects (Ingrassia et al. 2006; Yokosuka and Mimaki 2008; Peng et al. 2008) and cardiotonic activity (Nohara et al. 2007), and also because they act as antagonists for the proliferation stimulant hormone and display anti-herpes activity (Russell and Kahn 2007). In the course of a drug-screening project on medicinal plants in the search for biologically active compounds, we now report on the structure determination of tetrarhabinoside-12-tigloyl-14-hydroxy pregnane (1) (4.84%), tetrarhabinoside-12-(2′-amino)-benzoyl-14-hydroxy pregnane (2) (2.3%), tetrarhabinoside-12,14-dihydroxy-17α-pregnane (3) (0.9%), tetrarhabinoside-12,14-dihydroxy-17β-pregnane (4) (0.7%), and their derivatives 3-O-6-deoxy-3-O-methyl-2,4-diacetate-β-D-allopyranosyl-(1 → 4)-β-D-oleandropyranosyl-(1 → 4)-β-D-cymaropyranosyl-(1 → 4)-β-D-cymaropyranoside-12β-tigloyl-14β-hydroxy-17β-pregnane (5), 12-tigloyl-14-hydroxy pregnane (6), 12-(2′-amino)-benzoyl-14-hydroxy pregnane (7) and ramanone (8). We also demonstrate that 2D- NMR spectroscopy can be valuable for distinguishing 17α- and 17β-epimers.
Materials and methods
General Experimental Procedures
Melting points were determined on a Büchi B-540 apparatus and are uncorrected. Optical rotations were recorded in a Perkin-Elmer model 343 polarimeter. IR spectra were recorded using a Bruker model IFS-55 spectrophotometer and a Bruker model IFS-66/S spectrophotometer for ATR. 1H-and 13C-NMR spectra were obtained on a Bruker model AMX-500 spectrometer with standard pulse sequences operating at 500 MHz in 1H-and 125 MHz in 13C-NMR. CDCl3, C6D6, and CO(CD3)2 were used as solvents. Chemical shifts were expressed in δ (ppm) with TMS as internal standard. FAB and EIMS were taken on a Micromass model Autospec (70 eV) spectrometer. Column chromatography (CC) was carried out on silica gel (70–230 mesh, Merck), and 0.05 silica gel (Aldrich Chemical Company). Fractions obtained from CC were monitored by TLC (silica gel 60 F254).
Plant Material
Ceropegia fusca Bolle aerial parts were collected in August 2007 at Arico, Tenerife (Canary Islands) and were identified by Dr. M. C. Alfayate (University of La Laguna). Voucher specimens are deposited at the herbarium of that institution.
Extraction and isolation
The plant aerial parts were macerated in ethanol (8 L) for 30 days at room temperature and then subjected to reverse extraction with dichloromethane (3 × 4 L) and n-butanol in a Griffin Flask Shaker (3 × 500 mL). The dichloromethane soluble fraction was filtered off, dried (Na2SO4) and evaporated to dryness under reduced pressure with a Rotavapor at 40°C, yielding 26.97 g. Removal of the solvent from the dichloromethane extraction gave a residue which was subjected to column chromatography on silica gel using n-hexane–EtOAc of increasing polarity and then with increasing percentages of MeOH. The saponin fractions were rechromatographed on silica gel with 7:3 n-hexane/acetone.
3-O-6-Deoxy-3-O-methyl-β-D-allopyranosyl-(1 → 4)-β-D-oleandropyranosyl-(1 → 4)-β-D-cymaropyranosyl-(1 → 4)-β-D-cymaropyranoside-12-β-tigloyl-14-β-hydroxy-17-β-pregnane (1)
Rf 0.58 (1:1:1 n-hexane/CHCl3/acetone). [α] 20D +23.07 (c 0.52, CHCl3). IR (KBr): υmax 3,446, 2,970, 2,933, 1,699, 1,452, 1,369, 1,263, 1,162, 1,086, 1,002, 755 cm−1. λmax 201 nm. HRFABMS (positive ion mode) m/z: 1045.5680 [M + Na]+ (C54H86O18Na, calc. 1045.571). Anal.(C54H86O18) C, H.
