
Abstract
The present study describes the production of hyaluronan based porous microparticles by a semi-continuous gas anti-solvent (GAS) precipitation process to be used as a growth factor delivery system for in vivo treatment of ulcers. Operative process conditions, such as pressure, nozzle diameter and HYAFF11® solution concentrations, were adjusted to optimize particle production in terms of morphology and size. Scanning electron microscopy (SEM) and light scattering demonstrated that porous nano-structured particles with a size of 300 and 900 nm had a high specific surface suitable for absorption of growth factors from the aqueous environment within the polymeric matrix. Water acted as a plasticizer, enhancing growth factor absorption. Water contents within the HYAFF11® matrix were analyzed by differential scanning calorimetry (DSC). The absorption process was developed using fluorescence dyes and growth factors. Immunohistochemical analysis confirmed the high efficiency of absorption of growth factor and a mathematical model was generated to quantify and qualify the in vitro kinetics of growth factor release within the polymeric matrix. In vivo experiments were performed with the aim to optimize timed and focal release of PDGF to promote optimal tissue repair and regeneration of full-thickness wounds.
Similar content being viewed by others

Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
1 Introduction
Growth factors are polypeptides that transmit signals to modulate cellular activities. Growth factors can either stimulate or inhibit cellular proliferation, differentiation, migration, adhesion and gene expression [1]. One way of enhancing the in vivo efficacy of growth factors is to facilitate the sustained release of bioactive molecules over an extended time period by their incorporation into polymer carriers. Implantation of a drug delivery device directly into the tissue in need of treatment facilitates localized drug delivery. Delivery systems have been designed in a variety of configurations and have been fabricated from different types of natural and synthetic polymers (degradable, non-degradable) [2–4]. These devices have a common ability to control the release of bioactive proteins for extended periods of time by different mechanisms [5]. Through incorporation into polymeric devices, protein structure and thus biological activity can be stabilized, prolonging the length of time over which growth factors are released at the delivery site. The period of drug release from a polymer matrix can be regulated by the drug loading, type of polymer used and the processing conditions. Adverse processing conditions that cause protein aggregation or denaturation have to be avoided.
In biodegradable carriers, growth factor release is controlled by the polymer matrix’s rate of degradation, which causes changes in the morphological characteristics of the materials, such as porosity or permeability [6, 7]. The use of porous materials offers advantages that are particularly important for drug release systems: (i) higher specific surface for the adsorption/release of active components and (ii) enhancement of the drug release rate for erodible particulate systems. These two aspects are particularly relevant for particles that have nano-structured porosity [8, 9].
These particles are usually produced by double emulsion-solvent evaporation or spray drying techniques. The limits associated with these processes are excessive use of organic solvent, which leads to pollution of the product and waste disposal problems, toxicity for incomplete solvent removal and thermal and chemical degradation of substances [10]. Other techniques, such as spray drying, operate at temperatures that can thermally denature thermosensible compounds, such as proteins. New techniques based on high-pressure gas anti-solvent (GAS) have shown great potential [11], with the advantages of being environmentally safe and preserving the properties of thermally labile compounds. The GAS processes have been shown to produce different types of biopolymeric morphologies at mild temperatures (293–313 K) with an amount of residual organic solvent lower than that recommended by FDA.
In the present report, hyaluronan-based porous nano-particles, obtained with a high-pressure CO2 anti-solvent technique, were produced and used as growth factors delivery systems for in vivo treatment of skin ulcers. The particles had a nano-structured porosity that was particularly suitable for absorbing bioactive molecules. HYAFF11®, the benzyl ester of hyaluronic acid (Fidia advanced Biopolymer, Italy), is a biopolymer well known in tissue engineering applications such as in vitro reconstruction of skin, cartilage, and bone, and has been recently used for the in vivo regeneration of small arteries [12–16]. Microparticles, films or plugs prepared from hyaluronan esters have also been evaluated as a novel drug delivery system [17, 18]. In this study, PDGF was embedded in HYAFF microparticles as a delivery system designed to improve full-thickness wound repair. HYAFF microparticles have the ability to absorb different growth factors, cytokines and bioactive peptide fragments and to release them in a temporally and spatially specific event-driven manner. This timed and focal release of cytokines, enzymes and pharmacological agents should promote optimal tissue repair and regeneration of full-thickness wounds. We tested PDGF because it is a potent activator for cells of mesenchymal origin, and a stimulator of chemotaxis, proliferation and new gene expression in monocytes, macrophages and fibroblasts, accelerating ECM deposition [19, 20]. This family of growth factors exists in both homo- and heterodimeric forms and several authors reported that a single application of PDGF-BB to an incisional wound increased the neoangiogenesis through enhancement of endogenous PDGF-BB signalling [21]. Moreover, PDGF and its relative proteins were the first approved proteins for promoting diabetic foot healing and other chronic nonhealing ulcers. Since 1986, Knighton et al. reported their successful treatment of chronic ulcers with autologous platelet-derived wound healing formula (PDWHF) [22]. In the present study, our aim was to optimize timed and focal release of PDGF to promote optimal tissue repair and regeneration of full-thickness wounds.
2 Materials and methods
2.1 Materials
The biomaterial used in the present study was derived from the total esterification of hyaluronan (synthesized from 80 kDa to 200 kDa sodium hyaluronate) with benzyl alcohol, and is referred to as HYAFF11®. The final product is an uncrosslinked linear polymer with an undetermined molecular weight; it is insoluble in aqueous solution yet spontaneously hydrolyzes over time, releasing benzyl alcohol and hyaluronan. HYAFF11® was used to create non-woven meshes of 50 μm-thick fibers, with a specific weight of 100 g/m2. These devices were obtained from Fidia Advanced Biopolymers (FAB, Abano Terme, Italy) [23].
