
Abstract
Sequence specific resonance assignment constitutes an important step towards high-resolution structure determination of proteins by NMR and is aided by selective identification and assignment of amino acid types. The traditional approach to selective labeling yields only the chemical shifts of the particular amino acid being selected and does not help in establishing a link between adjacent residues along the polypeptide chain, which is important for sequential assignments. An alternative approach is the method of amino acid selective ‘unlabeling’ or reverse labeling, which involves selective unlabeling of specific amino acid types against a uniformly 13C/15N labeled background. Based on this method, we present a novel approach for sequential assignments in proteins. The method involves a new NMR experiment named, {12CO i –15N i+1}-filtered HSQC, which aids in linking the 1HN/15N resonances of the selectively unlabeled residue, i, and its C-terminal neighbor, i + 1, in HN-detected double and triple resonance spectra. This leads to the assignment of a tri-peptide segment from the knowledge of the amino acid types of residues: i − 1, i and i + 1, thereby speeding up the sequential assignment process. The method has the advantage of being relatively inexpensive, applicable to 2H labeled protein and can be coupled with cell-free synthesis and/or automated assignment approaches. A detailed survey involving unlabeling of different amino acid types individually or in pairs reveals that the proposed approach is also robust to misincorporation of 14N at undesired sites. Taken together, this study represents the first application of selective unlabeling for sequence specific resonance assignments and opens up new avenues to using this methodology in protein structural studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The first step towards three-dimensional (3D) structure determination of proteins by NMR involves sequence specific resonance assignments of its backbone and side-chain 1H, 13C and 15N nuclei (Wüthrich 1986). For proteins with molecular mass >10 kDa, this is accomplished using a suite of triple resonance spectra acquired with a doubly or triply labeled (13C, 15N or 2H, 13C, 15N) protein sample (Cavanagh et al. 1996). In this method, sequential connectivities between neighboring amino acid residues along the polypeptide chain (di-peptide links) are established and a stretch of assigned residues is mapped on to the primary sequence for assigning the resonances sequence specifically. Due to high sensitivity and resolution of triple resonance experiments, the spectra obtained are often amenable to automated assignments and several algorithms have been proposed in this direction (reviewed in Baran et al. 2004).
In the case of spectral overlap and/or larger molecular weight proteins, it becomes necessary to identify spin systems corresponding to the different amino acid types selectively to aid resonance assignments. This is accomplished in one of the two ways: (a) selective isotope labeling of amino acids (Goto and Kay 2000; Muchmore et al. 1989; Ohki and Kainosho 2008) or/and (b) use of NMR experiments which detect specific amino acid types selectively (Barnwal et al. 2008; Dötsch et al. 1996a, b; Dötsch and Wagner 1996; Schubert et al. 2005, 2001a, b, c, 1999). In the first approach, selective labeling of a specific amino acid type is achieved by feeding the host organism with the desired isotopically labeled (13C/15N/2H) amino acid, while supplying the rest of the amino acids in unlabeled form (Muchmore et al. 1989). There are two drawbacks of this method: (a) the use of expensive 13C/15N enriched amino acids, resulting in prohibitively high costs, especially if the protein yields are low and (b) the method provides 1HN/15N chemical shifts only for the amino acid residue being selectively labeled (residue i) and does not help in establishing a link between 1HN/15N resonances of residue i and residue i + 1 along the polypeptide chain, which is essential for sequence specific resonance assignments.
The second approach mentioned above involves design of NMR experiments which exploit the nature of the side-chain of different amino acids. The magnetization transfer pathways in these experiments are tuned so as to selectively detect a given amino acid type (Barnwal et al. 2008; Dötsch et al. a, b; Dötsch and Wagner 1996; Schubert et al. 1999, 2005, 2001a, b, c). A disadvantage of this approach is the rapid loss in sensitivity with increasing molecular weight of the proteins due to the long delay periods used in the pulse sequences. Further, these experiments often cannot be applied to deuterated proteins owing to the fact that the magnetization originates from the side-chain 1H of the different amino acids which are lost upon deuteration.
An alternative and inexpensive approach is the method of amino acid selective “unlabeling” or ‘reverse’ labeling (Atreya and Chary 2000, 2001; Vuister et al. 1994). This involves selective unlabeling of specific amino acid types against a uniformly or fractionally 13C or/and 15N labeled background. A particular or a set of amino acid type(s) in a protein is selectively unlabeled by feeding the host microorganism with 15NH4Cl or/and 13C (1H/2H) -D-glucose as the sole source of nitrogen and carbon, respectively, along with the desired amino acid(s) in unlabeled form (i.e., containing 12C and 14N). As a result, cross peaks due to these residues are not observed in the double and triple resonance spectra. A comparison of such a spectrum with that of a control spectrum involving uniformly 13C or/and 15N labeled protein enables one to distinguish peaks and assign the corresponding chemical shifts of nuclei belonging to the unlabeled residues. This method has the advantage of being relatively inexpensive (only unlabeled amino acids are required along with 15NH4Cl and/or 13C-D-glucose) and applicable to 2H labeled protein. It has been used in a range of applications such as spectral simplification (Kelly et al. 1999; Vuister et al. 1994), spin-system identification, (Atreya and Chary 2000; Mohan et al. 2008; Shortle 1995), stereospecific assignment of the pro-chiral methyl groups of Val and Leu in large molecular weight proteins (Tugarinov and Kay 2004) and measurement of residual dipolar couplings (RDC) (Mukherjee et al. 2005). Its application has been proposed for structural studies of membrane proteins (Hilty et al. 2002) and proteins with a paramagnetic centre (Turano et al. 2010). Despite such potential applications, a detailed study with respect to its robustness and its use particularly in the sequential assignment process is lacking.
