
Abstract
Star-shaped glycopolymers due to the attractive combination of the physicochemical, morphological, self-assembly properties along with biological activity have gained increased attention as innovative agents in novel cancer therapies. Unfortunately, the production of these highly desirable biomaterials remains a challenge in modern macromolecular chemistry. The main reason for that is the low polymerizability of ionic glycomonomers originated from their steric congestion and the occurrence of ionic interactions that generally negatively influence the polymerization progress and hinder controllable reaction pathway. In this work, the new ionic sugar monomer was (co)polymerized for the first time via Activator Generated by Electron Transfer Atom Transfer Radical Polymerization (AGET ATRP) using a three-arm resveratrol-based core to obtain star-like (co)polymers. The obtained products were examined in terms of their physicochemical properties and morphology. Aside from the synthesis of these new glycopolymers, also a thorough description of their thermal properties, ability to self-assembly, the formation of stable superstructures was studied in detail. It was found that examined (co)polymers did not show any heterogeneities and phase separation, while their variation of glass transition temperature (Tg) was strictly related to the change in the number of glycomonomer. Also, the stability and shapes of formed superstructures strictly depend on their composition and topology. Finally, we have shown that synthesized carbohydrate-based polymers revealed high antiproliferative activity against several cancer cell lines (i.e., breast, colon, glioma, and lung cancer). The cytotoxic activity was particularly observed for star-shaped polymers that were systematically enhanced with the growing concentration of amine moieties and molecular weight. The results presented herein suggest that synthesized star-shaped glyco(co)polymers are promising as drug or gene carriers in anticancer therapies or anti-tumor agents, depending on their cytotoxicity.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Saccharides are one of the most important and widespread chemical compounds in the world. Due to their non-toxic, biocompatible properties as well as an important role in several biochemical processes, crucial for the proper functioning of the living organisms [1] they caught a special attention of the scientists. As a consequence, different kind of native carbohydrates have been used in catalysis [2, 3], polymer chemistry [4, 5], or medicine [6, 7]. In this context, one can briefly mention about polysaccharides, the third major class of naturally occurring biomacromolecules (i.e., cellulose, starch, glycogen, chitin or its deacetylated derivative – chitosan), which are commonly applied in food, packaging, material, and biomedical industries. In fact, the latter application has become especially important since it was found that polysaccharides are biomimetic analogues being very active as mediators in a wide range of cell–cell recognition events. However, their use in a native form is not always convenient due to weak solubility or unsuitable hydrophilic/hydrophobic balance. For these reasons, many research groups work on the modifications of naturally occurring oligo or polysaccharides to enhance some important physical, chemical and mechanical properties without losing advantageous features of these materials.
An interesting alternative to native polysaccharides are glycopolymers defined as fully synthetic macromolecules possessing a non-carbohydrate backbone and pedant or terminal carbohydrate moieties [8]. It is worth noting that despite of the significant progress in macromolecular synthetic toolbox, the production of such macromolecules is challenging due to the steric effects occurring during synthesis. Generally, glycopolymers can roughly be prepared via two main synthetic routes: direct polymerization of carbohydrate-based monomers and the chemical modification of produced polymers with saccharide-containing compounds. Notably, the latter strategy is considered as more convenient than the first one, which often requires the multi-step process. For instance, this kind of reaction can be performed by common ‘click reactions’ using thiol-ene or CuACC chemistry [9,10,11]. Unfortunately, the main disadvantage of such a solution is the poor control over the degree of substitution, which is a result of the steric hindrance, density packing effects between long polymer chains and structurally complex large carbohydrate. In fact, much easier way is to use carbohydrates as polymerization initiators. The most popular pre-initiators used in this approach are dextrans [12], cellulose [13], starch, as well as simple sugars such as glucose [14,15,16].
In the light of the above, the best strategy to produce glycopolymers is a direct polymerization of saccharide-containing monomer that allows the production of materials with well-defined structures and varying type of topologies. This kind of synthetic pathway uses a variety of controlled polymerization methods including the atom transfer radical polymerization (ATRP) [17,18,19,20,21] and the reversible addition-fragmentation chain transfer polymerization (RAFT) [22,23,24,25,26]. In this context, one can recall the first historical reaction performed by Kobayashi who synthesized polystyrene-based glycopolymers with pendant lactose and maltose moieties [27]. However, the first controlled reaction using glycomonomer was reported by Ohno, who employed nitroxide mediated polymerization of N-vinyl-benzyl-O-b-D-galactopyranosyl-(1–4)-D-gluconamide [28]. Since then, controlled free-radical polymerizations have been used with increasing frequency, taking into account various variations and modifications as well as one-pot systems [4]. An example of the latter approach is Chen's work [29], which presents an approach combining RAFT and “click” reactions. A large proportion of the reports on glycopolymers are those ones using surface initiation techniques. Gold [30, 31], silver [32], or iron oxide [33, 34] particles obtained in this way were proven to act effectively as theranostic agents in many therapies. Moreover, it should be mentioned that little attention was paid to reports concerning the use of ATRP or its modifications in the polymerization of ionic glycomonomers. In general, only a few reports are devoted to polymerization of various ionic liquids via ATRP approach [35,36,37,38,39]. Nevertheless, ionic and nonionic glycopolymers' production seems to be important from the viewpoint of their wide applications. Interestingly, these types of macromolecules were characterized by enhanced adhesion to hepatocytes [40]. Besides, many of them were tested toward potential use as a new drug delivery systems [41], especially in chemotherapy [42,43,44,45], which is mostly related to the increased amount of saccharide sensitive receptors on the surface of cancer cells. Others are used to target specific cells in the body [46,47,48]. Finally, some glycopolymers show great antibacterial properties and are being developed as a new tool to fight drug-resistant microorganisms [49,50,51,52].
In this paper, a facile route toward novel tailored glycopolymers differing in structural and topological parameters (linear vs. star-shaped) is described. ATRP of glucose-bearing methacrylate monomer was performed on classical ATRP initiator, ethyl α-bromoisobutyrate, and a new one based on the resveratrol structure. Apart from this, a series of well-defined functional copolymers containing both carbohydrate and poly(N,N-dimethylaminoethyl methacrylate) segments were synthesized, aiming to screen the influence of different composition and topology on their thermodynamical parameters and aggregation behavior. It is also worth to stress that N,N-dimethylaminoethyl methacrylate was used as comonomer to enhance amphiphilic properties of the produced copolymers. It was shown that all tested macromolecules self-assemble in aqueous solutions and show poor pH-sensitivity. Finally, these materials demonstrated biological activity against several cancer cell lines, including breast, colon, glioma, and lung cancer. Additionally as a reference we performed cytotoxicity studies on normal human dermal fibroblasts.