2.2 Film production
Films were developed by solvent casting of 10% (w/w) HYAFF11®/DMSO solution: 0.5 ml were placed on a glass support and spread with a spatula; solvent was evaporated in an oven at 338 K for 30 min. The film was then peeled from the glass support.
2.3 Polymer particle production: process description
Production of microparticles was performed using a semi-continuous GAS process and the apparatus shown in Fig. 1. An exhaustive description of the process has been reported by Elvassore et al. [24]. Briefly, in the GAS precipitation process, a polymeric organic solution and the supercritical antisolvent were continuously added to the precipitation unit in co-current mode. The organic solution was atomized into small droplets within the high-pressure precipitation unit. The high-pressure GAS induced polymer precipitation from the organic solution, yielding nano- and microparticles that were collected at the bottom of the vessel. A washing step was carried out to extract the residual organic solvents to the desired amount.

Schematic view of experimental apparatus. P: precipitation vessel; P1: high-pressure liquid chromatographic pump; P2: CO2 pump; S: solution vessel; FT: filter; V1, V2: fine metering valves; V3, V4: on-off valves; V5, V6: expansion valves; V7: discharge valve; HE: heat exchangers; GC: CO2 cylinder; RF: refrigerating device; TB1, TB2: thermostatic baths; EU: expansion unit; ST: solvent recovering unit; FM: flow meters; R: rotator; F: aspiration system. (—) high pressure tubing; (---) water line for temperature control
A schematic description of the operative procedure follows: after steady conditions of pressure, temperature and CO2 flow rate were reached, the organic solution was atomized through a fused silica capillary nozzle into a 200 cm3 high pressure vessel by a high-pressure liquid chromatography (P1) pump. Temperature control was achieved by a heat exchanger (TB2) connected to an auxiliary bath (RF). Cell temperature was measured by two Pt 100Ω resistances placed at the top and the bottom of the vessel and CO2 flow rate was controlled by two fine metering valves (V5, V6) and measured with a dry flow-meter (FM).
The antisolvent (CO2) was fed by a reciprocating pump (P2) from the top of the cell. It was then vented through expansion valves (V5, V6) and expansion units (EU, ST). The valves and the outline of CO2 were heated by a thermostatic bath (TB1) to prevent freezing due to CO2 depressurization.
At the end of the experiment, the polymer microparticles were recovered at the bottom of the vessel on 0.22 μm-filters (Millipore, type GS).
2.4 Particle morphology analysis
Particle morphology was investigated by scanning electron microscopy (SEM) (Stereoscan 440, Leica Cambridge). The sample was dispersed in milli-Q water and sonicated for 45 min through an Ultrasonic cleaner (CP104, Vetrotecnica, Italy) in order to break particle aggregates. Samples were then centrifuged for 5 min at 2,000 rcf (Megafuge 1.0, Heraeus) and the supernatant was removed. This operation was repeated for 4 or 5 times in order to remove smaller fragments. 0.1 ml of water containing polymer particles was placed on a glass support and after natural evaporation of liquid, particles were gilded (Polaron, SEM coating system) and observed by SEM.
Particle size was evaluated by light scattering. 0.5 mg of microparticles was dispersed in milli-Q water and sonicated for 30 min (Ultrasonic cleaner, 65% power). The undivided material was discarded. The two phases were then separated by centrifuge at 2,000 rpm for 30 s. Afterwards, the diameter range in each phase was determined by light scattering (DLS Nicomp 380, Particle Sizing Systems, Inc. Santa Barbara, USA). Finally, water was evaporated under a vacuum and particles were weighed in order to determine the percentage of each phase.
2.5 Water content
Differential scanning calorimetric (DSC) spectra was performed in order to determine water content in HYAFF11® 50 μm-thick films. A total of 1 mg of polymer sample, previously immersed in pure water for 1 h and dried at room temperature for 3 h, was analysed by DSC (Q10, TA Instruments) with a ramp heating process from 303 K to 423 K at a rate of 10 K/min. The same analyses were performed on wetted samples dried in a vacuum at room temperature for 15 h, and on samples further dried in an oven at 378 K for 15 h. The quantity of evaporated water could be determined by analyzing the absorbed heat spectra.
2.6 Polymer impregnation
Impregnations were performed both in particles and in films with water soluble dyes (fluorescein [10 μg/ml]) and growth factors (PDGF [0.5 μg/ml]) by means of solution deposition on samples for different contact times (1 min, 5 min, 60 min, 24 h and 72 h).
2.7 Analysis of impregnation
2.7.1 Qualitative analysis
The depth of dye absorption was investigated by analyses of serial 7-μm-thick cryosections of the film. Briefly, films were embedded in medium for frozen tissue specimens (Tissue-Tek OCT, Sakura Finetek, USA) and frozen. Serial 7-μm-thick cryosections were cut by ultramicrotome, mounted on gelatin-covered slides and observed with microscopy.
Growth factor superficial impregnation on particles was qualitatively evaluated by immunohistochemical analysis.
Rabbit PDGF anti-human monoclonal antibodies (1/100) were used. The avidin-biotin complex technique (Vectastain-ABC kit; Vector, Burlingame, CA) was used to reveal the immunoreaction. Microparticles were developed for 20 min in peroxide substrate solution containing 3,3′-diaminobenzidine and hydrogen peroxide and at the end were counterstained with Mayer’s hematoxylin. Negative control specimens were prepared using sections incubated without the primary antibody. A brown coloration appeared if the growth factor had been absorbed.