3-O-6-Deoxy-3-O-methyl-β-D-allopyranosyl-(1 → 4)-β-D-oleandropyranosyl-(1 → 4)-β-D-cymaropyranosyl-(1 → 4)-β-D-cymaropyranoside-12-β-(2′-amino)-benzoyl-14-β-hydroxy-17-β-pregnane (2)
Rf 0.49 (1:1:1 n-hexane/CHCl3/acetone). [α] 20D −6.95 (c 2.1, CHCl3). IR (KBr): υmax 3,462, 2,969, 2,933, 1,694, 1,452, 1,369, 1,246, 1,162, 1,062, 1,002, 755 cm−1. λmax 220 nm. FABMS (positive ion mode) m/z: 1067 [M + Na-CH3]+. Anal. (C56H85NO18) C, H, N.
3-O-6-Deoxy-3-O-methyl-β-D-allopyranosyl-(1 → 4)-β-D-oleandropyranosyl-(1 → 4)-β-D-cymaropyranosyl-(1 → 4)-β-D-cymaropyranoside-12-β-14-β-dihydroxy-17-α-pregnane (3)
Mp: 137–139°C (CHCl3). Rf 3.5 (1:1:1 n-hexane/CHCl3/acetone). [α] 20D −34.37 (c 3.2, CHCl3). IR (KBr): υmax 3,430, 2,933, 1,690, 1,448, 1,368, 1,161, 1,056, 999 cm−1. λmax 201 nm. HRFABMS (positive ion mode) m/z: 963.5295 [M + Na]+ (C49H80O17Na+, calc. 963.5293). Anal. (C49H80O17) C, H.
3-O-6-Deoxy-3-O-methyl-β-D-allopyranosyl-(1 → 4)-β-D-oleandropyranosyl-(1 → 4)-β-D-cymaropyranosyl-(1 → 4)-β-D-cymaropyranoside-12-β-14-β-dihydroxy-17-β-pregnane (4)
Mp: 157–158°C (CHCl3). Rf 0.20 (1:1:1 n-hexane/CHCl3/acetone). [α] 20D −13.4 (c 7.16, CHCl3). IR (KBr): υmax 3,433, 2,970, 2,934, 1,692, 1,451, 1,370, 1,163, 1,087, 1,061, 1,003, 755 cm−1. λmax 201 nm. FABMS (positive ion mode) m/z: 963.5336 [M + Na]+ (C49H80O17Na, calc. 963.5293). Anal. (C49H80O17) C, H.
3-O-6-Deoxy-3-O-methyl-2,4-diacetyl-β-D-allopyranosyl-(1 → 4)-β-D-oleandropyranosyl-(1 → 4)-β-D-cymaropyranosyl-(1 → 4)-β-D-cymaropyranoside-14-β-hydroxy-12-β-tigloyl-17-β-pregnane (5)
A solution of 1 (34.7 mg, 0.034 mmol) in 1 mL of Ac2O and 1 mL of pyridine was allowed to stand for 66 h at room temperature, and poured into water (15 mL). The aqueous solution was extracted with CHCl3 (3 × 10 mL). After evaporation of the chloroform in vacuo the reaction was chromatographed on silica gel eluting with 9:1 CHCl3/acetone, to yield the corresponding diacetate (31.3 mg, 0.028 mmol, 83%). Mp: 127–128°C (MeOH). Rf 0.76 (1:1:1 n-hexane/CHCl3/acetone). [α] 20D +7.3 (c 6.7, CHCl3). IR (KBr): υmax 2,932, 1,740, 1,449, 1,371, 1,231, 1,159, 1,057, 1,001 cm−1. λmax 224 nm. HRFABMS (positive ion mode) m/z: 1129.5917 [M + Na]+ (C58H90O20Na, calc.1129.5923). Anal. (C58H90O20) C, H.
