
Abstract
The use of zinc oxide (ZnO) and titanium oxide (TiO2) nanoparticles in ultraviolet (UV) filters can cause serious health issues due to their genotoxicity and cytotoxicity effect. Recent research on cerium oxide (CeO2) nanoparticles has demonstrated their biocompatible nature, excellent antioxidant and ultraviolet–visible (UV–Vis) absorption properties. CeO2-based nanocomposite materials or nano-architectures can offer uniform absorptive properties, therapeutic skin effect and easy skin application. In this study, cerium oxide-polyethylene glycol (CeO2-PEG) nano-architecture was prepared by hydrothermal processing with the aim to improve stability, antioxidant properties, and enhance UV absorption for UV filtering applications. The morphology study of the developed nanocomposite revealed the formation of polymer bands with well-dispersed CeO2 nanoparticles. The nanocomposite structure has exhibited excellent UV absorption in the UV range 200–400 nm due to decrease in optical band gap in comparison to the naked CeO2 NPs. The evaluation of the photocatalytic activity (PCA) of the nanocomposite structure against P25 photocatalyst and ZnO nanoparticles showed no dye (crystal violet) degradation on UV irradiation. Hence, PCA assay verified the absence of ROS generation and increased stability of the nanostructure under UV irradiation. The investigation of antioxidant properties of the Ce-PEG nanocomposite by a dichlorofluorescein (DCF)-based assay revealed sufficient intracellular ROS scavenging in comparison to N-acetylcysteine (NAC) and H2O2 only assays. In vitro toxicity evaluation of Ce-PEG nanocomposite towards non-malignant human keratinocyte cell line (HaCaT) revealed a non-significant cell mortality ~ 10% of control at a dosage of 50 mg L−1. The nanocomposite structure also exhibited excellent UV protection for HaCaT cells under UV irradiation conditions compared to P25. Our findings suggest that the developed nanocomposite combined the antioxidant and UV absorption properties of CeO2 and skin emollient role of (PEG), and hence offered a novel biocompatible and multifunctional structure for sunscreens and therapeutic skin product.
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Extensive exposure to UV light can cause detrimental effects such as erythema (sunburn), premature skin ageing, skin wrinkles, cornea, actinic keratosis, solar elastosis, and liver spots [1, 2]. UV radiation is a part of electromagnetic spectrum, and divided into UVA (400–315 nm), UVB ((315–280) and UVC (280–100 nm) categories [2, 3]. UVC do not reach the earth surface as it is blocked by ozone layer. UVA and UVB are most prevalent radiations on earth surface that interact with biological matter. Exposure to UVB radiation cause long-term skin damage such as wrinkles and ageing. They also cause indirect damage to DNA. UVA radiation have deep skin penetration and directly cause DNA damage, genetic mutation and hence cancer [4]. UV rays generate ROS through activation of intracellular chromophores and photosensitization [5,6,7]. Overproduction of ROS cause oxidative stress that leads to many pathological conditions. Apart from direct/indirect DNA damage, UV radiation also cause other changes in the body such as imbalance in inflammatory response. To protect against the damaging effect of UV radiation, sunscreens/UV filters containing active inorganic and organic ingredients are used that provide protection through scattering, absorption and reflection of UV rays. Sunscreen is very effective to protect against UV-related pathologies, e.g. sunburn, actinic keratosis, carcinomas and melanomas [8]. The sunscreen formulation contains nanosize particles of inorganic oxide (typically TiO2 and ZnO) to impart higher UV absorption and transparent look after application. However, there are concerns in the use of TiO2 and ZnO in sunscreens due to their higher photocatalytic activity when exposed to UV radiation. Reactive oxygen species are produced due to electron/hole (e−/h+) generation upon UV irradiation [1, 9, 10]. Oxide nanoparticles (NPs) with reactive surfaces also react with polymeric ingredients to generate ROS. These ROS degrade the organic ingredients of a sunscreen and reduce the UV protection duration and efficiency [1, 11, 12]. Ginzburg et al. reported recently that nanosize ZnO degraded the sunscreen mixture after exposure to UV radiation and exhibited higher toxicity in embryonic zebrafish assay [11].
The production of ROS can cause oxidative stress in cells that leads to cellular organelle damage and premature cell death. Nanoparticles penetration into skin can cause ROS generation and cytotoxic effect. Studies showed a minimal epidermal penetration in healthy skin [13, 14]. However, transdermal penetration can increase in case of wound, sunburn and ape-stripped skin [13]. The cytotoxic effects in human body usually appear due to long or continuous exposure to NPs.
Various in vitro and in vivo studies on cytotoxic behaviour of ZnO and TiO2 NPs have been performed. Cytotoxicity study of ZnO NPs on different cell line such as human neuroblastoma (SH-SY5Y), human brain tumour (U87) and rat retinal ganglion cells (RCG-5) indicated an increase in ROS production, decreased cell viability, and mitochondrion and cellular membrane damage [15,16,17,18]. In animal study models, ZnO NPs-induced hippocampal pathological and neurobehavioural changes induced cognitive impairment and depressive behaviour [19, 20]. In vitro cytotoxicity study of TiO2 NPs on various cells lines such as human glial cells (U373), PC12 cell line and human neuroblastoma (SH-SY5Y) reported ROS augmentation, lipid peroxidation, cell membrane damage and inhibition of cell proliferation [21,22,23,24]. In vivo studies demonstrated a high burden on liver, spleen and lungs for one year after intravenous administration [25]. Similarly, intranasal administration for 90 consecutive days demonstrated high oxidative stress level, DNA peroxidation, high level of proteins, overproliferation of glial cells, tissue necrosis and alteration of 249 gene expression [26].
The chemical/organic sunscreen can easily penetrate through skin and cause more damage than physical sunscreens. There are several investigation reports on presence of chemical sunscreens in human body, detected through urine analysis. Philippat et al. reported a high concentration (average 200 ng mL−1, and up to 13,000 ng mL−1) of benzophenone-3 in the urine of Californian females [27]. Other studies by Calafat et al. and Bae et al. reported the presence of benzophenone-3 in the human urine samples with mean concentrations of 22.9 ng mL−1 and 0.05–8.65 ng mL−1, respectively [28, 29]. The presence of chemical sunscreens such as benzophenone-3, 4-methylbenzyli-dine, octyl methoxycinnamate, octocrylene and 3-benzylidene camphor decrease the cell viability and caused neurotoxicity [30,31,32,33,34]. They also showed toxicity in different body tissues and cellular organelles [35].