2.7.2 Quantitative analysis
Quantification of impregnated growth factor were tested by ELISA (SIGMA) analysis.
Growth factor release from HYAFF particles were detected in presence of Hyaluronidase (Sigma) [0.5% and 5%] or Na2CO3 [1, 5, 10 50 μg/ml] added to the medium.
2.8 Wounding and preparation of wound tissue
The protocol was approved by the Institutional Animal Care Committee of Padova University. A total of 18 male Wistar rats weighing 250–350 g were subjected to the surgical procedures under halothane anesthesia. The back was shaved and three circular excisional wounds of 1.0 cm diameter were generated that extended beyond the panniculus carnosus (full thickness wounds) using a surgical scalpel. The three wounds on the back of one animal were at least 1.5 cm apart from each other. All wounds were covered with a semi-occlusive polyurethane dressing (Tegadermd, 3 M, St. Paul, MN). Wounds were divided into three groups. One group was treated with PDGF Embedded Microparticles (PEMs) [2 mg microparticles embedded with PDGF 0.01%/ml of inert gel], the other two were treated with vehicle gel only (Inert gel: I) and inert gel plus untreated microparticles (M) [2 mg microparticles/ml of inert gel], respectively. All wounds received twice weekly a dose of 7.0 mg/cm2 of proper gel. At time intervals ranging from 3 days through 21 days after wounding, the rats were killed with an overdose of barbiturates (Nembutal sodium solution; Abbott Laboratories), and wounds were excised with a 2 mm rim of surrounding tissue. Samples were fixed in 10% buffered formalin or immediately frozen in liquid nitrogen for the following analysis. All surgical procedures were performed in an identical fashion by a single surgeon. No prophylactic antibiotic was administered and the animals were fed an unrestricted standard diet.
2.9 Wound photography and analysis
Standardized photography of the wound was performed prior to the initial dressing, following grafting, and daily thereafter. The entire bolster was changed at the time of daily photography. A single lens reflex camera with a macro lens was mounted on a camera stand. The macro lens setting was fixed at a 2:1 reproduction setting, and ISO 100 slide film used. A centimeter ruler was included in each photograph. The photographs were analyzed and an arbitrarily sized designated analysis area (DAA) was selected from a central area of each wound [25]. Using photographic slide scanning and digital planimetric software, the wounds were analyzed as reported by Harries et al. [25]. The areas remaining open within the DAA of each wound were measured. Neoepithelial areas (NEA) were calculated by subtracting daily wound areas from original wound areas. The percentage of neoepithelialization (%NE) was then determined for the DAA by the equation percentage NE = (NEA/DAA) × 100 [25]. This percentage NE was used as the percentage of healed wounds for determining treatment effect. Data from each day were compared among the four groups by one-way analysis of variance and between pairs of groups by Fisher’s least significance difference test with an alpha value of 0.05.
2.10 Histological and morphological analyses
For histological analyses, specimens were fixed in formalin, paraffin-embedded, and stained with haematoxylin and eosin. For immunohistochemical analyses, cryostatic sections (7 μm) were used. These were layered over gelatin-coated glass slides, fixed with absolute acetone for 10 min at room temperature, and cryopreserved at −20°C until use.
Collagen type I (Coll-1), fibroblasts (FU) and endothelial cells (CD 31) were visualized with the acid phosphatase anti-acid phosphatase (APAAP) procedure. Reactions were conducted in humidified chambers at room temperature. Briefly, after saturating non-specific antigen sites with 1:20 rabbit serum in 0.05 m maleate TRIZMA (Sigma) pH 7,6 for 20 min, the first antibody was added to samples (1:800 collagen type I-DAKO). After an incubation of 2 h, samples were rinsed with buffer solution, and the second antibody was added for 30 min (Link Ab-DAKO-, rabbit anti-mouse). After rinsing, the cryostatic sections were incubated for 30 min with 1:50 mouse APAAP Ab-DAKO, rinsed again, and lastly, reacted for 20 min with the Fast Red Substrate (Sigma). Counter staining was performed with haematoxylin (Sigma).
2.11 Semi-quantitative analysis of cells
In order to analyze the cellular response to treatments, masked microscopic examinations were performed on immunostained sections. Cells were identified by: haematoxylin and eosin staining for inflammatory cells; immunohistochemical staining for endothelial cells (positive for CD 31); fibroblasts and type I collagen. Briefly, two investigators analyzed in a masked fashion at least 3 slides for each experiment by light microscopy using 20× as the initial magnification. Each slide contained 3 sections of specimen and 5 fields of 322 μm2 each were analyzed for each tissue section. Experiments were performed at least three times and values were expressed as the mean ± SD.
3 Results
3.1 Particle characterization
In order to obtain micro- and nano-particles with a high specific surface, we performed different experiments investigating the influence of process variables, such as polymer-solvent mixture concentrations and injection flow rates, nozzle diameters and pressures. Table 1 summarizes the operative process conditions used in different experiments; the temperature at which the experiment was performed is reported for completeness.
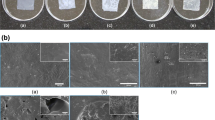
We initially observed that the 250 μm nozzle diameter (tests n. 1 and 2) produced large particles with a size around 100 μm (data not showed). These particles were compact and had a microporosity as reported in the scanning electron micrographs (Fig. 2).