Materials and methods
Glucose, α-bromoisobutyryl bromide, acetic anhydride, bromoethanol, N,N-dimethylaminoethyl methacrylate (DMAEMA), tris(2-pyridylmethyl)amine (TPMA), copper(II) bromide, boron trifluoride etherate, and sodium acetate were purchased from Sigma-Aldrich. Resveratrol was obtained from TCI Chemicals. Dichloromethane, methanol, tetrahydrofuran, ascorbic acid, and anhydrous pyridine were acquired from Chempur.
Synthesis of resveratrol-based trifunctional ATRP initiator
300 mg (1.3 mmol) of resveratrol was added to 20 mL of pyridine. The mixture was cooled down with ice bath and then 2 mL (3.72 g; 16.2 mmol) of α-bromoisobutyryl bromide was added. Reaction was carried out for 24 h. After that the mixture was concentrated under vacuum and poured into cold methanol in order to remove salt byproduct and unreacted resveratrol. White-yellow solid was then filtrated and washed with water three times. Then it was dried under vacuum. Yield 65%.
1H-NMR (500 MHz, DMSO-d6, ppm) 7.782 (2H, aromatic) 7.472 (2H, aromatic) 7.432 (2H, aromatic) 7.286 (1H, aromatic) 7.102 (2H, CH=CH) 2.115 (18H, -CH3).
Synthesis of penta-O-acetyl-β-D-glucopyranose (2)
The reaction was carried out according to the method described in the literature. [53] To boiling acetic anhydride (244 mL, 2.58 mol) containing sodium acetate (10.66 g, 0.13 mol) monosaccharide powder (0.26 mol) was added in small portions over 15 min. The reaction was carried out in a 500-mL-flask equipped with a reflux condenser, a mechanical stirrer, and an oil bath. After all the sugar had been added, the reaction was continued under reflux for a further 5 min and the reaction mixture was allowed to cool to room temperature. The reaction was terminated by adding plenty of cold water and shaking vigorously until a white precipitate formed. The mixture was then left to settle in the refrigerator overnight. The next day the precipitate was filtered and washed with water until the smell of acetic acid disappeared. The product thus formed did not require further purification after being well dried. Yield 93%.
1H NMR (600 MHz,CDCl3): δ ppm = 5.72 (d, 1H, J = 8.3 Hz), 5.26 (t, 1H, J = 9.4 Hz), 5.12–5.11 (m, 2H), 4.29 (dd, 1H, J = 4.6, 12.5 Hz), 4.12 (dd, 1H, J = 2.2, 12.5 Hz), 3.85 (ddd, 1H, J = 2.2, 4.6, 10.0 Hz), 2.12 (s, 3H), 2.09 (s, 3H), 2.04 (s, 6H), 2.02 (s, 3H). 13C NMR (150 MHz, CDCl3): δ ppm = 170.6, 170.1, 169.4, 169.3, 169.0, 91.8, 72.9, 72.8, 70.3, 67.9, 61.6, 20.9, 20.8, 20.6. ESI–MS: [M + Na]+ counted: 413.1060, found: 413.1056.
Synthesis of 2-bromoethyl-2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside (3)
The reaction was carried out according to the method described in the literature [54]. In a 1000-mL-round-bottom flask protected against light with aluminum foil and equipped with a dropping funnel, magnetic stirrer, and an ice bath, the protected monosaccharide (0.16 mol) was weighed and dissolved in anhydrous dichloromethane (250 mL). 2-Bromoethanol (13.9 mL, 0.19 mol) was added to the solution and the mixture was cooled to 0 °C. Then BF3·Et2O (100 mL, 0.81 mol) was added slowly. The reaction was carried out under a nitrogen atmosphere for 3 h maintaining the temperature at 0 °C and 20 h at room temperature. After completion of the reaction, the reaction mixture was diluted with dichloromethane (100 mL), 250 mL of cooled water were added and vigorously stirred. The organic layer was separated and washed repeatedly with saturated aqueous NaHCO3 solution. The organic layer was then dried over anhydrous magnesium sulfate, filtered, and the solvent was evaporated using a rotary evaporator. The residue was purified by column chromatography using 2% methanol in dichloromethane as eluent. Yield 50%. 1H NMR (600 MHz, CDCl3): δ ppm = 5.23 (t, 1H, J = 9.5 Hz), 5.09 (t, 1H, J = 9.4 Hz), 5.00–5.03 (m, 1H), 4.58 (d, 1H, J = 7.9 Hz), 4.25–4.28 (m, 1H), 4.11–4.18 (m, 2H), 3.80–3.84 (m, 1H), 3.72 (ddd, 1H, J = 2.4, 4.8, 10.0 Hz), 3.43–3.50 (m, 2H), 2.09 (s, 3H), 2.07 (s, 3H), 2.03 (s, 3H), 2.01 (s, 3H). 13C NMR (150 MHz, CDCl3): δ ppm = 170.5, 170.1, 169.3, 100.9, 72.5, 71.9, 71.0, 69.7, 68.3, 61.8, 29.8, 20.6, 20.5. ESI–MS [M + Na]+ counted: 477.0372, found: 477.0375.
Synthesis of N-[2-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyloksy)ethyl]-N-metacryloyloxyethyl-N,N-dimethylammonium bromide (GluAmMA) (4)
The amine was added dropwise to the fully acylated sugar (10.98 mmol) dissolved in ethanol (20 mL) in a substrate: amine molar ratio of 1: 0.9. The reaction was carried out for 48 h in the presence of an inert gas at the reflux temperature of the solvent. After completion of the reaction and after evaporation of the reactants, the residue was extracted with water: dichloromethane or washed with petroleum ether and a little chloroform for purification. Obtained white solid. Yield 68%. 1H NMR (400 MHz, CDCl3): δ ppm = 6.10 (d, J = 5.2 Hz, 1H), 5.88 (s, 1H), 5.62 (s, 1H), 5.16 (t, J = 9.5 Hz, 1H), 5.00 (t, J = 9.7 Hz, 1H), 4.93–4.83 (m, 1H), 4.71 (d, J = 8.0 Hz, 1H), 4.68–4.54 (m, 2H), 4.35–3.89 (m, 4H), 3.63 (s, 4H), 3.43 (s, 6H), 2.04 (s, 3H), 2.02 (s, 3H), 1.97 (s, 3H), 1.94 (s, 3H), 1.90 (s, 3H). 13C NMR (101 MHz, CDCl3): δ ppm = 170.66, 169.97, 169.62, 169.46, 166.30, 135.21, 127.40, 100.24, 72.42, 72.22, 70.99, 68.13, 64.15, 63.44, 61.48, 58.15, 52.78, 52.62, 50.30, 20.90, 20.85, 20.60, 20.56, 18.31. ESI–MS [M+] counted: 532.2394, found: 523.2396.