To minimize sunscreen degradation and ROS generation, the surface of the NPs must not be reactive towards the organic ingredients of a sunscreen and should be stable. There is more probability of aggregation in a sunscreen with physically dispersed NPs. The binding of NPs with polymer chains in the matrix can minimize the agglomeration. The stabilized nanocomposite with well-dispersed oxide NPs can reduce the ROS generation and exhibit excellent and uniform UV protection. Antioxidants or ROS scavenger materials can also be incorporated in the sunscreen formulation to supress the formation of ROS. Similarly, commercial chemical ingredients of a sunscreen must have reduced reactivity and stabilized surface to minimize skin penetration and interaction with the cells. The stabilized/unreactive surfaces of the NPs and the organic components can be achieved in a composite material synthesized under controlled processing parameters and selective organic and inorganic reagents.
Inorganic antioxidants such as CeO2 offer great advantage as an active ingredient in the sunscreen formulation because of requisite biocompatibility, and excellent antioxidant and UV absorptive properties [1, 36]. It has been reported in various published studies that CeO2 NPs exhibit enzyme mimicking activity like superoxide dismutase, and peroxidase due to its reversible switching between Ce+3 and Ce+4 oxidation states, and can effectively scavenge ROS and nitric oxide radicals [37,38,39]. Recently, it has been reported that CeO2 NPs can protect neurocytes and myocardial cells against damaging effect of ROS by scavenging them [40,41,42]. Li et al. have found that CeO2 NPs caused no apparent toxicity towards human skin fibroblast cells, and reduced the intracellular ROS level. Further, it inhibited the senescence-associated β-galactosidase condition, associated oxidative stress and DNA damage [43]. Morlando et al. reported the synthesis of CeO2 nanodots encrusted TiO2 nanocomposite, which showed lower phototoxic effect towards HaCaT cells in comparison with commercial TiO2. It also exhibited improved biocompatibility, increased UV protection and minimal formulation degradation [43].
Polyethylene glycol (PEG) has been used in the processing of the various composite materials because of its biocompatible properties and application in cosmetics. It has been widely employed as plasticiser, ointment, emollient and laxative agent in pharmaceuticals [30, 31].
In this research article, a novel nanocomposite structure of CeO2 NPs dispersed in polymer matrix (PEG) was synthesized by hydrothermal processing. The structural, morphological, UV absorption and biocompatibility of the nanocomposite structure were studied to evaluate its applications in sunscreens. The nanocomposite (Ce-PEG) exhibited improved biocompatibility, increased UV protection and ROS scavenging (Fig. 1). As per knowledge of the authors, the synthesized material has not been reported elsewhere.

CeO2 nanoparticles stabilized polyethylene glycol (PEG) nano-architecture with excellent UV absorption, ROS scavenging (extracellular and intercellular ROS) and emollient properties for application in sunscreens
Experimental
Reagents
Cerium nitrate hexahydrate (Ce(NO3)3·6H2O, ≥ 99.9%, Sigma-Aldrich), ethylenediaminetetraacetic acid (EDTA, C10H16N2O8, ≥ 99.1%, Sigma-Aldrich), polyethylene glycol 6000(C2nH4n+2On+1, ≥ 99%, Sigma-Aldrich), ammonium hydroxide (NH4OH, 28–30%, Sigma-Aldrich, triarylmethane dye, crystal violet (CV, C25H30ClN3, ≥ 90%, Sigma-Aldrich) and ethyl alcohol (CH3CH2OH, ≥ 99.5%, Chem-Supply, Australia) were purchased and used without any further treatment.
Methods
Synthesis of Ce-PEG nanocomposite was carried out by hydrothermal processing. For synthesis, solution A was prepared by dissolving 1 g of Ce(NO3)3·6H2O in 10 ml of DI water by magnetic stirring for 30 min. Solution B was prepared separately by dissolving 2 mg of EDTA in 10 ml of DI water. After complete dissolution of EDTA, 15 ml of PEG (15% w/v solution) and 2 ml of NH4OH solutions were added into the beaker and sonicated for 10 min. After formation of clear solution, the two solutions were mixed and magnetically stirred at 60 °C for 20 min. The resultant solution was then sealed into a Teflon-lined autoclave (50 ml) and maintained at 180 °C for 24 h inside an oven. After processing, the autoclave was cooled down to room temperature and precipitated particles (precursor) were collected by centrifuging at 6420×g for 10 min. The obtained precipitate was dried at 70 °C for 12 h inside an oven. After drying, the product was stored inside a cleaned glass vial. The obtained sample was also calcined under inert atmosphere (Ar gas) at 500 °C in a tube furnace to verify the oxidation state of cerium.
The process of hydrolysis and amination resulted in the formation of PEG bis(amine), which linked the cerium NPs (CeO2 and Ce2O3) to form Ce-PEG bis amine (abbreviated as Ce-PEG nanocomposite) composite, Figure S1. Schematic of the nanocomposite preparation is shown in Fig. 2.

Schematic for the preparation of Ce-PEG composite using hydrothermal processing
Characterization
X-ray diffraction (XRD) patterns of the samples were obtained using an Empyrean diffractometer, Malvern Panalytical with selected scanning angle 2θ = 10–80 degree (°) at 40 kV and 40 mA, scan rate 1.5 ° min−1, and step size 0.02° using a monochromatic CuKα radiation (λ = 1.5405 Ȧ) as emission source. The morphology of the Ce-PEG nanocomposite structure was observed by field emission scanning electron microscopy (FE-SEM, JSM 7500F, JEOL Ltd). Platinum (Pt) coating was applied to the particles on carbon tape attached to an appropriate size aluminium (Al) base.
The crystal morphology and the lattice fringes of the samples were observed and analysed using a high-resolution transmission electron microscopy (TEM, ARM200F, JEOL Ltd). For TEM specimens preparation, first, NPs suspensions were prepared by sonication, and then, 2–3 drops of suspension were poured on to carbon tape, and allowing it to dry.
X-ray photoelectron spectroscopy (XPS) was employed to determine the oxidation sate of Ce and other species on the surface of the samples. The spectra were recorded using a NEXA XPS (Thermo scientific™) with Al Kα radiations (hυ = 1.487 keV) as X-ray excitation source under high vacuum < 10–8 mbar. Avantage data analysis (Thermo scientific™) software package was used to analyse the XPS data.
UV–Vis absorption spectra were collected using a UV-1800 series spectrophotometer (Shimadzu) in the 200–800 nm wavelength range. Samples were suspended in CH3CH2OH by sonication (Branson 3800 brand, Ultrasonic Corp.) before UV absorption measurement. The optical band gap (Eg) of the samples was evaluated from the UV measurements using the Tauc relation [1, 44]:
where α is absorption coefficient, h the Planck’s constant, ν the photon’s frequency, A the proportionality constant and Eg the optical band gap. The value of the exponent (n) can vary between 0.5 and 3, depending upon the transition from the conduction to valence band in a semiconductor material.
