
Abstract
Invasion of cells from surrounding tissues is a crucial step for regeneration when using a-cellular scaffolds as a replacement of the nucleus pulposus (NP). The aim of current study was to assess whether NP and surrounding annulus fibrosus (AF) cells are capable of migrating into dense collagen scaffolds. We seeded freshly harvested caprine NP and AF cells onto scaffolds consisting of 1.5 and 3.0% type I collagen matrices, prepared by plastic compression, to assess cell invasion. The migration distance appeared both time and density dependent and was higher for NP (25%) compared to AF (10%) cells after 4 weeks. Migration distance was not enhanced by Hst-2, a peptide derived from saliva known to enhance fibroblast migration, and this was confirmed in a scratch assay. In conclusion, we revealed invasion of cells into dense collagen scaffolds and therewith encouraging first steps towards the use of a-cellular scaffolds for NP replacement.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Lumbar discectomy is an effective therapy for neurological decompression in patients suffering from a herniated nucleus pulposus (NP). Discectomies however, do not deal with the damaged intervertebral disc (IVD) and may even further aggravate existing damage [17]. In the majority of the patients, radiological signs of disc degeneration are present after 2 years [18]. It is therefore not surprising, that after a successful decompression, many patients suffer from persisting or progressive low back pain. After 10 years follow-up, almost one third of the patients are dissatisfied and a quarter underwent re-surgery in the meantime [5, 9, 16, 29]. During the last decade, increasing knowledge and technical advancements in the field of tissue engineering have resulted in numerous promising strategies to replace or regenerate the NP [17, 20]. Materials that have been used include collagen, chitosan, alginate and fibrin [1–3, 6, 7, 19, 32]. All of them have their own advantages, disadvantages, and clinical potential. In a previous study, we showed that with very dense collagen scaffolds (23% or ~230 mg/mL), prepared by plastic compression, the viscoelastic properties of native NP tissue can be approached [6]. Scaffold stiffness is increasingly recognized as a potent mechanical cue for the differentiation and biosynthetic response of (stem) cells [4, 6, 10–12, 14].
Our overall goal is to develop a-cellular collagen scaffolds that can be used to replace the NP in one surgical procedure combined with a discectomy. The scaffolds should prevent the degenerative cascade of the intervertebral disc and surrounding structures, which is initiated directly after disc herniation. Furthermore, restoration of the biomechanical properties should facilitate regeneration of the herniated disc. The advantage of using an a-cellular scaffold is the absence of time-consuming and expensive cell harvesting and culturing techniques during surgery. Instead, cells from the surrounding tissue are hypothesized to migrate into the scaffold thereby digesting the scaffold and secreting their own native extra cellular matrix (ECM) [6, 17]. The invasion of cells, also called ‘in situ seeding’, should occur from tissues that are in direct contact with the scaffold and thus only include the remnants of the NP and the surrounding annulus fibrosus (AF) [5].
The aim of the current study is to investigate if AF and NP cells are actually capable of migrating into dense collagen scaffolds. We investigate the migration of cells into scaffolds with densities of 1.5% (~15 mg/mL) and 3.0% (~30 mg/mL) collagen. Since the spaces between collagen fibers in dense collagen matrices are too small for cells to pass (Fig. 1), migration is expected to occur only in the presence of some matrix breakdown. We therefore studied the presence of Endo180, a receptor involved in the uptake for intra-cellular degradation of collagen and migration of the cells [31]. We also studied if migration can be enhanced by the use of an experimental chemotactic agent. For this purpose used Histatin-2, a peptide derived from saliva, which was recently discovered to have chemo attractive properties on fibroblasts [23, 24]. The migratory effects of Histatin-2 are finally confirmed in a scratch assay, which is a more sensitive and reproducible model to detect migration [8].