Polymer synthesis
AGET ATRP copolymerization of GluAmMA and DMAEMA (example for polymer IV).
GluAmMA (1.13 g, 1.846 mmol), DMAEMA (1 mL, 0.933 g, 5.93 mmol), resveratrol-based trifunctional initiator (6.9 mg, 0.0102 mmol), CuBr2 (3.2 mg, 0.0143 mmol), tris(2-pyridylmethyl)amine (TPMA) (11.6 mg, 0.0399 mmol), and dimethylsulfoxide (DMSO) (3 mL) were placed into the Schlenk reactor and the air was removed by three pump-thaw cycles. Then the 0.9 mg (0.0051 mmol) of ascorbic acid was added and the reactor was placed into the oil bath heated to 40 °C for 4 h. After this time the sample for NMR was taken and the mixture was poured into cold THF to precipitate polymer. Brown precipitate was then dissolved in methanol and placed into the dialysis bag (SpectraPor 3). The dialysis against methanol was carried out for 24 h. After that the polymer was dried under the reduced pressure.
Nuclear magnetic resonance
NMR spectra were recorded on using 500 MHz spectrometer (Bruker) for samples in commercially available CDCl3 and DMSO-d6.
High-resolution electrospray ionization mass spectroscopy
High-resolution electrospray ionization mass spectroscopy (ESI–MS) experiments were performed using a Waters Xevo G2 QTOF instrument equipped with an injection system (cone voltage 50 V; source 120 °C).
Gel permeation chromatography
Molecular weights (Mn, SEC) and dispersity indices (Ð) were determined by size-exclusion chromatograph (SEC, Thermo Scientific, Ultimate 3000) equipped with an ISO-3100SD isocratic pump, autosampler, degasser, thermostatic box for columns, and differential refractometer RefractoMax 521 Detector. Chromoleon 7.2 Chromatography Data System (CDS, Thermo Scientific) was used for data collecting and processing. The SEC calculated molecular weights were based on calibration applying linear poly(ethylene glycol) PEG/poly(ethylene oxide) PEO standards (Mp = 1020–504,000 g/mol). TSKgel SuperAW-L 4,6 mm I.D. × 3,5 cm, particle size 7 um guardcolumn, and TSKgel SuperAW3000 4um column were used for separation. The measurements were taken in distilled water as the eluent containing 0.05% NaN3, at T = 40 °C with a flow rate of 0.3 mL/min.
Differential scanning calorimetry
Calorimetric measurements were taken by using Mettler-Toledo DSC apparatus equipped with a liquid nitrogen cooling accessory and an HSS8 ceramic sensor (heat flux sensor with 120 thermocouples). Temperature and enthalpy calibrations were performed by using indium and zinc standards. The sample was prepared in an open aluminum crucible (40 μL) outside the DSC apparatus. The samples were measured on heating from 233 to 523 K at a constant heating rate of 10 K min−1.
Dynamic light scattering and zeta potential measurements
Hydrodynamic diameters dh and zeta potentials ZP of polymer particles were measured on Malvern Zetasizer Nano-ZS (4 mW He–Ne ion laser, λ = 633 nm) for samples in deionized water (0.5 mg/mL) at 25 °C ± 0.1 °C.
Transmission electron microscopy
Micelles were visualized by high-resolution transmission electron microscope (TEM, FEI Tecnai G2 Spirit BioTWIN) at 120 kV. The polymer samples in deionized water (0.5 mg/mL) placed on the carbon coated copper grids (200-mesh) were dried for 2 h.
Fourier-transformation infrared spectroscopy
FTIR spectroscopy measurements were taken on a Thermo Scientific Nicolet iS50 spectrometer equipped with a diamond attenuated total reflection (ATR) accessory. The samples were measured at room temperature (293 K) in the range of 4000–400 cm−1. Each spectrum was an average of 32 scans recorded at a resolution 4 cm−1.
Cell culture and antiproliferative assay
The human colorectal carcinoma (HCT 116), human breast carcinoma (MCF-7), and human lung carcinoma (A549) cell lines were purchased from ATCC. The human glioma (Hs683) cell line was kindly provided by prof. Gabriela Kramer-Marek from Institute of Cancer Research (London, UK). The normal human dermal fibroblasts (NHDF) were obtained from PromoCell. All of these adherent cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 12% of fetal bovine serum (Sigma) (for cancer cell lines) or 15% (for normal cell line) and a blend of standard antibiotics; 1% v/v of penicillin and streptomycin (Gibco). The cells were grown under standard conditions at 37 °C in a humidified atmosphere at 5% CO2. All cell lines were tested using PCR technique for mycoplasma contamination.
Antiproliferative assay was performed on 96-well plates (Nunc), where cells were seeded at a density of 5000 cells/well (for cancer cell lines) or 4000 cells/well (for normal cell line) and incubated at 37 °C for 24 h. Subsequently, the medium with various concentrations of the tested compounds was added and incubation was continued for the next 72 h. After incubation, the medium with the tested compounds was removed and the CellTiter 96® AQueous One Solution—MTS (Promega) solution in the medium without phenolic red was added (20µL MTS and 100µL DMEM per each well). The plates were incubated for 1 h at 37 °C and then the absorbance of the samples was measured at the 490 nm. The obtained results are expressed as a percentage of the untreated control. The IC50 values (inhibitory concentration) were determined using GraphPadPrism7. Each individual compound was tested in triplicate in a single experiment with each experiment being repeated three or four times.