We present here a novel approach to use the method of selective unlabeling in HN-detected double and triple resonance experiments based resonance assignments. Specifically, we have: (a) developed a method which will allow the assignment of 1HN and 15N resonances at the C-terminus (i.e., residue i + 1) of the amino acid residue being unlabeled (i.e., i) for establishing sequential connectivity (this results in a tri-peptide linkage from the knowledge of the amino acid types of i − 1, i and i + 1 residues) and (b) evaluated the extent of misincorporation 14N at undesired sites for the different amino acid types. For accomplishing the first objective above, we have devised a new NMR experiment, 2D {12CO i –15N i+1}-filtered HSQC to select residues in the protein that are 15N and 13C labeled and have an N-terminal neighbor that has been selectively unlabeled (i.e., containing 12C). These studies have been carried out on the protein Ubiquitin (8.6 kDa) and a intrinsically disordered protein, Tim23 (11 kDa) over-expressed in E. coli and can be combined with other methods for isotope labeling (Kainosho and Guntert 2009; Ohki and Kainosho 2008) and/or with completely/partially deuterated protein samples.
Materials and methods
Selection of amino acid types for selective unlabeling
Identification and assignment of resonances corresponding to the selectively unlabeled amino acids is done by comparing a given spectrum with that of a reference sample of the same protein which is uniformly 13C/15N labeled (Atreya and Chary 2000, 2001). In general, a single or combination of two or more amino acid types can be chosen for selective unlabeling at a time. Choosing more than one amino acid type in the same sample reduces the total number of samples required for analysis. In the case of one type of amino acid being unlabeled, its identification and assignment is straightforward (described below). However, if two or more amino acid types are chosen together for selective unlabeling in the same sample, the particular combination chosen depends on the relative ease with which the different amino acid types can be distinguished from one another in the spectrum and their relative abundance in the protein. Such discrimination can be made on the basis of 13Cα and 13Cβ chemical shifts which are well-known indicators of amino acid types (Atreya et al. 2002, 2000; Cavanagh et al. 1996). This is depicted in Fig. 1 using chemical shift statistics available in the BioMagResBank (BMRB, http://www.bmrb.wisc.edu). The 20 amino acids can be divided into 9 groups (labeled I–IX in Fig. 1) with each group having a distinct range of 13Cα and 13Cβ chemical shifts. For instance, Arg and Asn can be chosen together for selective unlabeling as their 13Cβ shifts resonate in different spectral regions (Fig. 1). Out of the 20 amino acid types, Gly, Ala, Ser and Thr are usually not required for selective detection owing to the fact their 13Cβ shifts occur in distinct spectral regions and can be identified directly from inspection of the 3D spectra. Further, the amino acid, Pro, is not observed in HN-detected double and triple-resonance experiments and hence omitted from selective unlabeling.

Classification of the 20 amino acids (indicated by single letter code) into 9 categories based on their 13Cα and 13Cβ chemical shifts (Atreya et al. 2000). The different categories are numbered I–IX and indicated on top or bottom of the dotted boxes drawn for each category of amino acids. Red and green circles denote 13Cα and 13Cβ chemical shifts, respectively
In the present study, we have chosen 8 amino acids in four pairs for selective unlabeling: (a) Gln, Ile (Q,I), (b) Arg, Asn (R,N) (c) Phe, Val (F, V) and (d) Lys, Leu (K, L). Each pair corresponds to one protein sample and together with the reference (uniformly 13C or/and 15N labeled), results in five samples. The four combinations have been chosen on the basis that the two amino acid types in each pair have distinct 13Cα and 13Cβ chemical shifts (Fig. 1). Furthermore, these are among the most abundant amino acid residues in a given protein. Together with Gly, Ala, Ser and Thr, they constitute ~80% of the amino acid residues found in proteins of prokaryotes and eukaryotes. However, in any given protein, it is possible to change the combination of amino acids chosen which is discussed below.
Identification of resonances corresponding to unlabeled amino acids
The signals/peaks corresponding to the selectively unlabeled amino acid residue in 2D [15N–1H] HSQC is usually identified by appropriate scaling and subtraction of the spectrum from that of the reference sample containing uniformly 15N labeled protein (hereafter referred to as the ‘difference spectrum’). The difference spectrum is comprised of peaks corresponding to the specific amino acid residue being unlabeled. A disadvantage of this method is the imperfect cancellation of undesired peaks upon subtraction, due to the fact the two protein samples (reference and the selectively unlabeled) may have different concentrations and hence different signal-to-noise (S/N) ratios. We have devised an alternative method for this process. It involves taking the ratio of two quantities, IRunlab and IRref (i.e., IRunlab/IRref), which are defined as follows: IRunlab is the ratio of the volume of each peak in the 2D HSQC spectrum of the selectively unlabeled protein sample to the sum total of intensities of all peaks in the same spectrum. That is, IRunlab,i = I unlab i /ΣI unlab i , where ‘I i ’ refers to the volume of a given peak (‘i’). Similarly in the case of the reference sample, IRref,i = I ref i /ΣI ref i . An alternative to using the total intensity is to choose a reference or ‘control’ peak in the spectrum which is known to be un-affected by unlabeling (for instance a Gly residue). The intensity of that peak in the two respective spectra can be used, replacing ΣI unlab i and ΣI ref i with I unlabcontrol and I refcontrol , respectively. The ratio, IRunlab/IRref, is thus independent of the differences in the signal-to-noise of the two spectra being compared and can be used in concert with the 2D difference spectrum. It also provides information on the extent to which the unlabeling of the particular amino acid has occurred. For instance, for a peak which is completely absent in the spectrum acquired for the selectively unlabeled sample IRunlab,i /IRref,i will be ~0, whereas IRunlab,i /IRref,i will be ~1 for any peak unaffected by selective unlabeling. In practice, a range of values from 0 to 1 are observed for the unlabeled residues reflecting the extent to which a given amino acid type has been unlabeled.