Transmission electron microscopic pictures of the collagen scaffolds; a Uncompressed 0.5% (~5 mg/mL), b Compressed 1.5% (~15 mg/mL) and c Compressed 3% (~30 mg/mL). The spaces between individual collagen fibers can be estimated from the in-picture bar shown on the right below
2 Materials and methods
2.1 Cell isolation and culturing
Cells of the AF and the NP were isolated from the thoracic spines of mature female Dutch milk goats. The IVDs were carefully excised from the endplates and separated into the AF and NP by knife. The tissues were minced and digested under gentle shaking at 37 °C in medium composed of Dulbecco’s Modified Eagle’s Medium (DMEM, Invitrogen, Carlsbad, Ca, USA) supplemented with 500 μg/mL streptomycin (Sigma-Aldrich, St. Louis, MO, USA), 600 μg/mL penicillin (Sigma-Aldrich, St. Louis, MO, USA) and 2.5 μg/mL amphotericin B (Fungizone, Sigma-Aldrich, St. Louis, MO, USA) in the presence of 2.5% (w/v) pronase E (Sigma-Aldrich, St. Louis, MO, USA) (digestion of the NP) or 5% (w/v) pronase E (digestion of the AF). After 1 h a solution of medium, fetal calf serum (FCS, HyClone, South Logan UT, USA) and liberase (Roche, Mannheim Germany) was added. The final concentration of liberase was 0.125% (w/v) for digestion of the NP and 0.25% (w/v) for digestion of the AF. The final concentration FCS was 5% for both types of tissue. Tissue was digested overnight (stirring, 37 °C). All digests were filtered through a cell strainer (100 μm pores, BD Falcon, San Jose, CA, USA). After centrifuging for 10 min at 600 RCF, the cells were rinsed in medium containing 10% FCS. Additionally, the cells that were used in the three-dimensional migration experiments were stained with 5 μM DiI (λabsorption = 553 nm, λemission = 570 nm, Molecular Probes, Carlsbad, CA, USA) for 20 min (37 °C, 5% CO2) and washed twice in phosphate buffered saline (PBS Invitrogen, Carlsbad, CA, USA). The cells were then re-suspended in medium containing 10% FCS and 50 μg/mL ascorbic acid (Merck Biosciences, Sandiago, CA, USA), which will be referred to as IVD cell medium.
2.2 Preparation of the collagen scaffolds
Dense collagen scaffolds were prepared by plastic compression, as described previously [6]. Briefly, a collagen solution (6 mg/mL, Arthro Kinetics, Esslingen, Germany) was neutralized, moulded in a cylinder with a cell culture insert on the bottom, and then polymerized in an incubator (37 °C, 5% CO2) for 90 min. Then a stamp was put on top of the matrix and the cylinder was put in the incubator overnight. The pores of the cell culture inserts allow the fluid to pass and retain the collagen matrix thereby increasing its density. The scaffold had a final height of 3.6 mm and a circular shape (diameter 25 mm, similar to the cylinders). Scaffolds were prepared in two densities, 1.5 and 3% collagen (Fig. 2).

Schematic presentation of the set up used for the migration experiments. First collagen scaffold are made by plastic compression in a cylinder with a cell culture insert used as a filter on the bottom (a). The filter is then transferred to a culture well and cells are seeded on top of the scaffolds (b)
2.3 Histatin-2 synthesis
Peptides were synthesized by solid-phase peptide synthesis by Fmoc chemistry with a MilliGen 9050 peptide synthesizer (Milligen-Bioresearch, Bredford, MA, USA). Purification by RP_HPLC and confirmation of authenticity by MS were conducted as described previously [23, 28]. The amino-acid sequence of Histatin 2 (Hst2) is RKFHEKHHSHREFPFYGDYGSNYLYDN. In addition, a dextro enantiomer, D-Hst2 was synthesized, which was shown to lack migratory effect. The sequence is the same, but made with d-amino acids.