Acid hydrolysis
Compound 1 (97.3 mg, 0.95 mmol) was dissolved in 10 mL of 1 M HCl solution (1:1 H2O-MeOH), and then the mixture was stirred at 70°C for 3 h. The solution was extracted with CHCl3 (5 × 10 mL) and then evaporated to dryness and chromatographed on silica gel eluting with 7:3 n-hexane/acetone, to give the aglycone 6 (20.7 mg, 0.048 mmol, 51%). Rf 0.24 (7:3 n-hexane/acetone). [α] 20D −9.13 (c 2.3, CHCl3). IR (KBr): υmax 3,434, 2,932, 1,708, 1,364, 1,272, 1,146 cm−1. λmax 219 nm. EIMS (70 eV) m/z: 430 [M]+. By the same method compound 2 was hydrolyzed, affording aglycone 7. Rf 0.20 (7:3 n-hexane/acetone). [α] 20D −68 (c 0.5, CHCl3). IR (KBr): υmax 3,448, 3,371, 2,929, 1,696, 1,059 cm−1. λmax 201 nm. HRMS (70 eV) m/z: 467.2668 [M]+ (C28H37NO5, calc. 467.2672).
Alkaline hydrolysis and subsequent acid hydrolysis
Saponins (77.2 mg) were dissolved in 4 mL of 5% MeOH-KOH, and the mixture was refluxed and stirred for 6 h. Subsequently 3 mL of 1 M HCl were added and the mixture was stirred at room temperature for 14 h, and then passed through a Dowex resin column. After evaporation of the MeOH in vacuo the reaction was chromatographed on silica gel eluting with 20:78:2 n-hexane/EtOAc/MeOH to yield compound 8, ramanone. Mp 197.5–198°C from 4:6 n-hexane/acetone (lit. 195–198°C) (Mitsuhashi and Nomura 1965). Rf 0.18 (20:78:2 n-hexane/EtOAc/MeOH). [α] 20D −6.49 (c 0.77, CHCl3). IR (KBr): υmax 3,407, 2,935, 1,692, 1,460, 1,361, 1,215, 1,166, 1,054, 757 cm−1. λmax 202 nm. HRMS (70 eV) m/z: 330.2191 [M-H2O]+ (C21H30O3Na, calc. 330.2195). HRFABMS (positive ion mode) m/z: 349.2376 [M + H]+ (C21H33O4, calc. 349.2379). Anal. (C21H32O4). C, H.
Results
The extract showed a positive Liebermann-Burchard reaction, indicating the presence of a steroid skeleton. The structure of the intact glycoside was established unambiguously by NMR spectroscopic methods (Coxon 2009; Agrawal 1992). Normally, the 1H-NMR spectrum of glycosides show well-resolved signals for the anomeric protons. Axial anomeric protons of 2-deoxy sugars (β-D-cymaropyranosyl, β-D-oleandropyranosyl) resonate as a double doublet at 4.4–5.05 ppm with coupling constants of 7.7–10.2 and 1.6–1.9 Hz indicating a β-glycosidic linkage (β-D-pyranoses in 4C1 conformation), whereas equatorial anomeric protons appear at 4.9–5.6 ppm. Small coupling constants of 3–4 and 1 Hz indicate an α-glycoside (α-D-pyranoses in 1C4 conformation). Axial anomeric protons of 2-hydroxy sugars resonate as a doublet at 4.7–5.05 ppm (3-O-methyl-6-deoxy-allopyranosyl). When both H-1 and H-2 are axial the coupling constant is large (7–8 Hz), due to its trans diaxial condition, which can be applied to firmly assign the anomeric configuration to β. Coupling constants between an axial and an equatorial or two equatorial protons are weaker (2–4 Hz) indicating an α-glycosidic linkage.