Sequential assignment strategy
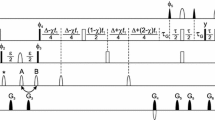
In the general sequential assignment process, a link between 1HN/15N resonances of residue i and residue i + 1 along the polypeptide chain is established by using, in concert, 3D HNCACB and 3D CBCA(CO)NH/HN(CO)CACB spectra (Cavanagh et al. 1996). Starting from a spin-system i in 2D [15N–1H] HSQC, a search is carried out in the 3D CBCA(CO)NH/HN(CO)CACB spectrum for a spin-system j such that the 13Cα and 13Cβ chemical shifts of residue i (obtained from HNCACB) are observed on j. The spin system j then corresponds to the C-terminal neighbor (i.e., i + 1) of i. In the case of spectral overlap and/or larger molecular weight proteins, 1Hα and/or 13CO shifts from suitable 3D experiments can also be used to establish this link (Baran et al. 2004; Tugarinov et al. 2004). This process is hindered if the search ends up with more than one choice for spin systems corresponding to i + 1. For unambiguous assignments, it is thus preferable to reduce the “search space” such that the spin-system i + 1 is easily tractable for a given spin-system i. Towards this end, we have devised a simple method based on a HSQC-type NMR experiment, which detects 1HN/15N resonances of the residue C-terminus (i + 1) of an amino acid unlabeled selectively (residue i). The selection scheme of the experiment, named {12CO i –15N i+1}-filtered HSQC, is depicted in Fig. 2 and the radio-frequency (r.f.) pulse scheme is shown in Fig. 3.

Schematic illustration of selection of magnetization in the 2D {12CO i –15N i+1}-filtered HSQC. The letters ‘t 1’ and ‘t 2’ indicate the chemical shift evolution periods in the RF pulse sequence (see Fig. 3)

R.f. pulse schemes of 2D {12CO i –15N i+1}-filtered HSQC. Rectangular 90° and 180° pulses are indicated by thin and thick vertical bars, respectively, and phases are indicated above the pulses. Where no r.f. phase is marked, the pulse is applied along x. High-power 90° pulse lengths are: 8.7 μs for 1H, 37 μs for 15N and 15.8 μs for 13C. The 1H r.f. carrier is placed at the position of the solvent line at 4.7 ppm. The 13C and 15N carrier position is set to 176 ppm and 118.5 ppm, respectively, throughout the sequence. GARP (Cavanagh et al. 1996) is employed to decouple 15N during acquisition. The shaped 180° pulses on 13C’ are of Gaussian cascade type (Cavanagh et al. 1996) with duration of 256 us. All pulsed z-field gradients (PFGs) are sinc-shaped with gradient recovery delay of 200 μs. The duration and strengths of the PFGs are: G1–G2 (1 ms, 26.8 G/cm); G3: (1 ms 42.9 G/cm) G4: (1 ms 4.3 G/cm). The delays are: τ1 = 2.3 ms, τ2 = 15.0 ms, τ3 = 2.3 ms, δ = 1.2 ms and δ1 = 1.2 ms. Phase cycling: ϕ1 = x, −x; ϕ2 = y; ϕrec = x, −x. Quadrature detection along ω1(15N) is achieved with sensitivity enhancement (Cavanagh et al. 1996) (G3 is inverted with a 180° shift for ϕ2)
The flow of magnetization is briefly explained as follows. After transfer of polarization from 1H i+1, the magnetization on 15N i+1 evolves under one-bond J-coupling to 13C’ i during the delay period τ (set to 1/2JNC’ ~30 ms) into anti-phase 15N i+1–13C i ’ magnetization. In the event of residue i being an unlabeled amino acid residue, 15N i+1 remains anti-phase only with respect to 1H N i+1 and gets frequency labeled during the chemical shift evolution period, t 1, similar to that in a 2D [15N–1H] HSQC. However, if the residue i is isotopically labeled (13C/15N), the resulting anti-phase 15N i+1–13C i ’ magnetization is dephased by the gradient, G2 (Fig. 3). Thus, the resulting 2D spectrum is comprised of peaks corresponding only to those residues which have selectively unlabeled N-terminal neighbors. This experiment is sensitive in nature and can be applied to deuterated proteins.