Results and discussion
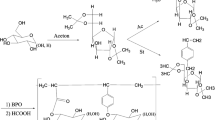
As previously mentioned, the synthesis of glycopolymers by direct polymerization of the carbohydrate-based monomers is a demanding and multi-step process. However, only in that way, full control over structural, topological, and morphological parameters of the formed polymer can be achieved. Herein, the glycomonomer was initially synthesized in the three steps starting with the commercially available β-D-glucopyranose (1) (Scheme 1). Intermediate (2) was prepared by per-acetylation of monosaccharide with yield of 93%, which was then converted to (3) by the BF3-etherate glycosidation method with 50% yield. The last step involved incorporating a methacrylic moiety into 2-bromoethyl glycoside (4), which was obtained with 68% yield. All structures were verified by NMR (See spectra in Supplementary information Figure S9-S15) and ESI MS. Next, the glycomonomer was used to obtain water-soluble star-shaped glycopolymers via Activators Generated by Electron Transfer Atom Transfer Radical Polymerization (AGET ATRP), which were then extensively tested to characterize their thermodynamical and solution properties. These investigations were also extended to star-shaped amphiphilic copolymers with precise composition and various morphologies adjusted by the content of poly(N,N-dimethylaminoethyl) methacrylate P[DMAEMA] domains. Star-like (co)polymers were synthesized via a ‘core-first’ approach using a three-functional resveratrol-based initiator previously obtained by a simple bromoacylation reaction with moderate yield of 65% (see Scheme 1). Of particular note is that the glycomonomers activity is often reduced due to their large size, which significantly lowers their polymerizability. What is more, in the present case, steric congestion was also enhanced by ionic character of monomer. With that in mind, polymerization of such kind of system should be performed using the most active catalytic agents /methodology. Hence, herein, we decided to use the AGET ATRP approach, known for its high efficiency and low sensitivity to the presence of oxygen. The ligand we used was highly active TPMA. The disadvantage of the chosen solution is, of course, a significant amount of the copper catalyst, which may significantly affect the potential biological applications. However, this catalyst can be effectively removed from the resulted polymer using dialysis in water (SpectraPor3, MWCO 3500 g/mol). It is worth noting that dialysis is commonly used to purify polymers prepared via ATRP variations or Cu-catalyzed 'click chemistry' ones [55,56,57]. Herein we adopted this methodology to eliminate copper content from the synthesized polymers below the threshold admission value given by the FDA for the food products [58]. The formation of (co)polymers was confirmed by NMR (Supporting information Figure S4, Figure S5) and SEC analysis (Supporting information Figure S1). FTIR analysis (Supporting information Figure S16) showed the presence of characteristic bands: around 1700 cm−1 for carbonyl groups from esters (both methacrylate and acetyl on sugar moieties) and amines around 1000–1200 cm−1 (both in ammonium grups in GluAmMA units and tertiary amines of DMAEMA units). The characteristics of resulted (co)polymers are outlined in Table 1.

Pathways outlining preparation of glycomonomers (a), linear (b), and star-shaped glycopolymers (c)
All AGET ATRP of glycomonomer ([glycomonomer]0/[initiator]0 = 100/1) were performed at 40 °C and with a high dilution of polymerizing system to avoid steric congestion effects and undesirable side reactions that can occur during the process. Also, relatively low conversions (10–30% glycomonomer consumption) were maintained in experiments that allowed us to synthesize well-defined star-shaped copolymers. Nevertheless, as shown in Table 1, the steric hindrance of the bulky glycomonomer units among the adjacent propagating chains strongly affects polymerization progress initiated from the resveratrol-based core. In that case, within an analogues timeframe (6 h) the conversion of glycomonomer was more than 2.5 times lower (I) compared to the system (VI) where a classical ATRP initiator, ethyl α-bromoisobutyrate was used.
As indicated by SEC traces (Supporting info, Figure S1), only one of the polymers (III) showed bimodality features, and hence, a likely lower initiation efficiency. This may be due to, for example, partial hydrolysis of the ester bonds of the initiator or the weaker control of the reaction in the synthesis of this polymer. Except for this single example, the remaining systems are characterized by very low dispersities. Thus, it can be concluded that they were synthesized in a controlled manner. Due to the difficulties related to a very weak laser signal, in the analysis of (co)polymers containing ionic and sugar groups at the same time, any attempt to determine their absolute weight was not possible. As a consequence of that accurate determination of the initiation efficiency for the tested polymerization cannot be performed and similar struggles were reported previously for ionic polymers [35, 59]. The results of molecular weights obtained from traditional SEC should therefore be treated as indicative. While there are methodologies to improve the reliability of results, such as side-chain solvolysis [60] or modification of the chromatography elution system [61], in practice, in a system similar to that studied herein, they are strongly demanding and do not warrant any advance in this matter.
Referring to the copolymerization process, it can be noted that the carbohydrate-based monomer turned out to be less-active and less-prone to incorporate into the polymer chains, which can also be attributed to steric hindrance effects (III–IV). Studies showed [62] that bulky monomers (to which we can include GluAmMA) create a steric congestion in the reactive end of growing polymer chain and thus are less-prone to participate in the process. Usually, in order to change the unfavorable kinetic conditions, it is necessary to apply additional modifications to the reaction system, e.g., the use of increased pressure or spatial restriction [63]. In contrast, the much higher conversion was observed during AGET ATRP of less sterically hindered DMAEMA from the resveratrol-based core (V). In particular, all glycopolymers were characterized by molecular weight in range Mn = 21.9–58 kg/mol, and these values in most cases, were in good agreement with the targeted ones. The discrepancy could be partially attributed to the SEC calibration using highly polar PEG/PEO standards. However, the molecular weight distributions Đ = 1.05–1.27 were all narrow and no peak attributed to star-star coupling was observed in the SEC traces (Supplementary Information), demonstrating that well-defined ionic polysaccharides were produced.
As the next step of our investigation, we characterized the thermodynamical properties of produced resveratrol-based and linear glycopolymers. Representative DSC curves obtained for the examined macromolecules are presented in the Supplementary Information (Figure S2), whereas values of the glass transition temperature, Tg, determined from these measurements are listed in Table 2.