The tigloyl pregnane glycoside (1) has the molecular formula C54H86O18 on the basis of its elementary analysis, high resolution FABMS and 13C DEPT NMR spectroscopy. The IR spectrum shows bands at 3446(OH), 2970, 2933 (CH), 1699 (C=O). It can be seen from the 13C-NMR spectrum that 1 has seven quaternary carbons. The 500 MHz 1H-NMR spectrum of 1 and spin–spin decoupling experiments show the characteristic signals for the tigloyl group at δ 6.90 (dq, J = 1.4, 7.3 Hz, 1H), 1.82 (3H), and 1.86 (3H) esterifying a C-21 pregnane glycoside. The glycosidation shifts of the aglycone carbon of 1 were observed at C-2 (−2.0 ppm), C-3 (+6.0), and C-4 (−3.4). The 1H-NMR anomeric proton signal at δ 4.46 (dd, J = 9.7, 1.9 Hz, 1H), 4.73 (dd, J = 9.7, 1.9 Hz, 1H), 4.77 (d, J = 8.0 Hz, 1H), 4.82 (dd, J = 9.6, 1.9 Hz, 1H) were consistent with a β-glycosyl linkage. The four secondary methyl signals at δ 1.19 (d, J = 6.2 Hz, 3H), 1.20 (d, J = 6.2 Hz, 3H), 1.25 (d, J = 6.2 Hz, 3H), and 1.33 (d, J = 5.7 Hz, 3H) suggest the presence of four 6-deoxy sugar moieties in the molecule. The four 2,6-deoxypyranose moieties are 3-methyl ethers, indicated by four singlet signals at δ 3.37, 3.42 (X2), and 3.64. The ROESY experiment for compound 1 showed a H-12α and a H-17α correlation (Fig. 1). The DQF-COSY and TOCSY spectra showed two similar groups of proton signals due to two cymarose moieties. These experiments also allowed the sequential assignment of the resonances for the four monosaccharides from the easily differentiated anomeric protons. The sequence of the sugar moieties was established by NOE in the ROESY spectrum. These methods provided unambiguous assignments for signals of the steroid and the sugar moieties (Fig. 2). Assignment of individual methoxy groups to each sugar was determined by HMBC of C-3 and the methoxy groups. The sequence and linkage sites of the sugar moieties were established as 3-O-6-deoxy-3-O-methyl-β-D-allopyranosyl-(1 → 4)-β-D-oleandropyranosyl-(1 → 4)-β-D-cymaropyranosyl-(1 → 4)-β-D-cymaropyranoside. The glycosidation shifts of the aglycone carbon for the remaining glycosides described here were also observed at C-2, C-3, and C-4 and hence the sugar moiety was linked to the C-3 hydroxyl group of the aglycone. The HMQC experiment leads to the full 13C- and 1H-NMR assignment of each sugar moiety.

Two-dimensional ROESY spectrum of compound 1 in CDCl3 as solvent, using a Bruker model AMX-500 spectrometer. The square shows H-12α and H-17α correlation. Chemical shifts were expressed in δ (ppm) with TMS as internal standard. 2D NMR plot parameters: CX2 20.00 cm, CX1 20.00 cm, F2PLO 7.021 ppm, F2LU 3,511.18 Hz, F2PHI 0.633 ppm, F2HI 316.58 Hz, F1PLO 7.042 ppm, F1LO 3,521.73 Hz, F1PHI 0.637 ppm, F1HI 318.38 Hz, F2PPMCM 0.31938 ppm/cm, F2HZCM 159.73032 Hz/cm, F1PPMCM 0.32025 ppm/cm, F1HZCM 160.16739 Hz/cm

The one-dimensional ROESY spectrum of 1 (500 MHz, CDCl3) was used to monitor the spatial environment of the selectively excited protons. This method provided unambiguous assignments for signals of the sugar moieties. 1D NMR plot parameters: CX 35.00 cm, F1P 7.467 ppm, F1 3,734.22 Hz, F2P −0.41 ppm, F2 −210.47 Hz, PPMCM 0.22535 ppm/cm, HZCM 112.70542 Hz/cm
The (2′-amino)-benzoyl pregnane glycoside (2) has the molecular formula C56H85NO18 on the basis of its elementary analysis, high resolution FABMS and 13C DEPT NMR. The (2′-amino)-benzoyl group was identified by 1H-NMR chemical shifts at δ 5.77 (s, 2H), 6.66 (ddd, J = 8.6, 7.0, 1.0 Hz, 1H), 6.70 (d, J = 8.6 Hz, 1H), 7.30 (ddd, J = 8.6, 7.0, 1.5 Hz, 1H), 7.87 (dd, J = 8.0, 1.5 Hz, 1H), the molecular ion peaks in FABMS at m/z 137, and from the positive ninhydrin reaction in TLC for nitrogen groups. A careful comparison of the 13C-NMR, DQF-COSY, TOCSY, ROESY, HSQC, and HMBC spectra of 1 and 2 reveals that, except for the esterifying group (Fig. 3), the remaining groups are identical with those of 1. Therefore the structure of 2 was confirmed to be 3-O-6-deoxy-3-O-methyl-β-D-allopyranosyl-(1 → 4)-β-D-oleandropyranosyl-(1 → 4)-β-D-cymaropyranosyl-(1 → 4)-β-D-cymaropyranoside-12-β-(2′-amino)-benzoyl-14-β-hydroxy-17-β-pregnane.