Photocatalytic activity
The photocatalytic activity of the synthesized nanostructures was determined from the degradation of the crystal violet dye with reference to P25 photocatalyst. A photochemical reactor (model RPR-200, Rayonet) lined with phosphor-coated lamps (300 nm and 350 nm wavelength) and fitted with 50 W (W) lamps was used for the dye degradation experiments. The chamber temperature was maintained at 25 ± 2 °C during the experiment. A 100 ml suspension containing NPs (10 mg L−1) and dye (5 mg L−1) in a quartz beaker was stirred for 5 min inside the UV reactor and then irradiated for a period of 60 min with 10 ml aliquots were collected periodically every 10 min. The process was repeated for NPs (5 mg L−1), control and P25 (5 mg L−1) for reference photocatalytic activity measurement. The degradation of dye after irradiation was measured using a UV–Vis spectrophotometer (Shimadzu) in the 200–800 nm wavelength range. The changes in absorption were calculated from the average peak absorbance at λ = 590 nm of three samples at the respective time interval. The plots of relative absorption and degradation kinetics were used to evaluate the photocatalytic activity.
Cell cultures
Adherent HaCaT (human keratinocyte) cell line (PMID: 2450098) was employed in this study for cytotoxicity evaluation. The identity of the HaCaT cells was verified by STRP (short tandem repeat profile at Garvan Institute of Medical Research, Australia). The cell cultures were grown and maintained in 75 cm2 tissue culture flasks (Greiner Bio-One) supplemented with Dulbecco’s modified Eagle (DMEM) (Thermo Fisher Scientific) media, 10% (v/v) foetal bovine serum (FBS, Bovogen Biologicals, Australia), which was heat-inactivated before use. Cells were incubated at 37 °C with 5% (v/v) CO2 and passaged using 0.5% trypsin solution (Thermo Fisher Scientific) when the cells approached confluency every 3–4 days. Negative mycoplasma test of cells was routinely checked using the MycoAlert™ detection kit (Lonza Biosciences).
Cell viability study
Cytotoxicity of Ce-PEG nanocomposite and Ce-NPs was evaluated using the MTS assay kit (Promega CellTiter 96™). In this assay, 100 μL of cells at a concentration of approximately 15 × 104 cells per well were seeded in a flat-bottomed 96-well plate and incubated at 37 °C with 5% (v/v) CO2 for 24 h to enable cell adhesion. Firstly, NPs were sterilized by exposing them to UVC light for 20 min, then suspended in a cell culture media at concentrations of 50 mg L−1 and 5 mg L−1 by sonication for 1 h. Then, the prepared NPs suspensions were added to respective cells containing wells at concentrations of 0, 5 and 50 mg L−1 and further incubated at 37 °C with 5% (v/v) CO2 for 24 h. After 20 h incubation time with NPs, MTS reagent (20 μL) was added to each well of the respective plates and then incubated for 4 h to enable the colour development. Then, the plates were centrifuged at 750×g for 10 min and a volume of 75 μL of the supernatant was transferred to the corresponding wells of a new flat-bottomed 96-well plate and the plate was read at λ = 490 nm using a microplate reader (SpectraMax Plus 384 type, Molecular Devices, USA). Each assay was performed in triplicate and repeated in three separate experiments (n = 3). The assays performed using the cells only were used as a negative control, whereas assays in the presence of ZnO NPs (< 100 nm, Sigma-Aldrich) and P25 were referred as positive controls. The viability (% of control) was obtained from the plots of the absorption data for each tested concentration.
The photo-induced toxicity on exposure to UVA/UVB radiation in the presence of Ce-PEG nanocomposite, Ce NPs with reference to TiO2 (P25) and control (cell only) was also evaluated. The cells were grown in a 96-well plate at a concentration 15–18 × 104 cells per well in a similar fashion as described in the procedure for cell viability assay without UV radiation exposure. The tested samples suspensions (50 mg L−1) were prepared in Dulbecco’s phosphate buffered saline (DPBS, without Ca+2, Mg+2) supplemented with 2% FBS by sonication for 1 h. After 24 h incubation period, the media from the wells was removed and replaced with NPs suspensions to make a total volume of 100 µl per well. The cells were incubated further for 1 h to allow the interaction of NPs/nanocomposite with cells. After 1 h, the cells were exposed to light source for a period of 15 min at an intensity of 6 mW cm−2 using a 300 W sunlamp (Ultra-Vitalux, OSRAM). During the exposure, the cell containing plates were placed on ice to avoid the heat-induced damage. After UVA/UVB exposure, the media from the cells was removed and a fresh media of 100 µL was added to each well, and cells were incubated for 24 h. After this period, cell viability was assessed in the same fashion as performed in the assay without UV irradiation.
Statistical analysis (One-way ANOVA with Tuckey’s post-hoc test) was performed on each assay using the software Prism 7.02 (GraphPad software Inc., USA). For analysis, a value of p < 0.05 was considered significant. The viability data of synthesized cerium nanostructures were compared with ZnO NPs and P25 because of their similar applications as UV filters.
Intercellular ROS measurement
The adhered HaCaT cell culture after 24 h of passage were treated with CeO2 NPs and Ce-PEG nanocomposite at a concentration of 50 mg L−1 in a flat-bottom 96-well plate and incubated further for 24 h. After 48 h mark, the cells were washed with Dulbecco’s phosphate-buffered saline (DPBS, Thermo Fisher Scientific) and incubated with fresh culture medium containing 0.5 mM H2O2 or/and 0.5 mM N-acetylcysteine amide (NAC) for 1 h. Then, cells were washed with DPBS and incubated with 20 µl of 0.5 mM 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) dye for 30 min in the dark. Finally, the cells were washed thrice with DPBS to remove the extracellular dichlorofluorescein (DCF) dye, and a volume of 100 µL of DPBS was added to each well and the dye fluorescence was measured using a plate reader (FLUOstar OPTIMA FL BMG LABTECH) at excitation/emission (Ex/Em) wavelength of 490/520 nm. Blank reading was subtracted from the respective cell containing wells readings of each individual assay. Each assay was repeated in three separate experiments in triplicate manner, and mean values were plotted as the change in DCF intensity (% of control).
Results and discussion
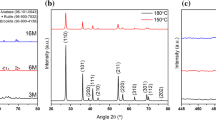
The XRD patterns of Ce-PEG nanocomposite and cerium oxide (CeO2) sample are shown in Fig. 3a, b.The XRD peaks for Ce-PEG bands are centered at 2θ = 14.65°, 15.08°, 19.16°, 23.25°, 26.19°, 26.92°, 27.85°, 30.90°, 32.67°, 36.13°, 39.69°, 42.94° and 45.15°, which is consistent with PEG XRD data given in the previous research reports [45,46,47]. Also, peaks corresponding to CeO2 and Ce2O3 were indexed with reference to powder diffraction file (PDF) cards (#01-089-8436) and (#96-101-0280), respectively, as shown in Fig. 3a. It can be seen (Fig. 3a) that peaks are not sharp, and indicating the growth phase of CeO2 and Ce2O3 particles. The average crystallite size was calculated by Scherrer equation [48, 49]:
where D = average crystallite size (nm), K = shape factor (usually 0.9 for spherical crystallite, λ = X-ray wavelength (0.15406 nm), β = full width half maximum (FWHM) of the peak, and θ = Bragg’s angle (°).