The next step is to map the 1HN/15N resonances observed in the difference spectrum with the corresponding resonances of their C-terminal neighbors in the 2D {12CO i –15N i+1}-filtered HSQC. This is depicted schematically in Fig. 4 and is accomplished by an inspection of the 3D HNCACB and 3D CBCA(CO)NH/HN(CO)CACB spectra acquired for the reference sample. For a given spin-system i in the 2D difference spectrum, the spin system corresponding to i + 1 in 2D {12CO i –15N i+1}-filtered HSQC is the one in which the 13Cα and 13Cβ chemical shifts in 3D CBCA(CO)NH/HN(CO)CACB spectrum matches that of the residue i in HNCACB (see Fig. 4). Based on the 13Cα and 13Cβ chemical shifts (Fig. 1), the amino acid types comprising the tri-peptide segment (residues i − 1, i, i + 1) can be identified, which can then be mapped on to the protein primary sequence for sequence specific resonance assignments. We have carried out a statistical analysis of 186 non-homologues primary sequences from the BMRB to estimate the extent to which such tri-peptide segments can be placed uniquely in a protein sequence (discussed below). Note the procedure can also be carried out in the reverse direction, that is, from residue i to i − 1. Further, the above process can be extended to longer stretches of amino acid residues by repeating the process further along the polypeptide chain.

Schematic illustration of the sequential assignment strategy used with selective unlabeling
Sample preparation
Ubiquitin containing plasmid (pGLUB) was transformed into E. coli BL21 cells. Cells were grown at 37°C in M9 minimal medium consisting of 1 g/L of 15NH4Cl and 3 g/L of 13C-Glucose. For selective unlabeling, 1.0 g/l each of desired unlabeled amino acid(s) (stock solution of 1.0 g in 50 ml of H2O was prepared and filter sterilized) was added to the growth medium. Protein expression was induced at midlog phase (O.D600 ~ 0.6) by addition of 1.0 mM isopropyl β-D-thiogalactoside (IPTG). Cells were harvested by centrifugation and solubilized in acetate buffer (5 mM EDTA, 50 mM Na acetate, pH 5). Following sonication, the supernatant containing the protein was loaded on to a pre-equilibrated ion exchange column (SP Sepharose fast flow from GE) and the protein eluted with a salt gradient of 0–0.6 M NaCl. The protein sample was further purified using size exclusion chromatography with Superdex 75 (Sigma). For NMR studies, a sample containing ~1.0 mM of protein in 50 mM Phosphate buffer (90% H2O/10% 2H2O, pH 6.0) was prepared. Four samples containing the following pairs of selectively unlabeled amino acids were prepared: (a) Gln, Ile, (b) Arg, Asn (c) Phe, Val and (d) Lys, Leu. Three other 15N (only) labeled samples containing the following selectively unlabeled amino acids were prepared: (a) Asp, (b) His and (c) Tyr.
The gene encoding N-terminal domain of Tim 23 (1–98) with the inclusion of a hexa-histidine tag at the C-terminus was cloned into a pET3a vector and transformed into the E. coli strain, Rosetta. The cells were grown at 30°C in 1-liter of a M9 minimal medium containing 4 g of 13C-glucose and 1 g of 15NH4Cl as the sole source of carbon and nitrogen, respectively. Lysine was selectively unlabelled by adding at a concentration of 1.0 g/L to the minimal medium. Cells were induced at A600 of 0.6 with 0.5 mM IPTG and grown for an additional 4 h. Cells were harvested by centrifugation at 4,000 rpm at 4°C and resuspended in 12 ml of lysis buffer (20 mM Tris–Cl, pH. 7.5, 20 mM imidazole, 0.5 M NaCl, 5% glycerol, 50 μM Leupeptin, 1 μM Pepstatin, 1 μg/ml Aprotinin and 1 mM PMSF). Cell lysis was carried out by sonication for 20 s on ice (Ultrasonic processor, 30% amplitude) for 5 times in an interval of 2 min. The lysate was clarified by centrifugation at 15,000 rpm (4°C) and applied to 1 ml of Ni–NTA column pre-equilibrated with the lysis buffer. The column was washed with 20 column volumes of a buffer A (20 mM Tris–Cl, pH 7.5, 20 mM imidazole, 0.5 M NaCl and 5% glycerol), followed by washing with 10 column volumes buffer B (20 mM Tris–Cl pH 7.5, 40 mM imidazole, 0.5 M NaCl, 5% glycerol, 1 mM ATP and 10 mM MgCl2). The protein was eluted with buffer C (20 mM Tris–Cl, pH 7.5, 200 mM imidazole, 0.5 M NaCl and 5% glycerol). To remove higher molecular weight impurities, the peak fractions were pooled and subjected to gel filtration on a GE Superdex Hiload-75 (16/60) column at a flow rate of 1 ml/min. The purified isotope labeled protein fractions were dialyzed (Spectra-Por dialysis membrane MWCO: 6–8 kDa) against 50 mM Phosphate buffer (pH 6.5) containing 50 mM NaCl and concentrated using Amicon Ultra-15 (Ultracel-5 K) centrifugal filter unit (Millipore). The purified protein was found to be 98% pure as checked on SDS–PAGE and confirmed as monomer (~12 kDa; with uniform 13C/15N labeling and including the hexahistidine tag) by mass spectrometry and NMR. For NMR studies, a sample of 1 mM concentration of protein in 50 mM phosphate buffer (pH 6.5) and 50 mM NaCl was prepared in 90% H2O/10% 2H2O.
NMR data collection, processing and analysis
All NMR experiments were performed at 25°C on Bruker 500 MHz and spectrometer equipped with a room temperature triple resonance probe with Z-axis gradients. Data were processed with NMRPipe (Delaglio et al. 1995) and analyzed using XEASY (Bartels et al. 1995).