Scanning electron micrographs of experiment 1 (Process conditions: 15 MPa, 0.3% (w/w) HYAFF11®/DMSO solution, 250 μm nozzle diameter): (a) 1,000×, (b) 3,000×
Experiments performed with the 100 μm diameter nozzle (experiment 3) and 0.7% (w/w) HYAFF11®/DMSO resulted in a large amount of stable micro-particle aggregates (data not shown). Experiments 4 and 5, performed with the same nozzle of experiment 3 (100 μm) but with a lower concentration of polymer solution (0.3% and 0.5% instead of 0.7% w/w), produced a fine, dry particulate matter. This powder was collected in large agglomerates that were easily dispersed in water by a sonication treatment of 20 min and observed by SEM. Figure 3 shows a fraction of large particles or stable particle agglomerates with dimensions ranging between 1 μm and 5 μm and a fraction of sub-micrometric particles. Figure 3 also shows that the particulate product obtained under the conditions of experiment 4 was formed by stable agglomeration of nano-particles. This finding was particularly important since micrometric or sub-micrometric particles have a nano-structured porosity and, consequentially, a very high specific surface.

Scanning electron micrographs of experiment 4 (Process conditions: 15 MPa, 0.5% w/w HYAFF11®/DMSO solution, 100 μm nozzle diameter): (a) 12,000×; (b) 21,020×
In order to further reduce particle dimension, we investigated the effect of decreasing the nozzle diameter to 50 μm and increasing the pressure of the process (experiments 6 and 7 in Table 1). With this small diameter (50 μm), nozzle occlusion problems during solution injection led to poor productivity and reproducibility. Conversely, higher pressure (200 bar; experiment 7) resulted in micro-particles with the same morphological structure as those produced in experiments 4 and 5 (data not shown).
Particle size distribution was studied by light scattering, and the results are summarized in Table 2. The operative conditions of experiments 1, 2 and 6 were not considered suitable for particle production.
An example of typical particle size distribution analyzed by light scattering is reported in Fig. 4. A typical bimodal distribution around 300–400 and 900 nm was obtained for all experiments reported in Table 2, which also reports the weight-based fraction of nano-particles belonging to the large (900 nm) and small (300–400 nm) size range.

Size distribution of nanoparticles of HYAFF11® obtained by GAS process (experiment 7, Process conditions: 20 MPa, 0.3% (w/w) HYAFF11®/DMSO solution, 100 μm nozzle diameter). The insert shows a canning electron micrograph of small size particles produced in experiment 7 (51,200×)
As reported in Table 2, the particles obtained with the 100 μm nozzle and with various polymer concentrations (experiments 3, 4 and 5) resulted in no change in particle size. However, under these operative conditions, a small change in HYAFF11®/DMSO concentration resulted in a dramatic change in the percentage of small and large particle fractions. For instance, experiment 4 (0.5% instead 0.7% or 0.3% w/w HYAFF11®/DMSO concentration) yielded only 17% particles of 900 nm size, whereas experiments 3 and 5 yielded approximately 100% large size particles.
The optimal experimental conditions in terms of morphology, particle dimension and fraction of small particles were those used in experiment 7. A higher operative pressure (200 bar) led to reproducible production of small particles with a typical size of 270 ± 78 nm. The weight-based fraction of large particles with a size of 900 ± 135 nm was only 25%. The light scattering analysis recorded for experiment 7 showed that 90–98% of particles had a diameter smaller than 1,000 nm. The nano-particles produced under the operative conditions of experiment 7 had a nano-structured porosity and high specific surface.
3.2 Absorption process
Because water can act as a plasticizing agent on the polymeric matrix, thereby enhancing the solute absorption from aqueous solution, the water content within the 50 μm thick polymeric film was evaluated by DSC analysis and found to be 8.5% (w/w). Samples dried in a vacuum at room temperature for 15 h had a 5.5% (w/w) water content. This amount was reduced to 3.7% (w/w) with further sample treatment in an oven at 378 K for 15 h. These results demonstrated the ability of the polymeric matrix to permanently absorb high amounts of water, which acted as a plasticizing agent that enhanced absorption of bioactive molecules.
In order to evaluate the time-scale of the absorption phenomena, dye diffusion within 50 μm thick polymeric films was studied. Figure 5 shows the images of a cross section of the HYAFF11® films impregnated with 10 μg/ml of sodium fluorescein for different incubation times: 5 min, 60 min, 24 h and 72 h. After 1 and 5 min, the surface was impregnated up to 25.4 ± 0.9 μm; after 10 to 30 min the coloured layer became thicker and after 60 min the dye started to be visible in the centre of the polymeric film. After 24 h, the color distribution was almost homogeneous (less signal was observed in the centre of films), whereas a uniform dye concentration was observed after 72 h.

Microphotographs (10× magnification) of 100 μm thick HYAFF11® films impregnated with 10 mg/l sodium fluorescein by an aqueous-solution contact method: (a) after 5 min, (b) after 60 min, (c) after 24 h and (d) after 72 h
An estimation of the diffusion coefficient (D dye) was obtained by fitting the penetration depth (z) of the dye estimated from the fluorescence images of the polymeric film cross section reported in Fig. 6. These experimental data were correlated using the following equation [26], which describes the time evolution of diffusing molecules within a polymeric matrix as a function of the axial coordinate, z:
where \( c_{\text{dye}} \) is the molar dye concentration, \( c_{\text{dye,0}} \) at baseline and z = 0, the depth (z) corresponds to the penetration length (distance from film surface to the depth where the sodium fluorescein started to be detected). This dye diffusion coefficient, D dye, was calculated to be 1.2 × 10−9 ± 0.2 × 10−9 cm2/s.