1 H-NMR. Signals for the esterifying group of compound 2. (500 MHz, acetone-d6): δ 6.58 (ddd, J = 7.1, 7.0, 1.1 Hz, 1H, H-5′), 6.68 (dd, J = 8.3, 1.0 Hz, 1H, H-3′), 7.25 (ddd, J = 7.0, 7.0, 1.6 Hz, 1H, H-4′), 7.89 (dd, J = 8.1, 1.6 Hz, 1H, H-6′)
The esterification shifts of the aglycone carbon of 1 in relation to 4 were observed at C-11 (−2.7 ppm), C-12 (+3.5), and C-13 (−1.4), respectively (Table 1). The glycosidation and esterification shifts of the aglycone carbon for the other glycosides described here were also observed at C-2, C-3, and C-4 for glycosidation and at C-11, C-12, and C-13 for esterification shifts, respectively.
Aglycones 6-8, 17α-epimers (Fig. 4), were more stable and were obtained as the main products.

Two-dimensional ROESY spectrum of compound 6 (500 MHz, CDCl3). Acid epimerization at C-17. The square shows H-12α and H-16α correlation. Chemical shifts were expressed in δ (ppm) with TMS as internal standard. The same 2D NMR plot parameters as Fig. 1
The pregnane glycosides 3 and 4 have the molecular formula C49H80O17 on the basis of their elementary analysis, high resolution FABMS and 13C DEPT NMR. The GROESY spectra of 3 obtained with selective inversion of the H-12 proton shows enhanced signals at H-16α, H-15α, H-11α, and H-9α, while the selective excitation of H-18 provided enhancement at H-17β. These experiments show NOE correlations between H-12α and H-16α. Thus, the structure of 3 was formulated as 3-O-6-deoxy-3-O-methyl-β-D-allopyranosyl-(1 → 4)-β-D-oleandropyranosyl-(1 → 4)-β-D-cymaropyranosyl-(1 → 4)-β-D-cymaropyranoside-12-β, 14-β-dihydroxy-17-α-pregnane. The GROESY experiment for compound 4 showed H-12α, H-17α, and H-16α correlations. Compound 4 was therefore characterized as 3-O-6-deoxy-3-O-methyl-β-D-allopyranosyl-(1 → 4)-β-D-oleandropyranosyl-(1 → 4)-β-D-cymaropyranosyl-(1 → 4)-β-D-cymaropyranoside-12-β, 14-β-dihydroxy-17-β-pregnane (Fig. 5).

Natural pregnane glycosides. Pregnane glycosides 1, 2, and 4 show 17β-configuration. Compound 3 shows the α configuration
The acetylation of 1 gave the diacetate 5. The IR spectra show an absorption band at 1,740 cm−1 corresponding to saturated esters. The 1H-NMR spectra show signals for two acetyl groups at 2.09 (s, 3H), and 2.10 (s, 3H). The 13C-NMR spectra show signals at δ 20.9 (X2), 169.6, and 169.8. The hydroxyl groups at C-2 and C-4 of 1 were found to be acetylated. Acid hydrolysis of 1 and 2 gave the semisynthetic aglycones 6 and 7. The hydrolyzation shifts of the aglycone carbon of 1 and 2 were observed at C-2 (+2.0 ppm), C-3 (−6.0), and C-4 (+3.4, +3.3), respectively (Table 1). The alkaline hydrolysis and subsequent acid hydrolysis of saponins gave ramanone (8) (Fig. 6).