Results
Figure 5a and b shows the 2D [15N–1H] HSQC-difference spectrum and 2D {12CO i –15N i+1}-filtered HSQC of the four selectively unlabeled samples, respectively. The efficacy of 2D {12CO i –15N i+1}-filtered HSQC to suppress peaks from residues having 13C/15N labeled N-terminal (sequential) neighbors was tested using the reference (uniformly 13C/15N) labeled sample of Ubiquitin. No peaks were observed in the spectrum (Figure S1 of Supporting Information) which is due to the fact that all residues in the reference sample have 13C/15N labeled N-terminal neighbors. Analysis of the intensities of peaks corresponding to the unlabeled amino acids (i.e., IRunlab/IRref) is shown in Fig. 5c. For calculating IRunlab and IRref, G75 was chosen as a control residue as it is unaffected by selective unlabeling in any of the samples. In all the four pairs of samples, complete peak yields were obtained for residues, i (i.e., the amino acid type being unlabeled) and i + 1. This implies that the extent of unlabeling of a desired amino acid type is always complete. Similar analysis for the 15N-labeled samples of the three other major selectively unlabeled amino acid types is provided in Supporting Information (Figure S2 of Supporting Information). Note that if two consecutive amino acids (i.e., both i and i + 1) are unlabeled, the experiment will not detect resonances corresponding to the i + 1 residue. This happens in the case of Q40–Q41, I2–I3, I30–Q31 and I61–Q62 in Ubiquitin.

Experimental (a) 2D difference spectrum and (b) 2D {12CO i –15N i+1}-filtered HSQC spectrum for the different selectively unlabeled samples of Ubiquitin. The difference spectrum was obtained by appropriate scaling and subtraction of 2D [15N, 1H] HSQC spectrum acquired for the selectively unlabeled sample from that of the reference sample. Amino acid pairs selectively unlabeled in each sample are indicated in each spectra. The assignments of peaks are indicated by the single letter code followed by the residue number. c A plot of IRunlab,i /IRref,i : IRunlab,i = I unlab i /I unlabcontrol and IRref, i = I ref i /I refcontrol where i denotes the residue number, I denotes volume of the peak and ‘control’ denotes a residue which does not undergo any effect of unlabeling in both selectively unlabeled and the reference sample. In this case the control residue chosen was G75. All residues that undergo the desired unlabeling are indicated as green bars and correspond to the pairs of amino acids indicated in the respective spectra in (a) and (b). Those residues which undergo unlabeling due to misincorporation of 14N are shown in red. In the case of (R, N) sample complete unlabeling of Arg and Asn was obtained (i.e., IRunlab,i /IRref ~ 0) and hence these residues are indicated in the top panel with the single letter code. E24 and G53 were not assigned and hence are absent along with Pro
Figure 6 illustrates the identification and assignment of a tri-peptide segment using the Arg, Asn unlabeled sample of Ubiquitin. As described above (Fig. 4), based on 3D HNCACB and 3D CBCA(CO)NH spectrum acquired for the reference (uniformly 13C/15N labeled) sample, the 1HN and the 15N resonances of the G75 were identified in 2D {12CO i –15N i+1}-filtered HSQC and the segment LRG could be assigned and placed uniquely in the primary sequence. A statistical analysis (described below) describes the extent to which such tri-peptide segments in general are present uniquely in a given protein sequence. Figure 7 illustrates larger segments of amino acid residues that can be assigned using different selectively unlabeled samples.

Illustration of sequence specific resonance assignment of a tri-peptide segment in Ubiquitin. Given a peak in the 2D difference spectrum of (R, N) selectively unlabeled sample, (e.g., R74), its C-terminal neighbour (i + 1) is identified using the procedure shown in Fig. 4 and its amino acid type (indicated as group I) is assigned using 13Cα and 13Cβ chemical shift based classification described in Fig. 1. The amino acid type of residue i − 1 (indicated as group VI) is assigned like-wise from 3D CBCA(CO)NH and the tri-peptide segment is then mapped on to the protein sequence shown for sequence specific assignments

Illustration of sequence specific resonance assignment of a penta-peptide segment in Ubiquitin. The procedure described in Fig. 4 is repeated for three contiguous tri-peptide segments: Y59–N60–I61, N60–I61–Q62 and I61–Q62–K63. First, the 15N and 1HN resonances of N60 and I61 are identified with the procedure described in Fig. 4 using the (R, N) selectively unlabeled sample. Subsequently, the procedure is repeated to obtain the 15N and 1HN resonances of Q62 and K63 using the (K, L) and (Q, I) selectively unlabeled samples, respectively
The methodology described above was particularly found to be useful for resonance assignment of an 11 kDa glycine rich and intrisincally disordered protein, Tim23 from S. cerevisiae (Cruz et al. 2010). Tim23 is a critical component of the protein machinary involved in the import of nuclear-encoded proteins into the mitochondrial membranes. Figure 8a shows an overlay of the 2D [15N–1H] HSQC acquired for uniformly 15N-labeled and selectively unlabeled sample of Tim23. Figure 8b shows the 2D {12CO i –15N i+1}-filtered HSQC acquired for the selectively unlabeled sample. The protein sequence has two D-K di-peptide segments (D7–K8 and D65–K66) resulting in ambiguity for assignment of the two lysines with the conventional selective labeling approach given further that all lysines had nearly degenerate 13Cα and 13Cβ chemical shifts. However, identification of G67 and T9 (i + 1 to K8 and K66, respectively) using 2D {12CO i –15N i+1}-filtered HSQC helped in removing this degeneracy and the tri-peptide segments D7–K8–T9 and D65–K66–G67 were uniquely identified (complete assignments are deposited in BMRB under accession number 16198).