Depth of sodium fluorescein penetration into HYAFF11® films as a function of contact time. The concentration of sodium fluorescein solution used was 10 μg/ml. The line was obtained by the fitting of data using Eq. 1
3.3 Growth factor release
In vitro release profiles give important information on the efficiency of the delivery system for the controlled release of drugs. An “in vitro” drug release study is indeed a prerequisite to obtaining correct predictions in order to design and test the “in vivo” activity of controlled drug delivery forms.
In the present study, particles are suspended in a small volume of receiving medium (2 ml) in order to reproduce topical administration of HYAFF11® microspheres (Fig. 7). The amount of drug released (expressed as ng of growth factors/mg microspheres) is plotted versus time. Data represent the mean of six independent experiments. Although native HA is able to dissolve rapidly in water, benzyl esters of hyaluronan show different behaviour possibly because of the different nature of the polymer. As reported in literature [27, 28] it is well known that in vitro growth factor release from HYAFF11® is not observable at early (namely within 15 days) stages in physiological (pH 7) conditions. Since they are composed of hyaluronan benzyl esters, HYAFF11 scaffolds,form a gellified network from which drug release can be controlled for long periods of time in culture medium [29].

Microphotographs (a) 5×, (b) 20× magnification) demonstrating the TGF-β immunohistochemical reaction on impregnated polymer particles
As reported in Fig. 8, the presence of hyaluronidase in the medium did not induce polymer degradation. These observations confirm that the carboxylic groups in the beta-glucoronic acid unit are the activation centre of this enzyme and that total blockage of these groups can restrict the cleavage of beta (1–>4) glycoside bonds by this enzyme [30–32]. Only in the presence of an alkaline environment (obtained by the addition of 5% Na2CO3) is a rapid degradation of HYAFF11 microspheres observable.

In vitro release of TGF-β (a) and PDGF (b) from impregnated microparticles as a function of time for different Hyaluronase concentrations
Analyzing the release profiles, one can observe that the progressive increase of Na2CO3 increased the release rate of the PDGF and TGF β (Fig. 9). In vitro PDGF and TGF β release profiles from impregnated micro and nano-particles are reported in Fig. 9 for different sodium carbonate concentrations. The maximum value of PDGF and TGF β released corresponded to the amount of growth factor absorbed in the micro-particles and was estimated to be 0.9 ng/mg.

In vitro release of TGF-β (a) and PDGF (b) from impregnated microparticles as a function of time for different Na2CO3 concentrations. The lines are only a visual guide
In order to analyze the release mechanisms and their dependence on Na2CO3, the experimental release profiles were fitted with the following equation developed for drug release from erodable polymeric particles with spherical shapes [33]:
where M t /M ∞ represents the drug fraction released at the time t; M t and M ∞ represent the cumulative absolute amount of drug released at time t (min) after the addition of Na2CO3 and at infinite time, respectively. K is a constant that depends on the surface erosion rate constant (k eros), the initial concentration of growth factor in the matrix (C 0) and the radius of the microspheres (R) at t = 0 as follows [31]:
In Fig. 10a, fair model correlations of experimental release data for PDGF/TGF β are reported; the same correlations were obtained for (data not shown). Polymer erosion was due to the action of Na2CO3. For this reason, the K values were plotted against the concentration of Na2CO3 and a linear correlation was observed (Fig. 10b). The values obtained for a Na2CO3 concentration of 50 μg/l were not considered because in this case a fast dissolution (less than 7 h) of growth factors was obtained and no experimental data were collected during this time period.

Experimental and mathematical comparisons: (a) experimental and mathematical modelling of in vitro cumulative TGF-β fractions released as a function of time for different Na2CO3 concentrations. The lines corresponded to data fitting by Eq. 2. (b) Linear correlation of K as a function of Na2CO3 concentration for both TGF-β and PDGF release
These results indicated that the growth factor release was mainly driven by the polymeric matrix erosion process. This is of fundamental importance in designing a proper release rate in vivo. In conclusion, the rate of polymer erosion and thus the growth factor release increased linearly with increasing Na2CO3 concentrations, thus it was possible to control the rate of release by changing the Na2CO3 concentration or the pH of the solution.
3.4 In vivo treatment
PDGF embedded microparticle treatment shows a trend in wound healing stronger than the vehicle control (inert gel: I) or microparticle (M) without growth factors at 7 days (Fig. 11). Epithelialization kinetics showing superiority of PDGF embedded microparticle at closing circular wounds. Percent healed wounds directly reflected the percentage of neoepithelialization of the wounds. Although breaking strength in all wounds increased over the entire 12-day period, PDGF embedded microparticle treatment still resulted in significantly stronger wounds on day 5 compared to the vehicle.

Epithelialization kinetics of vehicle control (inert gel: I) or Microparticles (M) with out growth factors treated wounds PEM (PDGF Embedded Microparticles) at closing interstices of rat split-thickness skin grafts. The percentage healed reflects the percentage neoepithelialization of the interstitial spaces. Bars represent mean ± SD
3.5 Cellular response to scaffolds
Cellular events involved in wound healing are summarised in Table 3.
At day 3, “I” treatment showed slow infiltration of granulocytes and macrophages whereas M and PEM showed a moderate amount of macrophages, fibroblasts and some granulocytes. No collagen fibers were observable in any wounds; scarce endothelial cells were present only in PEM-treated tissue.