a XRD pattern of Ce-PEG composite and reference PEG pattern, b XRD pattern of Ce-NPs with reference to CeO2 pattern, c deconvoluted Ce-3d spectrum of Ce NPs, d deconvoluted O1s region of Ce NPs spectrum
The average crystallite size of Ce-PEG sample and calcined Ce NPs using Scherrer formula were calculated as 51.5 nm and 6.42 nm, respectively. The crystallite size for these samples was also verified by William–Hall (W–H) method (Figure S2), which were estimated as 43.8 nm and 5.6 nm, respectively. The difference between crystallite size of nanocomposite and calcined sample resulted due to contribution of crystalline part of PEG. Calcination of the sample was performed to quantify the size of individual Ce NPs.
The XRD peaks in the calcined sample were indexed using the CeO2 reference patterns shown in Fig. 3b. Due to smaller crystallite size and broader nature of the peaks, the peaks cannot be clearly indexed to distinguish between CeO2 and Ce2O3. Further, XPS analysis was carried out to verify the oxidation states and their atomic per cent on the surface of calcined sample.
The presence of Ce+3 and Ce+4 on the surface of the sample can be determined using XPS. The deconvolution of core level spectra Ce 3d and O 1s was performed to determine the Ce+3/Ce+4 ratio in the sample as shown in Fig. 3c, d. In general, deconvolution of Ce 3d into 10 peaks is accepted model for the analysis. The sum of the peak areas was determined for each Ce+3 and Ce+4 state and the percentage of Ce+3 was calculated by [50]:
where [Ce+3] and [Ce+4] indicate integrated peak area obtained from the peak deconvolution corresponding to Ce+3 and Ce+4 states. It can be seen from Fig. 3c that peak deconvolution resulted in three doublet pairs of 3d orbital with spin states of j = 3/2 or 5/2 attributed to tetravalent (Ce+4) oxidation state and two doublet pairs linked to trivalent state (Ce+3), also in agreement with previous report by Morlando et al. [1].
The computed FWHM, atomic%, CPS at the respective binding energies of Ce+3 and Ce+4 states are given in Table S1. The atomic% was calculated from the area of the respective peak. The atomic% contents (40.32%) of Ce+3 in the calcined sample were obtained from its total peaks area after deconvolution. A higher Ce+3/Ce+4 contents on the surface of cerium oxide catalyst provide higher superoxide dismutase (SOD)-like activity. A disproportionate amount of superoxide (O2·−) is damaging to body tissues and cells. Superoxide dismutase (enzyme) help to catalyse the superoxide into H2O2 and O2, and reduce the damaging effect on cells and tissues. Various in vitro studies have shown SOD-like activity of cerium NPs, and proceed in the following fashion [51,52,53]:
The deconvolution of O1s region of XPS spectrum was performed to determine the oxygen species present on the surface of the cerium NPs, Fig. 3d. After adequate peak fitting, a BE value 529.05 eV was obtained for O1 peak, which is consistent with reported value of Ce+4 state of cerium [50, 54]. The second deconvoluted peak (O2) was obtained at BE of 530.09 eV, matched to the reported value of Ce+3 state [50]. A small shoulder peak was also obtained at BE of 531.45 eV which can be linked to either organic phase or oxygen deficiency phase [55]. Further, C 1 s region of CeO2 spectrum was deconvoluted to analyse the oxidation states of carbon in the structure, Figure S3. The obtained convoluted peaks C1, C2 and C3 at BE value 284.8 eV, 286.1 eV, and 288.30 eV correspond to C–C, C–O, and C–H (metal hydrocarbon) oxidation states, respectively, which confirmed the presence of organic impurities in the calcined sample.
The morphology of the Ce-PEG nanocomposite structure using SEM imaging is shown in Fig. 4a. SEM image observations revealed the formation of thin 1–4 µm size bands of the structure. However, it is difficult to resolve cerium NPs using SEM. Further, HR-TEM imaging was performed to analyse the structure at high resolution. It can be seen from dark- and bright-field TEM images Fig. 4c, d, f, g that cerium NPs with size range 10–40 nm are embedded in the structure. Also, TEM imaging, Fig. 4c, f shows the thin band-structure of Ce-PEG nanocomposite. HR-TEM image in Fig. 4e clearly showed the lattice fringes of cerium NPs in the PEG matrix. Selected area diffraction pattern (SAED) of the nanocomposite structure is shown in Fig. 4h. Diffused and circular ring pattern indicated the amorphous and polycrystalline nature of the nanocomposite, which confirmed the embedded NPs of cerium in PEG matrix. The diffraction spots also vary in brightness due to contribution of cerium and partly PEG crystallinity. Energy-dispersive x-rays spectroscopy (EDS) maps showed the presence of cerium in the Ce-PEG structure (Fig. 4b).

a SEM image of Ce-PEG nanocomposite, b EDS map, c, d dark-field and f, g bright-field TEM images, e HR-TEM image of lattice fringes, and h SAED pattern of Ce-PEG nanocomposite
UV–Vis absorption measurement of Ce-PEG nanocomposite in the wavelength range of 200–800 nm was performed in comparison to CeO2 NPs to assess the optical behaviour of the nanocomposite. The synthesized structure offered higher absorption as compared to CeO2 NPs as shown in Fig. 5a. Higher UV–Vis absorption of the nanocomposite structure was resulted due to uniform distribution and high dispersibility in the solution. Band gap (Eg) values of the samples were determined from their UV–Vis absorption values using Tauc plots and are shown in Fig. 5b. The measured Eg value of 3.35 ± 0.03 eV of CeO2 is in agreement with the reported value [1]. A lower value of Eg (2.78 ± 0.03 eV) was obtained for Ce-PEG nanocomposite. Due to lower Eg value, the nanocomposite material exhibited high UV–Vis absorption than CeO2 NPs.

a UV–Vis absorption spectra for Ce-PEG composite and Ce NPs, b Tauc plot for band gap determination, c degradation plots of Ce-PEG composite and Ce NPs at 5 and 10 mg L−1 concentrations with respect to control (dye only) and P25 (positive control), and d degradation kinetics of assays performed for dye degradation study
The photocatalytic properties of the synthesized nanostructures were evaluated by dye degradation experiment with reference to P25 and ZnO NPs (positive control, over a time period of 60 min, Fig. 5c. The degradation of CV alone was used as a negative control (control). UV–Vis absorption maximum values for the performed degradation experiments are given in Table S2.