a 2D [15N–1H] HSQC and (b) a selected region of 2D {12CO i –15N i+1}-filtered HSQC acquired for lysine selectively unlabeled sample of an 11 kDa intrinsically disordered protein Tim23. In (a) overlay of 2D [15N–1H] HSQC of the uniformly 15N labeled Tim23 (blue) with that of lysine selectively unlabeled (red) is shown. Assignments are indicated with the single letter code followed by the residue number. Note that the residue i + 1 of K25 is a Pro and hence not observed in the 2D {12CO i –15N i+1}-filtered HSQC
Analysis of misincorporation 14N at undesired sites
The extent of misincorporation of 14N at the undesired sites is presented in Fig. 5c, where the ratio: IRunlab/IRref has been plotted as a function of residue number. Residues which undergo the desired unlabeling are shown in green and residues with misincorporation of 14N are shown in red. A value of IRunlab/IRref < 0.5 is taken to indicate that the residue has undergone selective unlabeling. In the four selectively unlabeled samples prepared in the present study, it was found the while Arg, Asn and Lys did not undergo isotope scrambling, Phe, Tyr showed weak conversion to each other. On the other hand Ile, Leu, Val inter-converted strongly to each other. However, since these three amino acids belong to distinct categories (Fig. 1), such cross metabolism does not hamper the assignment process. Based on the intensity patterns of the peaks corresponding to the desired and undesired unlabeled amino acids, we have tabulated the extent of isotope scrambling for each amino acid type (Table 1). They are classified as strong or weak depending on the average value of IRunlab/IRref: a IRunlab/IRref < 0.5 implies strong and IRunlab/IRref > 0.5 signifies inefficient (weak) unlabeling. Such an analysis helps to choose the combination of amino acids for unlabeling. The overall nature of isotope scrambling is similar to that observed in the selective labeling approach (Muchmore et al. 1989), which is expected as the biosynthetic pathways involved are identical in the two methods. Notably, we observed that unlike in the selective labeling approach, unlabeling of glutamine was not detrimental. Its conversion to glutamic acid gets diluted by transamination of the latter to other amino acids. Only weak unlabeling of proline (via Glutamic acid) was observed in the (Q, I) sample (P19 and P38, Fig. 5b). On the other hand, glutamic acid was observed to strongly unlabel all other amino acid types.
Discussion
Sequence specific resonance assignments are often supported by selective identification of amino acid types. This is primarily done to simplify the ‘search problem’. Automated assignment approaches employ this information, if available, in their algorithms to reduce the ambiguity in linking amino acid residues (Baran et al. 2004). The objective of this study was to apply the method of selective unlabeling to sequential assignments and accomplish: (a) the identification and assignment of 1HN/15N resonances of given amino acid types selectively and (b) assignment of 1HN/15N resonances of their C-terminal neighbors. These together result in the assignment of tri-peptide segments from the knowledge of the amino acid types of residues: i − 1, i and i + 1, which can then be mapped on to primary sequence. The amino acid type information is obtained from 13Cα and 13Cβ chemical shifts (Fig. 1). Note that the following types of tri-peptide segments cannot be assigned using the approach described here: (a) the tri-peptide segment in which the residue Pro is the immediate C-terminal neighbor of the central (unlabeled) residue, (b) the tri-peptide segment in which the two consecutive amino acids i and i + 1 are selectively unlabeled and (c) if the selectively unlabeled residue is located at the C-terminus end of the protein.
We have carried out a statistical analysis of 186 non-homologues primary sequences from the TALOS database (Cornilescu et al. 1999) to estimate the extent to which such tri-peptide segments can be placed uniquely in a protein primary sequence. The analysis was carried out by assigning a unique number code to each of the 9 categories shown in Fig. 1 except for the residue being selectively unlabeled, which was assigned a separate code (given that its type is known exactly). Thus, both the tri-peptide segment and the polypeptide chain were converted into a new sequence of codes, which was then searched for multiple occurrences of the tri-peptide segment (this is further illustrated in Figure S3 of Supporting Information for a 99 amino acid residue protein HIVprotease). This approach was carried out for 15 amino acids (excluding Ala, Gly, Ser, Thr and Pro) in the central position of the tri-peptide segment and repeated for each protein in the database. The following two comparisons were made to judge the advantage of selective unlabeling: First, the number of proteins in the database was taken wherein all tri-peptide segments (comprising a given amino acid type in the centre) were rendered unique with and without selective unlabeling. This number brings out the extent of uniqueness obtained from the knowledge of the central and its C-terminal residue of the tri-peptide segment by selective unlabeling as compared to the conventional sequential assignment process without selective identification of amino acid types. The second calculation involved the remaining proteins in the database in which some of the tri-peptide segments had multiple occurrences. In such cases, the increase in the percentage of unique tri-peptide segments due to knowledge of the central amino acid residue was evaluated. The results are depicted in Fig. 9.