Interestingly as observed, there is only one end’othermic event, related to the glass transition, for both homopolymer and copolymer star-shaped macromolecules, independently on the concentration of DMAEMA. This indicates that interestingly, all produced glycopolymers do not undergo any phase separation. Similar behaviors were also observed in case of other star-shaped polymers, i.e., copolymers containing methyl (meth)acrylate and (meth)acrylic acid [64] and homopolymer choline-based polyionic liquid [36]. Note that the microphase separation is often observed for graft copolymers, when the degree of incompatibility of the two different parts is high enough [65,66,67]. In such case the formed microdomains are characterized by various dynamics and multiple Tgs appear, i.e., as observed for poly(vinyl alcohol)-graft-poly(methyl methacrylate) [68]. Furthermore, it is worth noting that Tg of synthesized star-shaped copolymers decreases with an increasing concentration of DMAEMA monomers from Tg = 137 °C to Tg = −12 °C for homopolymer star-shaped of P[GluAmA] and P[DMAEMA], characterized by comparable Mn. This can be easily explained by the increased mobility of chains resulting from decrease of steric congestion and spatial separation of bulky sugar moieties by smaller dimethylammonium groups. Surprisingly, both star-shaped homopolymers are characterized by remarkably different Tg, where the difference reached ΔTg ~ 120–140 °C. Taking into account chemical structures of comonomers (see Scheme 1), one can assume that aside of the steric hindrance and mer size effects the variation of Tg in star-shaped copolymers is strictly related to the change in the strength and populations of ionic interactions prevailing between ammonium cation containing units. Ionic repulsive interactions strongly affects (induce significant increase) of the glass transition temperature of low and high molecular weight soft materials. Such a behavior was already reported for linear poly(ionic liquids) [69]. Therefore, one can suppose that along with increasing concentration of DMAEMA the number of ionic interactions gets lower leading to the depression of the Tg [70]. Nevertheless, it should be emphasized that the glucopyranose-induced variation within the examined copolymers does not lead to any heterogeneities and phase separation within them. It is also worthwhile to add that in the case of P[GluAmA] homopolymers, surprisingly, we obtained similar value of Tgs for comparable Mn independently to their topology, see Table 2. Noteworthy, the glass transition temperatures of linear and star-shaped P[GluAmA] homopolymers reached Tg = 125 °C and Tg ~ 118–131 °C, respectively. One can expect that due to the presence of multi arms in star-shaped polymers instead of one singular polymer chain of linear macromolecules some differences between studied systems should occur [71,72,73]. However, the relatively small ΔTg might, in fact, imply that in the studied Mn range the topology-induced difference are neglectable. To explain this peculiar finding one can again refer to the ionic type interactions which have dominant influence and in fact control the Tg of linear and shaped polymers. Consequently the impact of different compatibility, steric hindrance of both kind materials is removed or strongly reduced.
Further, we also investigated supramolecular self-assembly, which seems to be an important factor contributing to the enhanced conductivity of the synthesized water-soluble glycopolymers. In the case of all investigated samples, efforts were made to induce aggregates by simple dissolution in both buffered solutions of pH 7.4 and 5 as well as the most popular cell culture medium (DMEM). It is worth noting that solutions of different pH were selected in order to study the pH sensitivity of PDMAEMA domains in the star-shaped (co)polymers. The self-assembly behavior of synthesized samples was also characterized by Zeta potential (ZP) measurements performed in the distilled water. All results are collected in Table 2.
In the case of all studied samples, the most hydrophobic parts of polymers are both the aromatic resveratrol core and backbones of arms. Both dimethylaminoethyl and ionic/sugar moieties present in side chains of mers display a high affinity to an aqueous environment. Taking this into account, it can be concluded that all of the studied polymers should form aggregates or micelles in which the hydrophobic part will be composed of aromatic cores and backbones of arms. On the other hand, the hydrophilic crown should be composed of hydrophilic groups of individual units exposed to the environment.
In our research, it was observed that all samples, regardless of the medium, were characterized by a certain degree of polydispersity (Fig. 1, Figure S3 Supporting Info). In order to investigate the behavior of the polymers obtained in more detail, the results obtained for both the intensity of the scattered light and the volume and number of particles suspended in the solutions were compared. The intensity analysis revealed the presence of highly aggregated particles with sizes close to 1 µm. However, as it is known, the presence of only a few large particles indicates a signal much stronger than that of a large population of small particles [74]. For this reason, the results obtained from the volume and particle number analysis seem to be much more convincing. As expected, the particle number analysis completely ignored the presence of large aggregates, and for most samples, indicated very small micelles, of diameters not exceeding 50 nm. This analysis showed that the created superstructures reach sizes larger than 100 nm only for polymer II. Interestingly, this result was similar to that obtained from the analysis of the intensity of the scattered light. Particle volume analysis showed results intermediate to those mentioned above, but with a focus on smaller particle sizes. In our opinion, this means that the tested solutions contain particles of various sizes, including a small portion of large aggregates with diameters close to 1 µm and a much larger population of small particles.

Dynamic light scattering analysis of sugar-containing star copolymers in PBS pH 7.4 (a), AcBS pH 5 (b) and DMEM medium (c). Comparative analysis of the size of particles formed by polymers IV (d), II (e), and III (f) in different solutions
DLS volume analysis (Fig. 1) revealed the biggest aggregates for star-shaped carbohydrate-based homopolymers with average hydrodynamic radii of dh = 35–43 nm. Interestingly, for star-shaped P[GluAmMA-co-PDMAEMA] and P[DMAEMA] samples, these values were more than two times lower, reaching ∼10–17 nm and 15 nm, respectively. In addition, slight pH sensitivity was observed for all DMAEMA containing polymers. Notably, for samples with amino groups, the increase in dh in acidic medium was 6 nm for III and 3 nm for V. The smaller increase for P[DMAEMA] could be a result of incomplete protonation of the amino units due to steric crowding effect. Surprisingly, for sample IV a slight decrease of the size (2 nm) was observed, what is contrary to the expected result. Such behavior may be due to the specific packing of polymer chains, in which units having sugar moieties are more exposed. This hypothesis is confirmed by the high ZP value measured for this copolymer. Additionally, a slight increase of the size was observed for star-shaped and linear homopolymers of P[GluAmMA]. Such an effect may be due to the possible occurrence of an ion exchange reaction. The bromide anions present in the monomer can be exchanged very easily with the acetate anions present in the acidic buffer, which in turn may cause an increase in particle size.
Concerning the particle sizes in the DMEM medium solution, they were much higher for all samples compared to those obtained for the buffer pH 7.4. The reason for such behavior may be a slight acidity of the medium, as well as additional interactions with its components (e.g., amino acids). The comparison of the particle size is best illustrated by the list of histograms depending on the fraction of DMAEMA or pH of the solution (Fig. 1). There is a little tendency to aggregation for all samples measured in DMEM, which might be caused by various components of that medium. The above histograms show that the homopolymer II forms the largest particles, regardless of the pH and medium in which it is dissolved. Again, this may be related to the fact that the sugar-containing units are large and further have a stable charge, leading to repulsion between each other. Thus, the size of the entire macromolecule is larger. The homopolymer I exhibits an even larger size, which is connected to its higher molecular weight. Interestingly, despite its higher DP, linear homopolymer VI exhibits a smaller size than both star-shaped ones. This may indicate the influence of the rigid core in the structure of star-shaped polymers on the ability of these polymers to fold and form particles and aggregates.