The degradation kinetics of the nanostructures were assessed from the plot of irradiation time (min) and ln(C0/C) as shown in Fig. 5b. These are pseudo-first-order rate reactions described by the Langmuir–Hinshelwood model. Rate constants (k) min−1 were determined from the linear plot of ln(C0/C) versus irradiation time using the expression [49]:
where t is the irradiation time (min), C0 is the initial concentration and C is the concentration at different irradiation times (mg l−1).
Both sample (Ce-PEG nanocomposite and CeO2 NPs sample) did not exhibited any significant dye degradation with reference to P25 and ZnO NPs (reference samples). Ce-PEG sample only exhibited 5% dye degradation compared to control and reference samples, Fig. 5c. This showed absence of ROS formation by the nanocomposite materials during irradiation, and hence, it showed good stability under UV–Vis environment, which play critical role in UV protection. From the degradation kinetic, it can be seen that P25 has highest degradation rate 5.07 × 10–2 ± 0.0026 followed by ZnO 1.68 × 10–2 ± 0.0012 compared to Ce-PEG @10 mg L−1 (9.4 × 10–4 ± 0.0077) and CeO2 NPs @10 mg L−1 (4.32 × 10–4 ± 0.00317), and even lower rate were recorded at 5 mg L−1 concentration of Ce-PEG and CeO2 NPs, given in Table S3. Degradation assay exhibited the ROS scavenging capabilities of the Ce-PEG nano-architecture in similar fashion as witnessed for CeO2 NPs. This validated the stability of the nanocomposite structure in UV–Vis environment.
The viability of HaCaT cells in the presence of 50 and 5 mg L−1 concentrations of Ce-PEG nanocomposite and CeO2 NPs with reference to the negative control (cells only) and positive controls (P25 and ZnO NPs) is shown in Fig. 6. The treatment with Ce-PEG showed highest cell viabilities of 90% and 94% (of control) at 50 mg L−1 and 5 mg L−1. Similar cell viabilities (90% and 95% of control) at 50 mg L−1 and 5 mg L−1 were observed in Ce NPs treated cells. The lowest cell viability (23% of control) was recorded in ZnO NPs (50 mg L−1) treated cells, which was employed as positive control. In P25 assay, cells treated at 50 mg L−1 showed cell viability of 78% of control, while all other treatments resulted in non-significant cell viabilities compared to the control.

a HaCaT cell viability after 24 h incubation with, Ce-PEG composite, CeO2 NPs, P25 and ZnO NPs at concentrations 50, 5 mg L−1. b HaCaT cell viability (% control) after treatment with NPs/nanocomposite and exposure of 15 min UV radiation at intensity of 6 mW cm−2 compared to unexposed cells. Cell viability (% of control) is normalized absorbance value for each concentration. One-way ANOVA and Tukey post-hoc tests were applied to check the statistically different data sets, where ****p ≤ 0.0001, ***p ≤ 0.001, **p ≤ 0.01 and *p ≤ 0.05, with reference to negative control and ####p ≤ 0.0001, ###p ≤ 0.001, ##p ≤ 0.01 and #p ≤ 0.05 with reference to corresponding non-irradiated cells. Each data set represents mean ± SD of three experiments (n = 3 per experiment)
The obtained data sets from performed assays in the presence of Ce-PEG nanocomposite and CeO2 NPs showed non-significant mortality with respect to the control (positive and negative). Hence, HaCaT cells viability data showed the biocompatible nature of the Ce-PEG nanocomposite and could be employed in UV filters and skin therapeutic applications.
The cytotoxic data of HaCaT cells in UU-Vis irradiation experiment at optimum concentration (50 mg l−1) of nanocomposite treated (irradiated) cells compared to non-irradiated, control (non-irradiated cells only) and (P25) (negative control) are shown in Fig. 6b. The reference P25 was selected for UV–Vis experiment because of less toxicity than ZnO for similar concentration. An overall decrease in cell viability was witnessed in UV–Vis irradiated cells compared to non-irradiated cells. A lowest cell viability (25%) was observed in cells exposed to UV–Vis irradiation without nanocomposite protection compared to unexposed cells. Similarly, in the case of P25 treated cells, a significant cell mortality was observed. In this instance, UV–Vis exposure of P25 caused ROS generation at a rate exceeding the natural scavenging by cellular processes. A higher level of ROS caused the oxidative stress which resulted in cell organelle damage, leading to cell death. Although some UV protection was provided by P25 through UV absorption, but increased ROS production outweighed the absorption effect. An increased in UV protection was observed in CeO2 NPs treated cells as cell viability of 55% was resulted after UV–Vis exposure. The highest UV protection (68% cell viability) was observed in C-PEG treated cells compared to control and unexposed cells. In the case of Ce-PEG and CeO2, high UV protection was linked to prevalent ROS scavenging, as ROS scavenging was witnessed in H2O2-based scavenging assay, Fig. 7. Also, UV absorption by the Ce-PEG nanocomposite enhanced the UV protection during irradiation. Hence, Ce-PEG nanocomposite aided against photo-induced ROS damage and oxidative stress by ROS scavenging and UV shielding.

Intracellular ROS in HaCaT cells using the CeO2 NPs and Ce-PEG composite in the presence of H2O2 with reference to the control (cells only) and positive control (H2O2), and amounts of ROS were quantified by recording the DCF fluorescence intensity using the microplate plate reader. One-way ANOVA and Tukey post-hoc tests were applied to check the statistically different data sets, where ****p ≤ 0.0001, ***p ≤ 0.001, **p ≤ 0.01 and *p ≤ 0.05, with reference to negative control and ####p ≤ 0.0001, ###p ≤ 0.001, ##p ≤ 0.01 and #p ≤ 0.05 with reference to positive control. Each data set represents mean ± SD of three experiments (n = 3 per experiment)
Intercellular ROS generation and scavenging of CeO2 NPs, Ce-PEG nanocomposite and NAC were evaluated in the presence of H2O2 with reference to negative control and positive control (H2O2) is shown in Fig. 7.
The ROS level was estimated from the measurement of DCF fluorescence intensity. It can be seen from Fig. 7 that H2O2 only treated cells generated highest level of ROS in cells, as there is no scavenging. In case of NAC assay, there is sufficient scavenging of ROS generated by H2O2, and the level of ROS matches to negative control. Similarly, CeO2 NPs showed 59% lower ROS level than H2O2 only assay, and 5% lower than negative control. Highest ROS scavenging was observed in case of Ce-PEG nanocomposite, which showed 63% lower ROS level than positive control and 15% lower than negative control. The results showed that CeO2 and Ce-PEG nanocomposite efficiently scavenged the ROS generated by H2O2 treatment.