Acetylation of compound 1, acid hydrolysis, alkaline hydrolysis and subsequent acid hydrolysis, respectively, of compounds 1 and 2
Discussion
We report here on a detailed 2D-NMR study of some natural oligosaccharides and their synthetic derivatives obtained from the aerial parts of the endemic plant Ceropegia fusca. The 1H-NMR spectrum for the 17α-epimer (compound 3) shows a chemical shift for the C-17 proton of 0.22 ppm (in deuteriochloroform) to a lower field, and the 13C-NMR spectrum a chemical shift of 3.5 ppm for the C-17 (Table 1, compounds 3 and 4). These values reveal small differences for the 17α- and 17β-epimers. Determination of the molecular structures by 2D-NMR spectroscopy enabled us to characterize the structure of these epimeric oligosaccharides. The 17α configuration was also confirmed by the observation that the carbonyl carbon of an α-linked methyl ketone at C-17 appears at δ 209.2, 209.3 and 214.5 ppm for the aglycones 6, 7 and 8, respectively, and at 214.4 ppm for the steroidal glycoside 3, when compared with δ 217 ppm for the β configuration (Table 1). 17-Epimers having an α-oriented side chain were isolated from the twigs of Pergularia pallida (Khare et al. 1984), the roots of Cynanchum wilfordii Hemsley (Tsukamoto et al. 1985), the roots of Calotropis gigantea Dryand (Shibuya et al. 1992), the roots of Cynanchum caudatum M. (Warashina and Noro 1995), the aerial part of Asclepias incarnata L. (Warashina and Noro 2000), the roots of Cynanchum auriculatum Royle ex Wight (Xhang et al. 2000), the aerial parts of Cynanchum aphyllum L. (Kanchanapoom et al. 2002), the stem of Marsdenia tenacissima (Deng et al. 2005), and from the pericarps, hairy seeds and leaves of Solenostemma argel Hayne (Perrone et al. 2008), the roots of Asclepias curassavica L. (Warashina and Noro 2008), the roots of Asclepias syriaca L. (Warashina and Noro 2009) (all of which species belong to the Asclepiadaceae family), and from Nerium oleander (Bai et al. 2007).
Conclusion
We show in this work that the epimerization of 20-keto pregnane glycosides can be determined by 2D-NMR spectroscopy. The ROESY experiment for 17β compounds shows a H-12α and a H-17α correlation, while this experiment shows correlations between H-12α and H-16α for 17α configurations. The anomeric proton of cymarose shows a NOE cross peak with H-3 of the aglycone in the ROESY spectrum. The H-4 proton of each sugar moiety has a NOE in the ROESY spectrum with the anomeric proton of the adjacent sugar. The structure of the intact glycoside can be established unambiguously by NMR spectroscopic methods.
References
Agrawal PK (1992) NMR spectroscopy in the structural elucidation of oligosaccharides and glycosides. Phytochemistry 31:3307–3330
Bai L, Wang L, Zhao M, Toki A, Hasegawa T, Ogura H, Kataoka T, Hirose K, Sakai J, Bai J, Ando M (2007) Bioactive pregnanes from Nerium oleander. J Nat Prod 70:14–18
Chabanel M (1990) Ionic aggregates of 1–1 salts in non-aqueous solutions: structure, thermodynamics and solvation. Pure Appl Chem 62:35–46
Coxon B (2009) Development in the Karplus equation as they relate to the NMR coupling constants of carbohydrates. In: Hoston B (ed) Advances in carbohydrate chemistry and biochemistry. Academic Press, Washington
Deng J, Liao Z, Chen D (2005) Three new polyoxypregnane glycosides from Marsdenia tenacissima. Helv Chim Acta 88:2675–2682
Ingrassia L, Nshimyumukiza P, Dewelle J, Lefranc F, Wlodarczak L, Thomas S, Dielle G, Chiron C, Zedde C, Tisnes P, van Soest R, Braekman J-C, Darro F, Kiss R (2006) A lactosylated steroid contributes in vivo therapeutics benefits in experimental models of mouse lymphoma and human glioblastoma. J Med Chem 49:1800–1807