2.4 Scaffold migration experiments
After the removal of the cell culture inserts together with the scaffolds from the cylinders, they were transferred to the wells of a six-well plate filled with 1.5 mL IVD cell medium. This arrangement is shown in Fig. 2. A plexiglass ring was placed inside the filter to prevent the leakage of medium from the upper side of the scaffold into the well. To obtain a sufficient number of cells, the cells acquired from two different donors were mixed and 300 K of these mixed cells were seeded on top of each scaffold. These cells were suspended in 0.5 mL IVD cell medium. Every day 0.5 mL IVD cell medium was added at top of the scaffold, compensating for the evaporation of the medium and also maintaining the chemotactic gradient. The medium inside the well was replaced twice a week. Cells were incubated for 2 or 4 weeks at 37 °C and 5% CO2. For the chemotactic experiments, the medium inside the well contained 50 μg/mL Hst2 or D-Hst2. During the incubation period, samples were weekly visualised using a Bio-Rad MRC-1000 UV confocal system attached to an inverted microscope (Leica Microsystems).
2.4.1 Histology
After the incubation period, the scaffolds were fixed with 4% formaldehyde and stored at 4 °C overnight. The scaffolds were cut in half and one half was dehydrated in a graded series of ethanol solutions, followed by xylene, and at last the scaffold was embedded in paraffin. Sections of 7 μm thickness were prepared orthogonal to the seeding plane. After deparaffinization of the sections with xylene substitute and hydration in a declining series of ethanol solutions, sections were stained with haematoxylin for 20 min, followed by eosin for 1 min. Sections were dehydrated in a series of ethanol solutions, transferred into xylene and coverslipped. A bright-field microscope (Leica Microsystems, Bannockburn, IL, USA) was used to visualize the sections. When the scaffold was too broad to be visualized in a single view, two pictures with sufficient overlap were taken. An overlay of these pictures was made using Photoshop (Adobe, San Jose, CA, USA). The pictures were read into Matlab (The MathWorks Inc., Eindhoven, The Netherlands). The level of migration M is defined by: \( M = \frac{d} {w} \times 100\%, \) where d is the distance travelled by the cells and w is the width of the scaffold (Fig. 3). Relative migration was used as a measure of migration because of deformations that where introduced in the scaffolds during the incubation period and fixation process. The migration in a scaffold is calculated as the mean of the four measures per scaffold. In turn, the migration for each different condition is the mean of the triple experiments. The standard deviation per condition is calculated as the root of the sum of the squared standard deviations of each individual scaffold. Only samples in which at least at two different positions migration could be determined are included in analysis.

Examples of 1.5% collagen scaffolds seeded with AF (a) and NP (b) cells after 28 days of migration (stain HE. Magnification ×20)
2.4.2 Immunohistology
A custom made rabbit polyclonal antibody directed against KLH-coupled linear synthetic peptide C-GTDVREPDDSPQGRRE corresponding to hinge domain between two adjacent C-type lectin-like domains (CTLDs) of ENDO 180 protein is used to verify the presence of Endo180 in the cells. Preliminary sections were deparaffinized with xylene substitute and hydrated in a declining series of ethanol solutions. The sections were then treated with proteinase K to retrieve the antigen (10 μg/mL, Merck, Darmstadt, Germany) for 30 min and washed three times with PBS. Subsequently, sections were incubated for 10 min with 3% hydrogen peroxide in methanol to block endogenus peroxidise activity and washed again with PBS. Sections were incubated with blocking solution (Zymed Laboratories, San Francisco, CA, USA) for 30 min and incubated with affinity purified primary antibody (1.3 μg/mL) for 30 min at room temperature followed by incubation overnight at 4 °C. Universal negative control rabbit (Dako) was used as negative control. Next day sections were washed with a solution of TBS, 0.25% BSA and 0.5% Triton X-100 and incubated for 30 min with 1:500 diluted HRP-labeled sheep anti-rabbit secondary antibody (Dako). After washing with PBS, sections were incubated for 15 min with 3,3′-diaminobenzidine (DAB) and hydrogen peroxide (peroxidase substrate kit DAB, Vector Laboratories, Burlingame, CA, USA) and washed in distilled water. Sections were counterstained with Mayer’s haematoxylin or 2 min.