At day 5 and 7, “I” treatment showed a moderate infiltration of granulocytes and macrophages whereas M and PEM showed larger amounts of macrophages, some granulocytes and few giant cells recruited to digest the microparticle polymer. The cellular response to M and PEM treatment at these time points, included a significative amount of endothelial cells and some fibroblasts. Collagen fibers were present overall in PDGF-treated tissue.
At day 14, M and PEM treated tissues showed a moderate amount of macrophages and fibroblasts throughout the scaffold and some non-phagocytic cells were present. A larger amount of macrophages and fibroblasts accompanied by some non-phagocytic cells were found throughout the wounds treated with M and PEM. At day 21, all the wounds were closed.
4 Discussion
This study aimed to produce HYAFF11® micro and nano-particles by a GAS technique. Appropriate experimental conditions resulted in the production of HYAFF11 based microspheres characterized by spherical shape, absence of aggregates and an almost perfect quantitative recovery.
Several authors obtained HYAFF11® microparticles for drug delivery systems by using solvent extraction methods and organic solvents [17]. These techniques are not flexible and the final products are often characterised by a high residual solvent content, low drug loading, drug degradation or denaturation, ineffective drug release and unsuitable physical and morphological properties. Supercritical or compressed fluid based techniques were used for the preparation of micro- and nanoparticulate products with pharmaceutical requisites (solvent free, suitable technological and biopharmaceutical properties, high quality). In the GAS precipitation process, the organic solution of the substances, which must be micronized, was sprayed into a vessel filled with dense CO2 at a suitable temperature. The use of CO2 diffusion within the small droplets of polymeric organic solution led to a reduction of solvent concentration. The CO2 acts as antisolvent, and its efficiency depends on the pressure inside the precipitation chamber. The minimum value of CO2 pressure at which there was precipitation of HYAFF11® was 100 bar at 313 K [11]. For this reason, all the experiments were carried out at a pressure equal to or higher than 160 bar.
When using high polymer concentrations (1% w/w), a large nozzle diameter (250 μm) and/or a low flow rate, the solution jet did not break into small droplets and sponges or other products with a smooth surface were obtained. The spray regime and jet break-up was favoured by a low solution viscosity, small nozzle diameter, high injection flow rate, and high density of the environment in which the solution was atomized [19]. This latter aspect was particularly relevant and was related to the CO2 density within the precipitation chamber.
Very small particles were obtained when a polymeric solution of 0.3% (w/w), a nozzle of 100 μm, a pressure of 200 bar and a flow rate of 5.6 ml/min were used. The distribution of the particle size was bimodal.
One experimental strategy used to obtain growth factor loaded micro-particles was the co-precipitation of growth factor and HYAFF11® by the semi-continuous GAS technique starting from a homogenous solution of protein and polymer. This technique has been previously used for insulin loading [19]. We found that the particles produced using this strategy possessed the correct morphology but the growth factor was poorly loaded and was not active (data not shown). This inactivation was probably due to growth factor denaturation resulting from high shear stress during the atomization process and/or the effect of the solvent: DMSO is able to denature protein molecules.
For this reason, we developed an alternative procedure to load bioactive growth factors within the polymeric nano-particles. The growth factors were absorbed into the microparticles only after their production, exploiting the qualities of water as a vehicle of transport for drugs inside the polymeric matrix. The high affinity between HYAFF11® and water was ascertained by DSC analysis. This fact was further demonstrated by the ease of the impregnation process with different dyes. HYAFF11® film and microparticles showed homogeneous absorption by simple contact with the impregnating substance at room temperature. This absorption process did not expose the growth factor to any denaturating agents and the procedure can be performed in completely sterile conditions.
In vitro release profiles gave important information on the efficiency of the delivery system for the controlled release of drugs. An “in vitro” drug release study is indeed a prerequisite to obtaining correct predictions in order to design and test the “in vivo” activity of controlled drug delivery forms [32].
The low release analyzed under physiological conditions was probably due to the high affinity of the growth factor for the polymer, thus it was necessary to use promoters to degrade the polymeric matrix in order to obtain complete release of growth factors.
The use of enzyme did not lead to release due to the structure of the HYAFF11®, so Na2CO3 was used as suggested in the literature [31, 32]. The salt was able to break the polymeric structure [32] and thus growth factor release depended on the degradation rate of the polymer. These observations confirm that the carboxylic groups in the beta-glucoronic acid unit are the activation centre of this enzyme and the total blockage of these groups restricts the cleavage of beta-glycoside bonds by this enzyme.
Dependence on degradation was demonstrated by the accurate fitting of K values obtained by the mathematical model of release. The coefficient correlated linearly with the salts concentration.
These results demonstrate that growth factor release from the HYAFF11® nano-particles produced by the GAS precipitation techniques was mainly driven by erosion phenomena. Other transport phenomena such as the diffusion process within the polymeric matrix and in the aqueous solution did not affect the release rate.
Bioactivity experiments designed to investigate in vivo the performances of growth factors embedded in HYAFF11 microspheres were performed. Because PDGF is known to promote reepithelialization, stimulate granulation tissue formation, and stimulate collagen deposition, these models were chosen to help identify its effect on those processes [19, 20]. The closure of circular wound defects requires both proliferation (mitosis) and migration of keratinocytes. Wounds showed an increased reepithelialization when treated with microparticles embedded with growth factors. The increased epithelialization kinetics shown by this rat wound model suggests that PDGF embedded microparticles might be useful in accelerating healing in wounds by facilitating the process of epithelialization for closure [34]. The fact that PDGF increased the breaking strength of wounds compared to both vehicle controls and microparticles at day six suggests that PDGF stimulates extracellular matrix and collagen deposition. These data are in agreement with Werner and Grose [35], who showed an increase in breaking strength and an increase in collagen content in PDGF treated incisions [35].