Ce-based nanocomposite structures have already been widely investigated by various groups for the development of efficient and biocompatible UV filters. Recently, Morlando et al. and Mueen et al. used cerium NPs for surface encrustation of TiO2 and ZnO in order to improve the biocompatibility and reduce photocatalytic activity [1, 2]. Our synthesized nanocomposite structure exhibited higher biocompatibility and absence of ROS formation compared to those encrusted nanostructures because of the absence of ZnO and TiO2. In another study, Ce-NPs were employed in the preparation of epoxy-based nanocomposite thin films for UV protection, reported by Dao et al. The reported structure can find application as UV reduction thin films to protect from the UV-induced damage [56]. Similarly, Wang et al. incorporated the Ce NPs in cellulose matrix for the development of transparent and hydrophobic thin films as UV anti-shields [57]. In comparison to above nanocomposite structures, a wide UV–Vis absorption range (200–480 nm), biocompatibility, flexibility of Ce-PEG nanocomposite structure were observed. In addition, hydrothermally synthesized Ce-PEG nanocomposite offered ease of applicability, and mixing with other skin protective formulations.
Conclusion
Hydrothermal processing of cerium and PEG in the presence of ammonium solution under suitable conditions of temperature and pressure formed the nanocomposite with cerium NPs dispersed in the matrix of PEG bands. The surface conjugation of cerium NPs by PEG resulted due to amine active component by oxygen linkage. XRD analysis showed the formation of (110) directed growth of PEG bands with embedded CeO2 NPs. SEM characterization revealed the band like structures of Ce-PEG nanocomposite. High-resolution morphology analysis by HR-TEM revealed the embedded CeO2 NPs in the Ce-PEG bands, and further study by SAED pattern revealed the diffused and circular ring pattern which verified the amorphous and polycrystalline nature of the nanocomposite. The nanocomposite material indicated the presence of mixed valence state of cerium (Ce+3 and Ce+4) as analysed by XPS study of Ce3d region. The deconvolution of O1s region provided the information about the presence of three types of oxygen species which were linked to Ce+3 and Ce+4 and organic impurities. The deconvolution and peak fitting of C 1s regions showed presence of three peaks linked to C–C, C–O, and C–H (metal hydrocarbon) species at the surface of the calcined sample, which confirmed the presence of organic phase. An increased UV–Vis absorption was observed for the Ce-PEG nanocomposite material compared to CeO2 NPs in 200–400 nm range. The nanocomposite material (Ce-PEG) reported decrease in band gap (2.76 ± 0.03 eV) with respect to CeO2 NPs (3.28 ± 0.03 eV). Photocatalytic behaviour study by dye (CV) degradation assay exhibited no significant dye degradation by CeO2 NPs and Ce-PEG nanocomposite compared to P25 photocatalyst. In vitro antioxidant properties evaluation of the Ce-PEG nanocomposite by DCF fluorescent-based ROS assay using HaCaT cells in the presence of H2O2 exhibited sufficient ROS scavenging, whereby observed the decreased ROS level (15% and 63%) with reference to negative control and positive control, respectively.
The cytotoxic assay on non-malignant HaCaT cells treated with Ce-PEG nanocomposite showed cell viability 90% and 94% (of control) at 50 mg L−1 and 5 mg L−1, respectively. The higher viability suggested the biocompatible nature of the Ce-PEG nanocomposite. UV irradiation of HaCaT cell significantly reduced the cell viability through ROS generation and oxidative stress augmentation. Particularly, in the case of irradiated P25 treated cells, only 40% cell viability was observed. A high protection against UV radiation was observed in Ce-PEG treated cells, and a viability of 69% was resulted.
Thus, Ce-PEG nanocomposite offered higher dispersibility of CeO2 NPs in the polymer matrix that constitute more uniform UV absorption properties. The flexible, semitransparent nano-architecture structure exhibited increased UV–Vis absorption, biocompatibility and protection against damaging effect of UV radiation. Thus, the synthesized nanocomposite can be suitably used as UV filter, antioxidant and skin therapeutic product due to soothing nature of the PEG matrix. Further, the nanostructure can be explored for applications in the field of drug delivery, imaging and regenerative medicine.
References
Morlando A, Borrás MC, Rehman Y et al (2020) Development of CeO2 nanodot encrusted TiO2 nanoparticles with reduced photocatalytic activity and increased biocompatibility towards a human keratinocyte cell line. J Mater Chem B 8:4016. https://doi.org/10.1039/D0TB00629G
Mueen R, Morlando A, Qutaish H, Lerch M, Cheng Z, Konstantinov K (2020) ZnO/CeO2 nanocomposite with low photocatalytic activity as efficient UV filters. J Mater Sci 55:6834. https://doi.org/10.1007/s10853-020-04493-x
Morlando A, McNamara J, Rehman Y, Sencadas V, Barker PJ, Konstantinov K (2020) Hydrothermal synthesis of rutile TiO2 nanorods and their decoration with CeO2 nanoparticles as low-photocatalytic active ingredients in UV filtering applications. J Mater Sci 55:8095. https://doi.org/10.1007/s10853-020-04598-3
Abebe B, Murthy HA, Amare E (2018) Summary on adsorption and photocatalysis for pollutant remediation: mini review. J Encapsul Adsorpt Sci 8:225. https://doi.org/10.4236/jeas.2018.84012
De Jager T, Cockrell A, Du Plessis S (2017) Ultraviolet light induced generation of reactive oxygen species. In: Ultraviolet light in human health, disease and environment, p 15. https://doi.org/10.1007/978-3-319-56017-5_2
Caputo F, De Nicola M, Sienkiewicz A et al (2015) Cerium oxide nanoparticles, combining antioxidant and UV shielding properties, prevent UV-induced cell damage and mutagenesis. Nanoscale 7:15643. https://doi.org/10.1039/C5NR03767K
Mueen R, Lerch M, Cheng Z, Konstantinov K (2020) Na-doped ZnO UV filters with reduced photocatalytic activity for sunscreen applications. J Mater Sci 55:2772. https://doi.org/10.1007/s10853-019-04122-2
Young AR, Claveau J, Rossi AB (2017) Ultraviolet radiation and the skin: photobiology and sunscreen photoprotection. J Am Acad Dermatol 76:S100. https://doi.org/10.1016/j.jaad.2016.09.038