2.5 Scratch assay
The effects of Hst2 on the migration of AF and NP cells were also examined in a 2D scratch assay. In 48-well plates, 25,000 cells were seeded and cultured until confluence in IVD cell medium. After 5 h of serum deprivation a scratch was made using a sterile blue pipette tip. The following conditions were tested: IVD cell medium without serum, IVD cell medium without serum containing 10, 50 or 100 μg/mL Hst2 or 50 μg/mL D-Hst2, and IVD cell medium. The scratch was photographed at the day of creation (day 0) and after 2 days. For each sample the surface area of the scratch was determined in the open source software ImageJ. Per donor the mean of the triple experiments was calculated. From these mean values relative closure (RC) was calculated as described by Oudhoff et al. [23] and given by: \( RC = \frac{{{X_0} - {X_2}}} {{S{F_0} - S{F_2}}}. \) X 0 is the mean surface area of the scratch in a specified condition at day 0 and X 2 is the mean surface area at the second day. SF 0 is the mean surface area of the scratch in serum free medium at day 0 and SF 2 is the mean surface area after 2 days of exposure to serum free medium. Cells were fixed for 20 min in 4% para-formaldehyde and stained with ALEXA-conjugated phalloidin (5 U/mL, Molecular Probes) for 1 h in a dark environment. In addition to the actin staining, the nuclei of the cells were stained by the addition of vectashield with DAPI (Vector Laboratories, Burlingame, CA, USA). Cells were viewed under a fluorescence microscope.
2.6 Statistical analysis
Statistical significance of the data was determined with a N-way ANOVA procedure. Additionally a t-test with Bonferroni correction was performed to determine significance between individual samples. A P value less than 0.05 was considered significant.
3 Results
3.1 Migration without chemoattractant
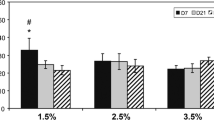
After 14 days, there were no significant differences in the migration distance between NP (white bars) or AF (black bars) cells (Fig. 4). In both series, the 1.5% and the 3% scaffolds, the mean migration distance was limited to approximately 5% (~180 μm) of the full scaffolds thickness. After 28 days, the migration distance of the NP cells had significantly increased compared to 14 days, while no differences were observed for the AF cells (Fig. 4). The mean migration distance of the NP cells after 28 days was significantly higher compared to the AF cells in both scaffolds densities. For the 1.5% collagen scaffolds the mean distance was 25% (~0.9 mm) for the NP cells compared to 9.5% (~0.35 mm) for the AF cells (P < 0.05). For both cell types, a significant higher migration distance was observed in the 1.5% collagen scaffolds compared to the 3% scaffolds (Fig. 4). The collagen scaffolds were too dense to visualise cells in the deeper layers of the scaffold. An example of the surface area of a scaffold is shown in Fig. 5.

Graphic showing the results of the migration experiments of NP (white bars) and AF (black bars) cells after 14 and 28 days in 1.5 and 3% collagen matrices. After 14 days, no significant differences are observed. After 28 days, NP cells show a significantly higher migration compared to the AF cells. For both cell types the migration is higher in the 1.5 compared to the 3% collagen scaffolds

Surface area of 1, 5% collagen type I scaffolds after 1 week of incubation. (a AF cells, b NP cells. Stain DiI)
3.2 Migration with Hst-2
The addition of Hst-2 did not result in increased migration distance. Instead, in a large fraction of the AF cell samples, no cells could be detected after 14 or 28 days, or too few to perform analysis. Although some cell death was also observed in a few of the NP cell experiments, these data was sufficient for analysis. After 14 days, no differences were observed for NP cells with our without chemo attractant. After 28 days however, NP cells in the samples supplemented with Hst-2 (Fig. 6, grey bars) showed significantly less migration when compared to samples without chemo attractant (Fig. 6, black bars) The migration without chemoattractant was significantly higher compared to the migration with Hst2 or D-Hst2 (Fig. 6, white bars). Between both peptides, no significant differences were found.