In conclusion, using GAS techniques, HYAFF11 porous nano-particles suitable for in vivo growth factor delivery were successfully developed.
References
S.P. Baldwin, W.M. Saltzman, Materials for protein delivery in tissue engineering. Adv. Drug. Deliv. Rev. 33, 71–86 (1998). doi:10.1016/S0169-409X(98)00021-0
R.C. Thomson, A.K. Shung, M.J. Yaszemski, A.G. Mikos, Polymer Scaffold Processing, in Principle of Tissue Engineering, ed. by R.P. Lanza, R. Langer, J. Vacanti (Elsevier, Amsterdam, 2000), pp. 251–262
J. Sohier, R.E. Haan, K. de Groot, J.M. Bezemer, A novel method to obtain protein release from porous polymer scaffolds: emulsion coating. J. Control Release 87, 57–68 (2003). doi:10.1016/S0168-3659(02)00350-4
D.W. Hutmacher, Scaffolds in tissue engineering bone and cartilage. Biomaterials 21, 2529–2543 (2000). doi:10.1016/S0142-9612(00)00121-6
K. Dash Alekha, C. Greggrey, I.I. Cudworth, Therapeutic applications of implantable drug delivery systems. J. Pharmacol. Toxicol. Methods 40, 1–12 (1998). doi:10.1016/S1056-8719(98)00027-6
E. Fattal, A.L. Gomes dos Santos, A. Bochot, A. Doyle, N. Tsapis, J. Siepmann et al., Sustained release of nanosized complexes of polyethylenimine and anti-TGF-β2 oligonucleotide improves the outcome of glaucoma surgery. J. Control Release 112, 369–381 (2006). doi:10.1016/j.jconrel.2006.02.010
D. Kaplan, V. Karageorgiou, Porosity of 3D biomaterial scaffolds and osteogenesis. Biomaterials 26, 5474–5491 (2005). doi:10.1016/j.biomaterials.2005.02.002
D.A. Edwards, J. Wang, A. Ben-Jebria, Inhalation of estradiol for sustained systemic delivery. J. Aerosol. Med. 121, 27–36 (1999)
H. Bernstein, J.A. Straub, D.E. Chickering, J.C. Lovely, H. Zhang, B. Shah et al., Intravenous hydrophobic drug delivery: a porous particle formulation of paclitaxel (AI-850). Pharm. Res. 22, 347–355 (2005). doi:10.1007/s11095-004-1871-1
F. Ungaro, G. De Rosa, A. Miro, F. Quaglia, M.I. La Rotonda, Cyclodextrins in the production of large porous particles: development of dry powders for the sustained release of insulin to the lungs. Eur. J. Pharm. Sci. 28, 423–432 (2006). doi:10.1016/j.ejps.2006.05.005
A. Bertucco, P. Pallado, Micronization of Polysaccharide by a Supercritical Anti-solvent Techniques, in Methods in Biotechnology, vol. 13: Supercritical Fluid Methods and Protocols, ed. by J.R. Williams, A.A. Clifford (Humana press Inc, Totowa, NJ, 2000), pp. 193–200
P. Brun, G. Abatangelo, M. Radice, V. Zacchi, D. Guidolin, D.D. Gordini et al., Chondrocyte aggregation and reorganization into three-dimensional scaffolds. J. Biomed. Mater. Res. 46, 337–346 (1999). doi:10.1002/(SICI)1097-4636(19990905)46:3<337::AID-JBM5>3.0.CO;2-Q
J. Aigner, J. Tegeler, P. Hutzler, D. Campoccia, A. Pavesio, C. Hammer et al., Cartilage tissue engineering with novel nonwoven structured biomaterial based on hyaluronic acid benzyl ester. J. Biomed. Mater. Res. 42, 172–181 (1998). doi:10.1002/(SICI)1097-4636(199811)42:2<172::AID-JBM2>3.0.CO;2-M
C. Tonello, B. Zavan, R. Cortivo, P. Brun, S. Panfilo, G. Abatangelo, In vitro reconstruction of human dermal equivalent enriched with endothelial cells. Biomaterials 24, 1205–1211 (2003). doi:10.1016/S0142-9612(02)00450-7
S. Lepidi, G. Abatangelo, V. Vindigni, G.P. Deriu, B. Zavan, C. Tonello et al., In vivo regeneration of small-diameter (2 mm) arteries using a polymer scaffold. FASEB J. 20, 103–105 (2006)
L. Benedetti, R. Cortivo, T. Berti, A. Berti, F. Pea, M. Mazzo et al., Biocompatibility and biodegradation of different hyaluronan derivatives (HYAFF) implanted in rats. Biomaterials 15, 1154–1160 (1993). doi:10.1016/0142-9612(93)90160-4
E. Esposito, E. Menegatti, R. Cortesi, Hyaluronan-based microspheres as tools for drug delivery: a comparative study. Int. J. Pharm. 288, 35–49 (2005). doi:10.1016/j.ijpharm.2004.09.001
M. Singh, M. Briones, D.T. O’Hagan, A novel bioadhesive intranasal delivery system for inactivated influenza vaccines. J. Control Release 70, 267–276 (2001). doi:10.1016/S0168-3659(00)00330-8
G.F. Pierce, J.E. Tarpley, D. Yanagihara, T.A. Mustoe, G.M. Fox, A. Thomason, Platelet-derived growth factor (BB homodimer), transforming growth factor-beta 1, and basic fibroblast growth factor in dermal wound healing. Neovessel and matrix formation and cessation of repair. Am. J. Pathol. 140(6), 1375–1388 (1992)