Statistical analysis of the uniqueness of tri-peptide segments in proteins rendered with selective unlabeling. The numbers/percentages corresponding to the selective unlabeling approach are shown in pink colour, whereas those corresponding to the non-selective method are indicated in blue. a The number of proteins in the database of 186 non-homologues proteins which have unique tri-peptide sequences containing a given amino acid residue in the central position. In such tri-peptide segments, the residue preceding and following the central residue is given a number code according to its amino acid type (Fig. 1) and the central residue is given a unique code (see Figure S3 of Supporting Information). The entire polypeptide chain (also converted to a sequence of codes) is then searched for multiple occurrences of such tri-peptide segments. b In proteins which had more than one occurrences of a given tri-peptide segment, the fraction of tri-peptide segments (expressed as percentage) that were unique using the criterion described in (a). c The ratio of number of tri-peptide segments that had multiple occurrences without the knowledge of the central amino acid residue as against multiple occurrences with selective identification
It is evident that the information of amino acid type significantly reduces the multiple occurrences of tri-peptide segments. For instance, nearly one-third of the proteins in the database had all tri-peptide segments containing lysine as the central residue unique given the knowledge of its amino acid type (shown in dark blue) (Fig. 9a). Further out of the remaining two-thirds of the protein, about 60% of tri-peptide segments containing lysine as the central residue were unique compared to 30% without selective identification of lysine (Fig. 9b). Thus, the selective unlabeling approach described here not only helps in reducing the search space for sequential assignments (with 2D {12CO i –15N i+1}-filtered HSQC) to link two adjacent amino acid residues, but also increases the uniqueness of the tri-peptide segment identified, resulting in unambiguous assignments. Larger stretches of assgined residues (Fig. 7) further helps in reducing the ambiguities facilitating accurate assignments. The advantage with selective unlabeling is more in the case of Group V and Group VI amino acid residues, which are composed of a large number of amino acid types (Fig. 1). In the case of Ala, Gly, Ser and Thr, while selective unlabeling approach is not needed for spin system identification, it will nevertheless benefit for assignment of 1H/15N resonances of their C-terminal neighbors.
Figure 9c indicates the ratio of number of tri-peptide segments that had multiple occurrences without the knowledge of the central amino acid residue as against multiple occurrences with selective identification. Thus, selective identification of residues reduces multiple occurences of tri-peptide segments 5–20 times in comparison to the situation wherein such selective identification is not carried out.
As in the case of selective labeling, misincorporation of the isotopic label at the undesired sites also occurs in selective unlabeling (Table 1). It leads to incorporation of 14N in the undesired amino acids resulting in diminution of their intensity. This can be alleviated by use of E. coli strains that are auxotrophic for a given amino acid type (Muchmore et al. 1989) or by employing the cell-free protein synthesis approach (Apponyi et al. 2008; Kainosho et al. 2006; Vinarov et al. 2004). However, we have observed that with an appropriate choice of the amino acid type, the misincorporation is not detrimental to the analysis. For instance, selective unlabeling of Leu results in significant unlabeling of Ile and Val (Fig. 5). However, the 13Cα and 13Cβ chemical shifts of these occur in distinct spectral regions (Fig. 1), enabling their distinction. Thus, by avoiding selective unlabeling of amino-acid types belonging to the same group, the effect of misincorporation of 14N at the undesired sites can be safely ignored.
Conclusion
In summary, we have developed a new method for sequence specific NMR assignments in proteins based on the method of amino acid selective unlabeling. Using a new pulse sequence proposed in combination with 2D [15N–1H] HSQC, the method involves the detection of tri-peptide segments along the polypeptide chain. The method achieves the dual objective of reducing the search space for sequential assignments and conferring uniqueness to short segments of linked amino acid residues. This will directly benefit automated assignment algorithms for unambiguous and accurate resonance assignments. The method can be combined with other approaches for isotope labeling (Kainosho and Guntert 2009; Ohki and Kainosho 2008) and/or with completely/partially deuterated samples prepared for structure determination of larger molecular weight proteins. Taken together, this study opens up new avenues to using this methodology in protein structural studies.
References
Apponyi MA, Ozawa K, Dixon NE, Otting G (2008) Cell-free protein synthesis for analysis by NMR spectroscopy. Methods Mol Biol 426:257–268
Atreya HS, Chary KVR (2000) Amino acid selective ‘unlabeling’ for residue-specific NMR assignments in proteins. Curr Sci 79:504–507
Atreya HS, Chary KVR (2001) Selective ‘unlabeling’ of amino acids in fractionally C-13 labeled proteins: an approach for stereospecific NMR assignments of CH3 groups in Val and Leu residues. J Biomol NMR 19:267–272
Atreya HS, Sahu SC, Chary KVR, Govil G (2000) A tracked approach for automated NMR assignments in proteins (TATAPRO). J Biomol NMR 17:125–136
Atreya HS, Chary KVR, Govil G (2002) Automated NMR assignments of proteins for high throughput structure determination: TATAPRO II. Curr Sci 83:1372–1376