Graphic showing the results of the migration experiments of NP in a 1.5% collagen matrix with and without the addition of Hst-2 as a chemo attractant. The results with the negative enantiomer DHst-2 are also shown. The samples with Hst2 and Dhst-2 reveal a significantly decreased migration after 28 days
3.3 Expression of endo180
The expression of endo180 by both NP and AF was assessed using an antibody directed to a hinge domain in the protein. Figure 7 shows that endo180 was expressed on both cell types after 14 days of migration in 1.5% collagen scaffolds. Similar results were obtained after 28 days of incubation and in 3% scaffolds.

Images showing the results of Endo180 staining (HE counterstain) of 1.5% collagen scaffolds after 14 days of migration. a AF cells stained for Endo180 (brown color), b AF cells, negative control, c NP cells stained for Endo180 (brown color), d NP cells negative control
3.4 Scratch assay
The scratches had a mean width of 666 μm (+/− 77). No statistically significant differences between AF and NP cells were observed in any of the assays (Fig. 8). The addition of Hst-2 to the medium had no significant effect on scratch closure compared to the negative control (D-Hst-2) or serum free medium. The addition of 10% serum to the medium resulted in complete closure of the scratch in both, assays with NP and AF cells (7). Staining of the F-actin filaments, to visualize the morphology of the cells, revealed different cell shapes for AF and NP cells (Fig. 9). In addition, differences were found between cells inside and outside the scratch. Non-migrated NP cells outside the scratch had a round, chondrocytic, morphology (Fig. 9a), while NP cells that had migrated into the scratch were characterized by long dendritic processes (Fig. 9b) Cells of the AF outside the scratch had a more flattened, fibroblast like, appearance (Fig. 9c), whereas cells inside the scratch were more elongated and aligned with each other (Fig. 9d). To confirm Hst-2 activity, a scratch assay was performed with human oral mucosa derived cells (HO-1-N-1 cell line), known to be sensitive for Hst-2 stimulation [24]. This assay showed a significant increased scratch closure after addition of Hst-2 (Fig. 10).

Graphic showing the results of the scratch assay. No significant differences are observed between the samples supplemented with Hst-2 compared to the negative control (D-Hst2) or serum free medium. The addition of 10% serum to the medium resulted in a significant increase in closure of the scratches

Images of cells during the scratch assay stained for F-actin (green) and the nuclei are stained with DAPI (blue). a NP cells outside the scratch, b NP cells inside the scratch, c AF cells outside the scratch, d AF cells inside the scratch

A confirmation scratch assay with human squamous carcinoma HO-1-N-1 cells reveals a significant enhanced cell migration after the addition of Hst-2
4 Discussion
The aim of the current study was to assess the migration of native cells into collagen scaffolds intended for functional replacement of the NP. Migration distance proved to be both, time and density dependent. After 14 days, the observed migration was very limited for all cell types and collagen densities. Although current conditions are not fully comparable to other studies, since migration is usually studied from high towards low density matrices, a certain ‘lag phase’ before migration occurs has been recognised [25]. A few explanations have been suggested for this phenomenon [25]. Firstly, cells require some time to overcome the differences in matrix stiffness [15, 25]. This could also explain the relative long lag period (14 days) in the current study compared to the reported periods (16 h), since in current study cells were expected to migrate into a high stiffness matrix from the outside [15, 25]. Secondly, cells need some time to upregulate biosynthetic features such as actin–myosin activity necessary for migration [22, 25]. This again seems to apply for the current condition, since we used freshly harvested cells for the experiments. Prior to harvesting, these cells are surrounded by their own pericellular matrix, interacting with neighbouring cells and subjected to tensile forces (AF cells) or hydrostatic pressure (NP cell) [5, 13].