In addition, ZP values were also determined within this work, and, as expected, they were high. This indicates a relatively low aggregation capacity of the obtained polymers, which is in-line with the obtained dh results (Table 2). Besides, ZP values estimated for synthesized herein homopolymers correspond with those obtained for several glycopolymers containing glucose, mannose, and galactose units [75]. Interestingly, the highest values of ZP were obtained for copolymer samples. This may be an effect of the specific self-assembly of these macromolecules favoring the exposure of charged sugar units, which in turn results in a higher surface charge. The literature data demonstrated a similar scenario in glycopolymers modified by the incorporation of alkaline (i.e., lysine55 or oligoethylene amine56) or acidic domains (e.g., poly(acrylic acid))53,54, where ZP values exhibited higher and much lower values, respectively, compared to carbohydrate-based homopolymers. We are aware that the small number of tested samples (3) did not allow us to determine the key relationships between polymer molecular weight, topology, composition, and ZP values. Nevertheless, it seems that a star-shaped sample of similar Mn to the linear one revealed much higher values for both size and charge (Fig. 2A). Both these values were also higher for star-shaped copolymer samples than for homopolymer ones (Fig. 2B). We assume that a specific arrangement of macromolecules could cause such phenomena, as already mentioned.

Dependencies of dh and ZP values on molecular weight (a) and fraction of DMAEMA units (b)
As complementary to the DLS studies, we also carried out transmission electron microscopy (TEM) analysis that is a suitable tool to investigate the structure of the aggregates. Images of the formed polymeric superstructures in water are shown in Fig. 3.

TEM analysis of particles formed by II (a), IV (b), I (c), III (d), VI (e), and V (f’)
Clear aggregation caused by fast water evaporation was observed for all star-shaped (co)polymers, which formed relatively regular and spherical superstructures. As can be seen in Fig. 3, homo- and copolymer samples form hollow spherical particles with obvious contrast between central and other parts that is characteristic for vesicular structures (Fig. 3A, C). Interestingly, linear homopolymer VI (Fig. 3E), in addition to forming spherical structures, exhibited the affinity to create more regular cubic structures (see Supplementary information Figure S17). Thus it may show the influence of topology on the shape of formed particles. It is particularly worth noting that the rigid resveratrol-based core most likely forces the self-organization of the polymer chains and this phenomenon contributed to the formation of spherical aggregates/polymerosomes. On the other hand, in linear glycopolymer (lack of resveratrol core) the regular ordering processes are not suppressed, what may also contribute to different way of self-assembly.
In contrast, PDMAEMA formed particles that resemble flattened spheres or red blood cells. Such a different appearance of the particles is a result of the presence of only amine groups in the polymer chains.
Additionally to the described above tests, we also carried out measurements of the electrophoretic mobility of synthesized samples in dilute solutions. We set out to probe the impact of P[DMAEMA] domains in star-shaped homo- and copolymers on their behavior and stability in suspended media. As expected, the first value is higher for copolymers and drops for both carbohydrate and amine-based homopolymers (Figure S7, Supporting Information).
Such results are connected with the surface charge, and thus, copolymers having the highest values and the best exposition of charged moieties, which always exhibit higher mobility. Noteworthy, there is no influence of MW on that value. Both star-shaped homopolymers exhibited almost the same mobility despite the difference in weight of a few thousand Daltons (Table 2). We assume that the molecular weight independence of mobility in dilute polyelectrolyte solutions could result from the formation of counterion cloud [76]. Moreover, it is also worth noting that the linear homopolymer showed the lowest electrophoretic mobility among all (co)polymers containing GluAmMA units, which suggests that the value of this physical quantity is also significantly influenced by topology.
Since many glycopolymers were obtained and applied as anti-tumor agents or drug delivery systems for anti-tumor therapies [44, 77, 78] as a final point of our studies, we conducted cytotoxicity tests on selected human cancer cell lines differing in origin and genetic landscape. The selection of the studied cell lines was significantly influenced by the popularity of the occurrence (HCT116, MCF-7), malignancy (Hs683), and frequent difficulties in the treatment of a given tumor (A549). Additionally, we conducted experiments on normal human dermal fibroblasts (NHDF), which are a popular model for testing the selectivity of new materials. It can be stressed that additional analysis of the correlation between the composition and topology of new glycopolymers and their antiproliferative properties was also performed. It was found that the studied (co)polymers showed anticancer activity against all tested cell lines, with the most significant effect noted for Hs683 line.
In the case of other cell lines, there were no differences (within experimental uncertainty) in the anticancer mode of action of the applied materials. Additionally, the results indicated that copolymers III and IV showed the highest antiproliferative activity against all cell lines that were tested. Within this group of glycopolymers, an increase in the antiproliferative effect correlates with the amount of DMAEMA moieties (Table 3, Fig. 4A). Pure saccharide-based macromolecules (x of GluAmA = 1) were characterized by reduced activity on the studied cell lines. That simply means that amine moieties in the polymer structure are mainly responsible for the cytotoxic effects of the polymers. This hypothesis was confirmed by the very high antiproliferative activity of P[DMAEMA] homopolymer (V), which showed the highest cytotoxic effect in comparison to other polymers. These results are in accordance with the literature reports, which also indicated that P[DMAEMA] exhibits a cytotoxic effect [79, 80]. Herein, one can point out that some of our polymers, particularly these containing amine moieties, revealed significant influence on the lung cell lines, which seems to be one of the most resistant cancers [81] toward the majority of the tested compounds so far.

Dependency of the IC50 parameter on the fraction of amine units in polymers (a), ZP (b), and dh (c). More readable figure without error bars is included in Supporting Information, as Figure S8
Our systematic cytotoxicity studies also revealed several quite interesting correlations worth further commenting. It was clearly seen that one of the most important characteristics influencing anti-tumor activity was the topology of the polymer. Comparison of star-shaped glycopolymers (I and II) with the linear one (VI) clearly indicate that star-shaped polymers are much more active than the linear (Table 3). This observation is in-line with the previous reports emphasizing that star topology is beneficial for anti-tumor activity on MCF-7 cell lines82. Another finding is connected to the relationship between the cytotoxic activity enhancement with increasing molecular weight of the polymer. In this context, we can recall that Glycopolymer I, with the highest molecular weight, showed a greater cytotoxic effect than glycopolymer II characterized by lower Mn. Similar results were reported in the previous studies [83].