Rehman Y, Cheng Z, Wang X, Huang X-F, Konstantinov K (2021) Theranostic two-dimensional superparamagnetic maghemite quantum structures for ROS-mediated cancer therapy. J Mater Chem B 9:5805. https://doi.org/10.1039/D1TB01036K
Tyagi N, Gambhir K, Kumar S, Gangenahalli G, Verma YK (2021) Interplay of reactive oxygen species (ROS) and tissue engineering: a review on clinical aspects of ROS-responsive biomaterials. J Mater Sci 56:16790. https://doi.org/10.1007/s10853-021-06338-7
Ginzburg AL, Blackburn RS, Santillan C, Truong L, Tanguay RL, Hutchison JE (2021) Zinc oxide-induced changes to sunscreen ingredient efficacy and toxicity under UV irradiation. Photochem Photobiol Sci 20:1273. https://doi.org/10.1007/s43630-021-00101-2
Kong L, Rau A, Yang N, Lu K (2021) Flexible ZnO nanoparticle-poly(methyl methacrylate) hybrid films and their ultraviolet shielding behaviors. JOM 73:432. https://doi.org/10.1007/s11837-020-04454-4
Lin LL, Grice JE, Butler MK et al (2011) Time-correlated single photon counting for simultaneous monitoring of zinc oxide nanoparticles and NAD (P) H in intact and barrier-disrupted volunteer skin. Pharm Res 28:2920. https://doi.org/10.1007/s11095-011-0515-5
Raphael AP, Sundh D, Grice JE, Roberts MS, Soyer HP, Prow TW (2013) Zinc oxide nanoparticle removal from wounded human skin. Nanomedicine 8:1751. https://doi.org/10.2217/nnm.12.196
Valdiglesias V, Costa C, Kiliç G et al (2013) Neuronal cytotoxicity and genotoxicity induced by zinc oxide nanoparticles. Environ Int 55:92. https://doi.org/10.1016/j.envint.2013.02.013
Guo D, Bi H, Liu B, Wu Q, Wang D, Cui Y (2013) Reactive oxygen species-induced cytotoxic effects of zinc oxide nanoparticles in rat retinal ganglion cells. Toxicol In Vitro 27:731. https://doi.org/10.1016/j.tiv.2012.12.001
Kim J-H, Jeong MS, Kim D-Y, Her S, Wie M-B (2015) Zinc oxide nanoparticles induce lipoxygenase-mediated apoptosis and necrosis in human neuroblastoma SH-SY5Y cells. Neurochem Int 90:204. https://doi.org/10.1016/j.neuint.2015.09.002
Wahab R, Kaushik NK, Verma AK et al (2011) Fabrication and growth mechanism of ZnO nanostructures and their cytotoxic effect on human brain tumor U87, cervical cancer HeLa, and normal HEK cells. J Biol Inorg Chem 16:431. https://doi.org/10.1007/s00775-010-0740-0
Tian L, Lin B, Wu L et al (2015) Neurotoxicity induced by zinc oxide nanoparticles: age-related differences and interaction. Sci Rep 5:1. https://doi.org/10.1038/srep16117
Xie Y, Wang Y, Zhang T, Ren G, Yang Z (2012) Effects of nanoparticle zinc oxide on spatial cognition and synaptic plasticity in mice with depressive-like behaviors. J Biomed Sci 19:1. https://doi.org/10.1186/1423-0127-19-14
De Simone U, Lonati D, Ronchi A, Coccini T (2016) Brief exposure to nanosized and bulk titanium dioxide forms induces subtle changes in human D384 astrocytes. Toxicol Lett 254:8. https://doi.org/10.1016/j.toxlet.2016.05.006
Hu Q, Guo F, Zhao F, Fu Z (2017) Effects of titanium dioxide nanoparticles exposure on parkinsonism in zebrafish larvae and PC12. Chemosphere 173:373. https://doi.org/10.1016/j.chemosphere.2017.01.063
Márquez-Ramírez SG, Delgado-Buenrostro NL, Chirino YI, Iglesias GG, López-Marure R (2012) Titanium dioxide nanoparticles inhibit proliferation and induce morphological changes and apoptosis in glial cells. Toxicology 302:146. https://doi.org/10.1016/j.tox.2012.09.005
Ratnasekhar C, Sonane M, Satish A, Mudiam MKR (2015) Metabolomics reveals the perturbations in the metabolome of Caenorhabditis elegans exposed to titanium dioxide nanoparticles. Nanotoxicologly 9:994. https://doi.org/10.3109/17435390.2014.993345
Disdier C, Devoy J, Cosnefroy A et al (2015) Tissue biodistribution of intravenously administrated titanium dioxide nanoparticles revealed blood-brain barrier clearance and brain inflammation in rat. Part Fibre Toxicol 12:1. https://doi.org/10.1186/s12989-015-0102-8
Ze Y, Zheng L, Zhao X et al (2013) Molecular mechanism of titanium dioxide nanoparticles-induced oxidative injury in the brain of mice. Chemosphere 92:1183. https://doi.org/10.1016/j.chemosphere.2013.01.094
Philippat C, Bennett D, Calafat AM, Picciotto IH (2015) Exposure to select phthalates and phenols through use of personal care products among Californian adults and their children. Environ Res 140:369. https://doi.org/10.1016/j.envres.2015.04.009
Calafat AM, Wong L-Y, Ye X, Reidy JA, Needham LL (2008) Concentrations of the sunscreen agent benzophenone-3 in residents of the United States: National Health and Nutrition Examination Survey 2003–2004. Environ Health Perspect 116:893. https://doi.org/10.1289/ehp.11269
Bae J, Kim S, Kannan K, Louis GMB (2016) Couples’ urinary concentrations of benzophenone-type ultraviolet filters and the secondary sex ratio. Sci Total Environ 543:28. https://doi.org/10.1016/j.scitotenv.2015.11.019
Zucchi S, Blüthgen N, Ieronimo A, Fent K (2011) The UV-absorber benzophenone-4 alters transcripts of genes involved in hormonal pathways in zebrafish (Danio rerio) eleuthero-embryos and adult males. Toxicol Appl Pharmacol 250:137. https://doi.org/10.1016/j.taap.2010.10.001
Axelstad M, Boberg J, Hougaard KS et al (2011) Effects of pre-and postnatal exposure to the UV-filter octyl methoxycinnamate (OMC) on the reproductive, auditory and neurological development of rat offspring. Toxicol Appl Pharmacol 250:278. https://doi.org/10.1016/j.taap.2010.10.031
Broniowska Ż, Pomierny B, Smaga I, Filip M, Budziszewska B (2016) The effect of UV-filters on the viability of neuroblastoma (SH-SY5Y) cell line. Neurotoxicology 54:44. https://doi.org/10.1016/j.neuro.2016.03.003
Faass O, Schlumpf M, Reolon S et al (2009) Female sexual behavior, estrous cycle and gene expression in sexually dimorphic brain regions after pre-and postnatal exposure to endocrine active UV filters. Neurotoxicology 30:249. https://doi.org/10.1016/j.neuro.2008.12.008
Blüthgen N, Meili N, Chew G, Odermatt A, Fent K (2014) Accumulation and effects of the UV-filter octocrylene in adult and embryonic zebrafish (Danio rerio). Sci Total Environ 476:207. https://doi.org/10.1016/j.scitotenv.2014.01.015