Migration was not enhanced by the addition of Hst-2, a peptide present in human saliva, which was described to be an important wound closure-stimulating factor [23]. The peptide was shown to enhance the activity of oral and non-oral fibroblasts in vitro [24]. The exact receptor, however, to which the protein binds, remains unknown [23, 24], hardening to hypothesize why current IVD cells were insensitive to the pro-migratory effects of Hst-2 and cell death was even increased. A scratch assay, which is a more sensitive model to detect chemotaxis [23], confirmed that NP and AF cells are insensitive to Hst-2. Migration of both, AF and NP cells was not enhanced by Hst2 or D-hst2 compared to the negative control (serum free medium). All samples showed significantly less migration than the positive control (medium with 10% serum), in which full closure of the scratches was observed. The absence of any chemo attractive effects of Hst2 on current IVD cell populations might be related to differences between human and caprine cells. We used cells derived from goats, while human cells were used in earlier studies [23, 24]. Interestingly, the scratch assay allowed visualising the phenotypical differences between NP and AF cells, both before and during migration (Fig. 9). These findings are of importance since specific phenotypical markers for both cell types are still being studied and not generally accepted [26, 27, 30].
We studied collagen densities of 1.5% (~15 mg/mL) and 3% (~30 mg/mL), which are both higher compared to the free floating collagen matrices most often studied (up to 0.5% or 5 mg/mL) [6]. In the latter, cells adhere to the matrix and transmit forces to the collagen fibers resulting in increased stiffness and contraction [25]. For stiffer matrices, as currently studied, cells have to undergo major cytoskeletal reorganisation in order to induce the formation of stress fibers and focal adhesions. Furthermore, microtubules play an important role in the remodelling of 3D matrices. In stiff matrices, the microtubules determine cell polarity, while they mainly participate in spreading in soft matrices [22, 25]. Miron et al. [22] hypothesized that if the collagen matrix can resist the cellular traction force, cells can move. Reversely, if the matrix cannot resist the traction forces, remodelling occurs first. However, the matrices studied by Miron et al. had a much lower collagen density (1.5 mg/mL) compared to our study (15 and 30 mg/mL). Figure 1 shows that the spaces between individual collagen fibers (approximately ~100 Nm) are too small for cells (~10–20 μm) to pass. For this reason, some collagen degradation will be necessary to allow cell migration. We therefore stained one specimen of every series with an antibody directed to the hinge domain of the collagen internalisation receptor Endo180 (Fig. 7). Endo180 binds and internalises collagen for lysosomal degradation and was shown to be important for ECM remodelling and cell migration [21]. Interestingly, expression of the receptor was found on both, NP and AF cells. The presence of the antibody indicates that cell invasion in stiff collagen matrices may occur via collagen breakdown. This may be an important explanation why stimulation of migration by a chemokine alone does not have any effect. However, the differences in invasion and migration between AF and NP cells cannot be explained, since endo180 was present on both (Fig. 7). Other mechanisms, either via intra-cellular uptake or via extra-cellular collagen degradation (by matrix metalloproteinase’s) might be responsible [21].
An important limitation of our study is that the stiffness of the tested scaffolds (1.5 and 3%) is lower than the stiffness of the NP itself, which was found to agree with 23% collagen [6]. We did not study such high concentrations because preliminary studies in our laboratory showed that this would require culturing times in the order of years (unpublished data). However, as scaffolds within the repaired intervertebral disc will continuously be loaded and deformed, the migration speed of native cells into the scaffolds might actually be much higher in vivo. Furthermore, long culturing times, which limited our choice for higher collagen densities, are of course no problem in vivo. In vivo studies in a large animal model are required to address these issues. Another potential limitation of current study is the use of scaffolds consisting of Collagen type I, since the major collagen component of the NP is type II collagen. However, the fabrication of dense collagen scaffold requires large amounts of collagen, making collagen type II unattractive. Furthermore, the technique of plastic compression has not yet been applied to type II collagen.