The other interesting fact that can be deduced from the collected data is that value of IC50 drastically decreased with decreasing ZP (Fig. 4B). Consequently, P[DMAEMA] (V) characterized by the lowest ZP turned out to be the most active polymer. Importantly, no clear correlation between biological activity and size of the particles formed by studied (co)polymers (Fig. 4C) was observed. This result is quite non-intuitive since one can expect that size of the particles is a crucial step allowing or preventing diffusion of the anti-tumor agent inside the cells leading to either enhancement or reduction of their activity. However, it should be noted that in case of studied polymers of moderate molecular weight, particle sizes were rather large, making impossible penetration through the cell membrane. Therefore, we can suppose that antiproliferative features of the synthesized new glycopolymers are related to some surface effects between cells and polymers. However, identification of the molecular mechanism responsible for the enhanced toxic activity of the applied materials is yet to be clarified in the future.
The selectivity of the tested compounds is differentiated according to the cell line being investigated. In Supporting Information, we included selectivity indexes (SI) calculated based on toxicity toward normal and cancer cell lines (Table S1, Figure S6, Supporting Information). In order to take a benchmark, we compared the results to the well-known and commonly used in anticancer therapies cytostatic drug Doxorubicin. The lowest selectivity was obtained for Hs683 line, for which the highest cytotoxicity among the investigated cancer lines was also recorded. For MCF-7 and HCT 116 cell lines, SIs were comparable to Doxorubicin. However, tested compounds showed good selectivity toward A549 cell line compared to Doxorubicin (depending on the polymer tested, one to four times higher).
Hence, based on the results presented and discussed above, it can be concluded that the glycopolymers P[GluAmMA], which show the lowest cytotoxicity, may in the future be used as drug or gene carriers in anticancer therapies. Only copolymers containing amine units could be used alone as potential anti-tumor agents.
Conclusions
In summary, this paper describes the synthesis of a new ionic sugar monomer and proposes a method of their direct polymerization using the popular AGET ATRP technique. Importantly, a simple technique (dialysis) was also proposed to remove up to 98% of the copper catalyst. The use of the facile AGET ATRP method performed on resveratrol-based and classical ATRP initiators was critical in this effort, as glycopolymers of desired structural, morphological, and topological parameters were obtained. It should be noted that similar results could not be achieved when applying methods relying on the lower catalyst loading considering low activity and specific interactions dominating in the ionic monomers. Produced glycopolymers were extensively characterized to probe their thermodynamical, morphological properties, and the behavior in the aqueous solutions. Due to self-assembly properties, they formed microstructures that stability and shapes were strictly dependent on their composition and topology.
The tested materials showed anti-tumor activity, which was verified for a few selected cell lines. As it turned out, the cytotoxic effect was stronger for the copolymers containing DMAEMA units. The obtained results were compared with those obtained for the tests on healthy cells (NHDF), and it was calculated that for at least one of the lines (A549), the proposed polymers showed good selectivity. It is also worth noting that this result is better than for the model cytostatic agents commonly used in anticancer therapies—doxorubicin.
References
Garegg PJ (1992) Saccharides of biological importance: challenges and opportunities for organic synthesis. Acc Chem Res 25:575–580. https://doi.org/10.1021/ar00024a005
Erfurt K, Wandzik I, Walczak KZ et al (2014) Hydrogen-bond-rich ionic liquids as effective organocatalysts for Diels-Alder reactions. Green Chem 16:3508–3514
Erfurt K, Markiewicz M, Siewniak A et al (2020) Biodegradable surface active D-glucose based quaternary ammonium ionic liquids in the solventless synthesis of chloroprene. ACS Sustain Chem Eng 8:10911–10919. https://doi.org/10.1021/acssuschemeng.0c03239
Pramudya I, Chung H (2019) Recent progress of glycopolymer synthesis for biomedical applications. Biomater Sci 7:4848–4872. https://doi.org/10.1039/c9bm01385g
Miura Y, Hoshino Y, Seto H (2016) Glycopolymer nanobiotechnology. Chem Rev 116:1673–1692. https://doi.org/10.1021/acs.chemrev.5b00247
Sunasee R, Narain R (2013) Glycopolymers and glyco-nanoparticles in biomolecular recognition processes and vaccine development. Macromol Biosci 13:9–27. https://doi.org/10.1002/mabi.201200222
Housni A, Cai H, Liu S et al (2007) Facile preparation of glyconanoparticles and their bioconjugation to streptavidin. Langmuir 23:5056–5061. https://doi.org/10.1021/la070089n
Miura Y (2012) Design and synthesis of well-defined glycopolymers for the control of biological functionalities. Polym J 44:679–689. https://doi.org/10.1038/pj.2012.4
Slavin S, Burns J, Haddleton DM, Becer CR (2011) Synthesis of glycopolymers via click reactions. Eur Polym J 47:435–446. https://doi.org/10.1016/j.eurpolymj.2010.09.019
Otman O, Boullanger P, Drockenmuller E, Hamaide T (2010) New amphiphilic glycopolymers by click functionalization of random copolymers - Application to the colloidal stabilisation of polymer nanoparticles and their interaction with concanavalin A lectin. Beilstein J Org Chem 6:1–7. https://doi.org/10.3762/bjoc.6.58
Vinson N, Gou Y, Becer CR et al (2011) Optimised “click” synthesis of glycopolymers with mono/di- and trisaccharides. Polym Chem 2:107–113. https://doi.org/10.1039/c0py00260g
Ferji K, Venturini P, Cleymand F et al (2018) In situ glyco-nanostructure formulation via photo-polymerization induced self-assembly. Polym Chem 9:2868–2872. https://doi.org/10.1039/c8py00346g