D. Shure, R.M. Senior, G.L. Griffin, T.F. Deuel, PDGF AA homodimers are potent chemoattractants for fibroblasts and neutrophils, and for monocytes activated by lymphocytes or cytokines. Biochem. Biophys. Res. Commun. 186(3), 1510–1514 (1992). doi:10.1016/S0006-291X(05)81577-3
M. Ikeda, M. Kohno, T. Horio, K. Yasunari, K. Yokokawa, H. Kano et al., Effect of thrombin and PDGF on endothelin production in cultured mesangial cells derived from spontaneously hypertensive rats. Clin. Exp. Pharmacol. Physiol. Suppl. 22(1), S197–S198 (1995). doi:10.1111/j.1440-1681.1995.tb02879.x
D.R. Knighton, K.F. Ciresi, V.D. Fiegel, L.L. Austin, E.L. Butler, Classification and treatment of chronic nonhealing wounds. Successful treatment with autologous platelet-derived wound healing factors (PDWHF). Ann. Surg. 204(3), 322–330 (1986). doi:10.1097/00000658-198609000-00011
L. Benedetti, R. Cortivo, T. Berti, A. Berti, F. Pea, M. Mazzo et al., Biocompatibility and biodegradation of different hyaluronan derivatives (HYAFF) implanted in rats. Biomaterials 14(15), 1154–1160 (1993). doi:10.1016/0142-9612(93)90160-4
N. Elvassore, A. Bertucco, P. Caliceti, Production of insulin-loaded poly(ethylene glicol)/Poly(l-lactide) (PEG/PLA) nanoparticles by gas antisolvent techniques. J. Pharm. Sci. 90, 1628–1636 (2001). doi:10.1002/jps.1113
R.H. Harries, B.G. Rogers, I.O. Leitch, M.C. Robson, An in vivo model for epithelialization kinetics in human skin. Aust. N. Z. J. Surg. 65(8), 600–603 (1995). doi:10.1111/j.1445-2197.1995.tb01705.x
J. Crank J (ed.), The Mathematics of Diffusion (Clarendon Press, Oxford, 1956)
J.B. Leach, C.E. Schmidt, Characterization of protein release from photocrosslinkable hyaluronic acid-polyethylene glycol hydrogel tissue engineering scaffolds. Biomaterials 26(2), 125–135 (2005). doi:10.1016/j.biomaterials.2004.02.018
T. Avitabile, F. Marano, F. Castiglione, C. Bucolo, M. Cro, L. Ambrosio, Biocompatibility and biodegradation of intravitreal hyaluronan implants in rabbits. Biomaterials 22(3), 195–200 (2001). doi:10.1016/S0142-9612(00)00169-1
E. Esposito, E. Menegatti, R. Cortesi, Hyaluronan-based microspheres as tools for drug delivery: a comparative study. Int. J. Pharm. 288(1), 35–49 (2005). doi:10.1016/j.ijpharm.2004.09.001
D. Campoccia, J.A. Hunt, P.J. Doherty, S.P. Zhong, M. O’Regan, L. Benedetti et al., Quantitative assessment of the tissue response to films of hyaluronan derivatives. Biomaterials 17(10), 963–975 (1996). doi:10.1016/0142-9612(96)84670-9
E. Milella, E. Brescia, C. Massaro, P.A. Ramires, M.R. Miglietta, V. Fiori et al., Physico-chemical properties and degradability of non-woven hyaluronan benzylic esters as tissue engineering scaffolds. Biomaterials 23, 1053–1063 (2002). doi:10.1016/S0142-9612(01)00217-4
C. Nastruzzi, E. Esposito, R. Cortesi, R. Gambari, E. Menegatti, Kinetics of bromocriptine release from microspheres: comparative analysis between different in vitro models. J. Microencapsul. 11, 565–574 (1993). doi:10.3109/02652049409034995
D.Y. Arifin, L.Y. Lee, C.H. Wang, Mathematical modelling and simulation of drug release from microspheres: implications to drug delivery systems. Adv. Drug Deliv. Rev. 58, 1274–1325 (2006). doi:10.1016/j.addr.2006.09.007
M.C. Zweers, J.M. Davidson, A. Pozzi, R. Hallinger, K. Janz, F. Quondamatteo et al., Integrin alpha2beta1 is required for regulation of murine wound angiogenesis but is dispensable for reepithelialization. J. Invest. Dermatol. 127(2), 467–478 (2007). doi:10.1038/sj.jid.5700546
S. Werner, R. Grose, Regulation of wound healing by growth factors and cytokines. Physiol. Rev. 83(3), 835–870 (2003). Review
Acknowledgements
We gratefully acknowledge the FIRB (Fondo per gli investimenti per la ricerca di base—MIUR) for financial support.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Zavan, B., Vindigni, V., Vezzù, K. et al. Hyaluronan based porous nano-particles enriched with growth factors for the treatment of ulcers: a placebo-controlled study. J Mater Sci: Mater Med 20, 235–247 (2009). https://doi.org/10.1007/s10856-008-3566-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10856-008-3566-3