In conclusion, in the current study we showed that IVD cells are capable of migrating into dense collagen scaffolds and that intra-cellular uptake and digestion of collagen are involved. The migration speed was both time and density dependent and was higher for NP compared to AF cells. Migration speed could not be enhanced by the use of Hst-2, a peptide derived from human saliva that was recently described to have chemo attractive properties. Although the densities currently studied have a lower stiffness compared to the NP, current results underscore the potential of “in situ” seeding concept of scaffolds for intervertebral disc engineering. However, the thickness of the final implant should be kept small to facilitate invasion and remodelling.
References
Abbushi A, Endres M, Cabraja M, Kroppenstedt SN, Thomale UW, Sittinger M, Hegewald AA, Morawietz L, Lemke AJ, Bansemer VG, Kaps C, Woiciechowsky C. Regeneration of intervertebral disc tissue by resorbable cell-free polyglycolic acid-based implants in a rabbit model of disc degeneration. Spine. 2008;33:1527–32.
Alini M, Li W, Markovic P, Aebi M, Spiro RC, Roughley PJ. The potential and limitations of a cell-seeded collagen/hyaluronan scaffold to engineer an intervertebral disc-like matrix. Spine. 2003;28:446–54.
Boyd LM, Carter AJ. Injectable biomaterials and vertebral endplate treatment for repair and regeneration of the intervertebral disc. Eur Spine J. 2006;15(Suppl 3):S414–21.
Breuls RG, Jiya TU, Smit TH. Scaffold stiffness influences cell behavior: opportunities for skeletal tissue engineering. Open Orthop J. 2008;2:103–9.
Bron JL, Helder MN, Meisel HJ, van Royen BJ, Smit TH. Repair, regenerative and supportive therapies of the annulus fibrosus: achievements and challenges. Eur Spine J. 2009;18:301–13.
Bron JL, Koenderink GH, Everts V, Smit TH. Rheological characterization of the nucleus pulposus and dense collagen scaffolds intended for functional replacement. J Orthop Res. 2009;27:620–6.
Cloyd JM, Malhotra NR, Weng L, Chen W, Mauck RL, Elliott DM. Material properties in unconfined compression of human nucleus pulposus, injectable hyaluronic acid-based hydrogels and tissue engineering scaffolds. Eur Spine J. 2007;16:1892–8.
Cory G. Scratch-wound assay. Methods Mol Biol. 2011;769:25–30.
Dai LY, Zhou Q, Yao WF, Shen L. Recurrent lumbar disc herniation after discectomy: outcome of repeat discectomy. Surg Neurol. 2005;64:226–31.
Damianova R, Stefanova N, Cukierman E, Momchilova A, Pankov R. Three-dimensional matrix induces sustained activation of ERK1/2 via Src/Ras/Raf signaling pathway. Cell Biol Int. 2008;32:229–34.
Dikovsky D, Bianco-Peled H, Seliktar D. Defining the role of matrix compliance and proteolysis in three-dimensional cell spreading and remodeling. Biophys J. 2008;94:2914–25.
Discher DE, Janmey P, Wang YL. Tissue cells feel and respond to the stiffness of their substrate. Science. 2005;310:1139–43.
Duncan NA. Cell deformation and micromechanical environment in the intervertebral disc. J Bone Joint Surg Am. 2006;88(Suppl 2):47–51.
Engler AJ, Sweeney HL, Discher DE, Schwarzbauer JE. Extracellular matrix elasticity directs stem cell differentiation. J Musculoskelet Neuronal Interact. 2007;7:335.
Grinnell F, Ho CH, Tamariz E, Lee DJ, Skuta G. Dendritic fibroblasts in three-dimensional collagen matrices. Mol Biol Cell. 2003;14:384–95.
Hakkinen A, Kiviranta I, Neva MH, Kautiainen H, Ylinen J. Reoperations after first lumbar disc herniation surgery; a special interest on residives during a 5-year follow-up. BMC Musculoskelet Disord. 2007;8:2.
Hegewald AA, Ringe J, Sittinger M, Thome C. Regenerative treatment strategies in spinal surgery. Front Biosci. 2008;13:1507–25.