Baran MC, Huang YJ, Moseley HNB, Montelione GT (2004) Automated analysis of protein NMR assignments and structures. Chem Rev 104:3541–3555
Barnwal RP, Rout AK, Atreya HS, Chary KVR (2008) Identification of C-terminal neighbors of amino acid residues without an aliphatic 13Cγ as an aid to NMR assignments in proteins. J Biomol NMR 41:191–197
Bartels C, Xia TH, Billeter M, Güntert P, Wüthrich K (1995) The Program XEASY for computer-supported NMR Spectral-Analysis of Biological Macromolecules. J Biomol NMR 6:1–10
Cavanagh J, Fairbrother WJ, Palmer AG, Skelton NJ (1996) Protein NMR spectroscopy. Academic Press, San Diego
Cornilescu G, Delaglio F, Bax A (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR 13:289–302
Cruz L, Bajaj R, Becker S, Zweckstetter M (2010) The intermembrane space domain of Tim23 is intrinsically disordered with a distinct binding region for presequences. Protein Sci 19:2045–2054
Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe—a multidimensional spectral processing system based on unix pipes. J Biomol NMR 6:277–293
Dötsch V, Wagner G (1996) Editing of amino acid type in CBCA(CO)NH experiments based on the 13Cβ–13Cγ coupling. J Magn Reson B 111:310–313
Dötsch V, Matsuo H, Wagner G (1996a) Amino-acid type identification for deuterated proteins with a β-carbon edited HNCOCACB experiment. J Magn Reson B 112:95–100
Dötsch V, Oswald RE, Wagner G (1996b) Amino acid type selective triple resonance experiments. J Magn Reson B 110:107–111
Goto NK, Kay LE (2000) New developments in isotope labeling strategies for protein solution NMR spectroscopy. Curr Opin Struct Biol 10:585–592
Hilty C, Fernandez C, Wider G, Wüthrich K (2002) Side chain NMR assignments in the membrane protein OmpX reconstituted in DHPC micelles. J Biomol NMR 23:289–301
Kainosho M, Guntert P (2009) SAIL—stereo-array isotope labeling. Q Rev Biophys 42:247–300
Kainosho M, Torizawa T, Iwashita Y, Terauchi T, Ono AM, Güntert P (2006) Optimal isotope labeling for NMR protein structure determinations. Nature 440:52–57
Kelly MJS, Krieger C, Ball LJ, Yu Y, Richter G, Schmieder P, Bacher A, Oschkinat H (1999) Application of amino acid type-specific 1H and 14N labeling in a 2H-, 15N-labeled background to a 47 kDa homodimer: potential for NMR structure determination of large proteins. J Biomol NMR 14:79–83
Mohan PMK, Barve M, Chatteljee A, Ghosh-Roy A, Hosur RV (2008) NMR comparison of the native energy landscapes of DLC8 dimer and monomer. Biophys Chem 134:10–19
Muchmore DD, McIntosh LP, Russell CB, Anderson DE, Dahlquist FW (1989) Expression and nitrogen-15 labeling of proteins for proton and nitrogen-15 nuclear magnetic resonance. Methods Enzymol 177:44–73
Mukherjee S, Mustafi SM, Atreya HS, Chary KVR (2005) Measurement of 1J(N,Cα), 1J(Ni, Ci-1‘), 2J(Ni, C αi-1 ), 2J(HNi,Ci-1’) and 2J(HNi, C αi ) values in C-13/N-15-loabelled proteins. Magn Reson Chem 43:326–329
Ohki S, Kainosho M (2008) Stable isotope labeling methods for protein NMR. Prog Nucl Spectrosc 53:208–226
Schubert M, Schmalla M, Schmeider P, Oschkinat H (1999) MUSIC in triple-resonance experiments: amino acid type-selective 15N/1H correlations. J Magn Reson 141:34–43
Schubert M, Oschkinat H, Schmeider P (2001a) Amino acid type-selective backbone 15N/1H correlations for Arg and Lys. J Biomol NMR 20:379–384
Schubert M, Oschkinat H, Schmeider P (2001b) MUSIC and aromatic residues: amino acid type-selective backbone 15N/1H correlations, part III. J Magn Reson 153:186–192
Schubert M, Oschkinat H, Schmeider P (2001c) MUSIC, selective pulses and tuned amino acid type-selective 15N/1H correlations, part II. J Magn Reson 148:61–72
Schubert M, Labudde D, Leitner D, Oschkinat H, Schmeider P (2005) A modified strategy for sequence specific assignment of protein NMR spectra based on amino acid type selective experiments. J Biomol NMR 31:115–128
Shortle D (1995) Assisgnment of amino acid type in 1H–15N correlation spectra by labeling with 14N-amino acids. J Magn Reson B105:88–90
Tugarinov V, Kay LE (2004) Stereospecific NMR assignments of prochiral methyls, rotameric states and dynamics of valine residues in malate synthase G. J Am Chem Soc 126:9827–9836
Tugarinov V, Hwang PM, Kay LE (2004) Nuclear magnetic resonance spectroscopy of high-molecular-weight proteins. Ann Rev Biochem 73:107–146
Turano P, Lalli D, Felli IC, Theil EC, Bertini I (2010) NMR reveals pathway for ferric mineral precursors to the central cavity of ferritin. Proc Natl Acad Sci USA 107:545–550
Vinarov DA, Lytle BL, Peterson FC, Tyler EM, Volkman BF, Markley JL (2004) Cell-free protein production and labeling protocol for NMR-based structural proteomics. Nat Methods 1:149–153
Vuister GW, Kim SJ, Wu C, Bax A (1994) 2D and 3D NMR-study of phenylalanine residues in proteins by reverse isotopic labeling. J Am Chem Soc 116:9206–9210
Wüthrich K (1986) NMR of proteins and nucleic acids. Wiley, New York
Acknowledgments
The facilities provided by NMR Research Centre at IISc supported by Department of Science and Technology (DST), India is gratefully acknowledged. HSA acknowledges support from DST-SERC research award. PDS acknowledges support from the Wellcome Trust International Senior Research Fellowship in Biomedical Science, WT081643MA. GJ acknowledges fellowship from Council of Scientific and Industrial Research (CSIR) and AT acknowledges support from University Grants Commission (UGC). We thank Dr. John Cort, Pacific Northwest National Laboratory, for providing the Ubiquitin plasmid.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Krishnarjuna, B., Jaipuria, G., Thakur, A. et al. Amino acid selective unlabeling for sequence specific resonance assignments in proteins. J Biomol NMR 49, 39–51 (2011). https://doi.org/10.1007/s10858-010-9459-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-010-9459-z