Ruszkiewicz JA, Pinkas A, Ferrer B, Peres TV, Tsatsakis A, Aschner M (2017) Neurotoxic effect of active ingredients in sunscreen products, a contemporary review. Toxicol Rep 4:245. https://doi.org/10.1016/j.toxrep.2017.05.006
El-Toni AM, Yin S, Sato T (2008) UV Shielding performance enhancement of CaO doped ceria by coupling with plate-like K0.8Li0.27Ti1.73O4. J Mater Sci 43:2411. https://doi.org/10.1007/s10853-007-2110-z
Alizadeh N, Salimi A, Sham T-K, Bazylewski P, Fanchini G (2020) Intrinsic enzyme-like activities of cerium oxide nanocomposite and its application for extracellular H2O2 detection using an electrochemical microfluidic device. ACS Omega 5:11883. https://doi.org/10.1021/acsomega.9b03252
Jiang H, Li X, Chen S, Wang H, Huo P (2020) g-C3N4 quantum dots-modified mesoporous CeO2 composite photocatalyst for enhanced CO2 photoreduction. J Mater Sci Mater Electron 31:20495. https://doi.org/10.1007/s10854-020-04568-0
Xue Y, Balmuri SR, Patel A, Sant V, Sant S (2018) Synthesis, physico-chemical characterization, and antioxidant effect of PEGylated cerium oxide nanoparticles. Drug Deliv Transl Res 8:357. https://doi.org/10.1007/s13346-017-0396-1
Kim CK, Kim T, Choi IY et al (2012) Rücktitelbild: ceria nanoparticles that can protect against ischemic stroke (Angew. Chem. 44/2012). Angew Chem 124:11334. https://doi.org/10.1002/anie.201203780
Pagliari F, Mandoli C, Forte G et al (2012) Cerium oxide nanoparticles protect cardiac progenitor cells from oxidative stress. ACS Nano 6:3767. https://doi.org/10.1021/nn2048069
Karakoti A, Singh S, Dowding JM, Seal S, Self WT (2010) Redox-active radical scavenging nanomaterials. Chem Soci Rev 39:4422. https://doi.org/10.1039/B919677N
Li Y, Hou X, Yang C et al (2019) Photoprotection of cerium oxide nanoparticles against UVA radiation-induced senescence of human skin fibroblasts due to their antioxidant properties. Sci Rep 9:1. https://doi.org/10.1038/s41598-019-39486-7
Escobedo-Morales A, Ruiz-López I, Md Ruiz-Peralta L, Sánchez-Cantú M, Tepech-Carrillo, Moreno-Orea J (2019) Automated method for the determination of the band gap energy of pure and mixed powder samples using diffuse reflectance spectroscopy. Heliyon 5:e01505. https://doi.org/10.1016/j.heliyon.2019.e01505
Jayaramudu T, Raghavendra GM, Varaprasad K et al (2016) Preparation and characterization of poly(ethylene glycol) stabilized nano silver particles by a mechanochemical assisted ball mill process. J Appl Polym Sci. https://doi.org/10.1002/app.43027
Hussein MA, Abu-Zied BM, Asiri AM (2014) Preparation, characterization, and electrical properties of ZSM-5/PEG composite particles. Polym Compos 35:1160. https://doi.org/10.1002/pc.22763
Wan L, Cao X, Xue X et al (2021) Self-healing and flexible ionic gel polymer electrolyte based on reversible bond for high-performance lithium metal batteries. Energy Technol 10:2100749. https://doi.org/10.1002/ente.202100749
Rehman Y, Copet C, Morlando A, Huang X-F, Konstantinov K (2020) Investigation of ROS scavenging properties and in vitro cytotoxicity of oxygen-deficient La2O3−x nanostructure synthesized by spray pyrolysis method. J Nanostruct Chem 10:347. https://doi.org/10.1007/s40097-020-00356-8
Rehman Y, Morlando A, Chaki Borras M et al (2021) Defect-rich La2O3 nanoparticles with antioxidant activity for human keratinocytes. ACS Appl Nano Mater 4:6345. https://doi.org/10.1021/acsanm.1c01177
Barth C, Laffon C, Olbrich R, Ranguis A, Parent P, Reichling M (2016) A perfectly stoichiometric and flat CeO2 (111) surface on a bulk-like ceria film. Sci Rep 6:1. https://doi.org/10.1038/srep21165
Korsvik C, Patil S, Seal S, Self WT (2007) Superoxide dismutase mimetic properties exhibited by vacancy engineered ceria nanoparticles. Chem Commun. https://doi.org/10.1039/B615134E
Xu C, Qu X (2014) Cerium oxide nanoparticle: a remarkably versatile rare earth nanomaterial for biological applications. NPG Asia Mater 6:e90. https://doi.org/10.1038/am.2013.88
Das S, Dowding JM, Klump KE, McGinnis JF, Self W, Seal S (2013) Cerium oxide nanoparticles: applications and prospects in nanomedicine. Nanomedicine 8:1483. https://doi.org/10.2217/nnm.13.133
Hezam A, Namratha K, Drmosh QA et al (2019) CeO2 nanostructures enriched with oxygen vacancies for photocatalytic CO2 reduction. ACS Appl Nano Mater 3:138. https://doi.org/10.1021/acsanm.9b01833
Jiang D, Wang W, Zhang L, Zheng Y, Wang Z (2015) Insights into the surface-defect dependence of photoreactivity over CeO2 nanocrystals with well-defined crystal facets. ACS Catal 5:4851. https://doi.org/10.1021/acscatal.5b01128
Dao NN, Dai Luu M, Nguyen QK, Kim BS (2011) UV absorption by cerium oxide nanoparticles/epoxy composite thin films. Adv Nat Sci Nanosci Nanotechnol 2:045013. https://doi.org/10.1088/2043-6262/2/4/045013
Wang W, Zhang B, Jiang S, Bai H, Zhang S (2019) Use of CeO2 nanoparticles to enhance UV-shielding of transparent regenerated cellulose films. Polymers 11:458. https://doi.org/10.3390/polym11030458
Acknowledgements
We would like to thank technical and administration staff of “Institute of Super Conducting & Electronic Materials (ISEM)”,” Australian Institute for Advanced Materials (AIIM)”, and the “Illawarra Health & Medical Research Institute (IHMRI)” at University of Wollongong (UOW) Australia for providing technical assistance and guidance to carryout this research.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. This work was supported by University of Wollongong (UOW) Global challenge program “Next Generation Sunscreens”, carried out at the Institute of Super Conducting and Electronic Materials (ISEM), Australian Institute for Advanced Materials (AIIM), and the Illawarra Health & Medical Research Institute (IHMRI), UOW. In addition, financial support was also provided under the joint scholarship program of Higher education commission (HEC) Pakistan and UOW, Australia.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial or personal interests.
Additional information
Handling Editor: Yaroslava Yingling.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rehman, Y., Qutaish, H., Kim, J.H. et al. Nanoarchitectonics of (110) directed polyethylene glycol stabilized cerium nanoparticles for UV filtering applications. J Mater Sci 57, 12848–12864 (2022). https://doi.org/10.1007/s10853-022-07437-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-022-07437-9