Jonsson B, Stromqvist B. Repeat decompression of lumbar nerve roots. A prospective two-year evaluation. J Bone Joint Surg Br. 1993;75:894–7.
Leone G, Torricelli P, Chiumiento A, Facchini A, Barbucci R. Amidic alginate hydrogel for nucleus pulposus replacement. J Biomed Mater Res A. 2008;84:391–401.
Leung VY, Chan D, Cheung KM. Regeneration of intervertebral disc by mesenchymal stem cells: potentials, limitations, and future direction. Eur Spine J. 2006;15(Suppl 3):S406–13.
Messaritou G, East L, Roghi C, Isacke CM, Yarwood H. Membrane type-1 matrix metalloproteinase activity is regulated by the endocytic collagen receptor Endo180. J Cell Sci. 2009;122:4042–8.
Miron-Mendoza M, Seemann J, Grinnell F. Collagen fibril flow and tissue translocation coupled to fibroblast migration in 3D collagen matrices. Mol Biol Cell. 2008;19:2051–8.
Oudhoff MJ, Bolscher JG, Nazmi K, Kalay H, van’t Hof W, Amerongen AV, Veerman EC. Histatins are the major wound-closure stimulating factors in human saliva as identified in a cell culture assay. FASEB J. 2008;22:3805–12.
Oudhoff MJ, van den Keijbus PA, Kroeze KL, Nazmi K, Gibbs S, Bolscher JG, Veerman EC. Histatins enhance wound closure with oral and non-oral cells. J Dent Res. 2009;88:846–50.
Rhee S. Fibroblasts in three dimensional matrices: cell migration and matrix remodeling. Exp Mol Med. 2009;41:858–65.
Rutges J, Creemers LB, Dhert W, Milz S, Sakai D, Mochida J, Alini M, Grad S. Variations in gene and protein expression in human nucleus pulposus in comparison with annulus fibrosus and cartilage cells: potential associations with aging and degeneration. Osteoarthr Cartil. 2010;18:416–23.
Sakai D, Nakai T, Mochida J, Alini M, Grad S. Differential phenotype of intervertebral disc cells: microarray and immunohistochemical analysis of canine nucleus pulposus and anulus fibrosus. Spine. 2009;34:1448–56.
Veerman EC, Valentijn-Benz M, Nazmi K, Ruissen AL, Walgreen-Weterings E, van Marle J, Doust AB, van’t Hof W, Bolscher JG, Amerongen AV. Energy depletion protects Candida albicans against antimicrobial peptides by rigidifying its cell membrane. J Biol Chem. 2007;282:18831–41.
Videman T, Nurminen M. The occurrence of anular tears and their relation to lifetime back pain history: a cadaveric study using barium sulfate discography. Spine. 2004;29:2668–76.
Vonk LA, Kroeze RJ, Doulabi BZ, Hoogendoorn RJ, Huang C, Helder MN, Everts V, Bank RA. Caprine articular, meniscus and intervertebral disc cartilage: an integral analysis of collagen network and chondrocytes. Matrix Biol. 2010;29:209–18.
Wienke D, MacFadyen JR, Isacke CM. Identification and characterization of the endocytic transmembrane glycoprotein Endo180 as a novel collagen receptor. Mol Biol Cell. 2003;14:3592–604.
Wilke HJ, Heuer F, Neidlinger-Wilke C, Claes L. Is a collagen scaffold for a tissue engineered nucleus replacement capable of restoring disc height and stability in an animal model? Eur Spine J. 2006;15(Suppl 3):S433–8.
Acknowledgments
The authors like to thank Prof. Dr. E. C. I. Veerman for his kind donation of the histatins and attributions to the design of the experiments.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bron, J.L., Mulder, H.W., Vonk, L.A. et al. Migration of intervertebral disc cells into dense collagen scaffolds intended for functional replacement. J Mater Sci: Mater Med 23, 813–821 (2012). https://doi.org/10.1007/s10856-011-4545-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10856-011-4545-7