Junquera E, Olmos D, Aicart E (2002) Carbohydrate-water interactions of p-nitrophenylglycosides in aqueous solution. Ultrasonic and densitometric studies. Phys Chem Chem Phys 4:352–357
Kanchanapoom T, Kasai R, Ohtani K, Andriantsiferana M, Yamasaki K (2002) Pregnane and pregnane glycosides from the malagasky plant, Cynanchum aphyllum. Chem Pharm Bull 50:1031–1034
Khare NK, Khare MP, Khare A (1984) Two pregnane ester glycosides from Pergularia pallida. Phytochemistry 23:2931–2935
Mitsuhashi H, Nomura T (1965) On the structure of ramanone. Chem Pharm Bull 3:1332–1340
Nohara T, Ikeda T, Fujiwara Y, Matsushita S, Noguchi E, Yoshimitsu H, Ono M (2007) Physiological functions of solanaceous and tomato steroidal glycosides. J Nat Med 61:1–13
Peng Y, Li Y, Liu X, Zhang J, Duan J (2008) Antitumor activity of C-21 steroidal glycosides from Cynanchum auriculatum Royle ex Wight. Phytomedicine 15:1016–1020
Perrone A, Plaza A, Hamed A, Pizza C, Piacente S (2008) Solenostemma argel: a rich source of very unusual pregnane and 14, 15-secopregnane glycosides with antiproliferative activity. Curr Org Chem 12:1648–1660
Russell SJ, Kahn CR (2007) Possible regulation of lifespan by steroid hormones in mice. Nat Rev Mol Cell Biol 8:681–691
Shibuya H, Zhang R, Park JD, Takeda Y, Yoshikawa M, Kitagawa I (1992) Indonesian medicinal plants. V. Chemical structures of calotroposides C, D, E, F, and G, five additional new oxypregnane-oligoglycosides from the root of Calotropis gigantea (Asclepiadaceae). Chem Pharm Bull 40:2647–2653
Skvortsov D, Lee SJ, Choi MY, Vilesov AF (2009) Hydrated HCl clusters, HCl(H2O)1–3, in helium nanodroplets: studies of free OH vibrational stretching modes. J Phys Chem 113:7360–7365
Smith A, Vincent MA, Hillier IH (1999) Mechanism of acid dissociation in water clusters: electronic structure studies of (H2O) n HX (n = 4, 7; X = OH, F, HS, HSO3, OOSO2H, OOH·SO2). J Phys Chem A 103:1132–1139
Tsukamoto S, Hayshi K, Mitsuashi H (1985) The structures of six glycosides, wilfoside C1N, C2N, C3N, C1G, C2G and C3G, with novel sugar chain containing a pair of optically isomeric sugars. Tetrahedron 41:927–934
Wang S, Bianco R, Hynes JT (2009) Depth-dependent dissociation of nitric acid at an aqueous surface: Car-Parrinello molecular dynamics. J Phys Chem A 113:1295–1307
Warashina T, Noro N (1995) Steroidal glycosides from the root of Cynanchum caudatum M. Chem Pharm Bull 43:977–982
Warashina T, Noro N (2000) Steroidal glycosides from the aerial part of Asclepias incarnata. Phytochemistry 53:485–498
Warashina T, Noro N (2008) Steroidal glycosides from the roots of Asclepias curassavica. Chem Pharm Bull 56:315–322
Warashina T, Noro N (2009) Acylated-oxypregnane glycosides from the roots of Asclepias syriaca. Chem Pharm Bull 57:177–184
Xhang R, Ye Y-P, Shen Y-M, Liang H-L (2000) Two new cytotoxic C-21 steroidal glycosides from the root of Cynanchum auriculatum. Tetrahedron 56:3875–3879
Yokosuka A, Mimaki Y (2008) Steroidal glycosides from the underground parts of Trillium erectum and their cytotoxic activity. Phytochemistry 69:2724–2730
Acknowledgments
This work was supported by grants from the Ministerio de Ciencia y Tecnología (PTQ2002-0529) and Luciano Reverón e Hijos S. L. We would like to thank M. Carmen Mederos, Juan A. Suárez, Manuel de León, Domingo Acosta and Sergio Suárez for technical assistance, Dr. John Lowry, Humboldt State University, for helpful comments on the manuscript and Pauline Agnew for assistance with language revision.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
García, V.P. Acid epimerization of 20-keto pregnane glycosides is determined by 2D-NMR spectroscopy. J Biomol NMR 50, 91–97 (2011). https://doi.org/10.1007/s10858-011-9499-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-011-9499-z