Majoinen J, Haataja JS, Appelhans D et al (2014) Supracolloidal multivalent interactions and wrapping of dendronized glycopolymers on native cellulose nanocrystals. J Am Chem Soc 136:866–869. https://doi.org/10.1021/ja411401r
Neugebauer D, Maksym-Bȩbenek P, Mielańczyk A, Biela T (2013) Branched copolymers with biodegradable core and pendant oxirane groups. Polym Eng Sci 53:1146–1153. https://doi.org/10.1002/pen
Mielańczyk A, Skonieczna M, Neugebauer D (2017) Cellular response to star-shaped polyacids. Solution behavior and conjugation advantages. Toxicol Lett 274:42–50. https://doi.org/10.1016/j.toxlet.2017.03.022
Mielańczyk A, Neugebauer D (2015) Designing drug conjugates based on sugar decorated V-shape and star polymethacrylates: influence of composition and architecture of polymeric carrier. Bioconjug Chem 26:2303–2310. https://doi.org/10.1021/acs.bioconjchem.5b00533
Pfaff A, Shinde VS, Lu Y et al (2011) Glycopolymer-grafted polystyrene nanospheres. Macromol Biosci 11:199–210. https://doi.org/10.1002/mabi.201000324
Ladmiral V, Melia E, Haddleton DM (2004) Synthetic glycopolymers: an overview. Eur Polym J 40:431–449. https://doi.org/10.1016/j.eurpolymj.2003.10.019
Vázquez-Dorbatt V, Lee J, Lin EW, Maynard HD (2012) Synthesis of glycopolymers by controlled radical polymerization techniques and their applications. ChemBioChem 13:2478–2487. https://doi.org/10.1002/cbic.201200480
Lou S, Zhang X, Zhang M et al (2017) Preparation of a dual cored hepatoma-specific star glycopolymer nanogel via arm-first ATRP approach. Int J Nanomed 12:3653–3664. https://doi.org/10.2147/IJN.S134367
Fleet R, Van Den Dungen ETA, Klumperman B (2011) Novel glycopolymer brushes via ATRP: 1. Synthesis and characterization Macromol Chem Phys 212:2191–2208. https://doi.org/10.1002/macp.201100288
Matsumura S, Hlil AR, Lepiller C et al (2008) Stability and utility of pyridyl disulfide functionality in RAFT and conventional radical polymerizations. J Polym Sci Part A Polym Chem 46:7207–7224. https://doi.org/10.1002/pola
Piloni A, Walther A, Stenzel MH (2018) Compartmentalized nanoparticles in aqueous solution through hierarchical self-assembly of triblock glycopolymers. Polym Chem 9:4132–4142. https://doi.org/10.1039/c8py00792f
Piloni A, Wong CK, Chen F et al (2019) Surface roughness influences the protein corona formation of glycosylated nanoparticles and alter their cellular uptake. Nanoscale 11:23259–23267. https://doi.org/10.1039/c9nr06835j
Bernard J, Hao X, Davis TP et al (2006) Synthesis of various glycopolymer architectures via RAFT polymerization: from block copolymers to stars. Biomacromolecules 7:232–238. https://doi.org/10.1021/bm0506086
Stenzel MH, Zhang L, Huck WTS (2006) Temperature-responsive glycopolymer brushes synthesized via RAFT polymerization using the Z-group approach. Macromol Rapid Commun 27:1121–1126. https://doi.org/10.1002/marc.200600223
Kobayashi K, Sumitomo H, Ina Y (1985) Synthesis and functions of polystyrene derivatives having pendant oligosaccharides. Polym J 17:567–575
Ohno K, Tsujii Y, Miyamoto T et al (1998) Synthesis of a well-defined glycopolymer by nitroxide-controlled free radical polymerization. Macromolecules 31:1064–1069. https://doi.org/10.1021/ma971329g
Lu J, Zhang W, Richards SJ et al (2014) Glycopolymer-coated gold nanorods synthesised by a one pot copper(0) catalyzed tandem RAFT/click reaction. Polym Chem 5:2326–2332. https://doi.org/10.1039/c3py01526b
Dykman L, Khlebtsov N (2012) Gold nanoparticles in biomedical applications: recent advances and perspectives. Chem Soc Rev 41:2256–2282. https://doi.org/10.1039/c1cs15166e
Marradi M, Chiodo F, García I, Penadés S (2013) Glyconanoparticles as multifunctional and multimodal carbohydrate systems. Chem Soc Rev 42:4728–4745. https://doi.org/10.1039/c2cs35420a
Veerapandian M, Lim SK, Nam HM et al (2010) Glucosamine-functionalized silver glyconanoparticles: characterization and antibacterial activity. Anal Bioanal Chem 398:867–876. https://doi.org/10.1007/s00216-010-3964-5
Ei-Boubbou K, Zhu DC, Vasileiou C et al (2010) Magnetic glyco-nanoparticles: a tool to detect, differentiate, and unlock the glyco-codes of cancer via magnetic resonance imaging. J Am Chem Soc 132:4490–4499. https://doi.org/10.1021/ja100455c
El-Boubbou K, Gruden C, Huang X (2007) Magnetic glyco-nanoparticles: a unique tool for rapid pathogen detection, decontamination, and strain differentiation. J Am Chem Soc 129:13392–13393. https://doi.org/10.1021/ja076086e
Bielas R, Mielańczyk A, Siewniak A, Neugebauer D (2016) Trimethylammonium-based polymethacrylate ionic liquids with tunable hydrophilicity and charge distribution as carriers of salicylate anions. ACS Sustain Chem Eng 4:4181–4191. https://doi.org/10.1021/acssuschemeng.6b00690
Maksym P, Tarnacka M, Wolnica K et al (2018) How does the type of counterion influence the polymerization rate and thermal properties of tailored choline-based linear- and star-shaped poly(ionic liquid)s PILs? J Polym Sci Part A Polym Chem 56:2681–2691. https://doi.org/10.1002/pola.29252
Ding S, Tang H, Radosz M, Shen Y (2004) Atom transfer radical polymerization of ionic liquid 2-(1-butylimidazolium- 3-yl)ethyl methacrylate tetrafluoroborate. J Polym Sci Part A Polym Chem 42:5794–5801. https://doi.org/10.1002/pola.20423
He H, Luebke D, Nulwala H, Matyjaszewski K (2014) Synthesis of poly(ionic liquid)s by atom transfer radical polymerization with ppm of Cu catalyst. Macromolecules 47:6601–6609. https://doi.org/10.1021/ma501487u
He X, Yang W, Pei X (2008) Preparation, characterization, and tunable wettability of poly(ionic liquid) brushes via surface-initiated atom transfer radical polymerization. Macromolecules 41:4615–4621
Kobayashi A, Akaike T, Kobayashi K, Sumitomo H (1986) Enhanced adhesion and survival efficiency of liver cells in culture dishes coated with a lactose-carrying styrene homopolymer. Die Makromol Chemie, Rapid Commun 7:645–650. https://doi.org/10.1002/marc.1986.030071005
Lin HM, Behera K, Yadav M, Chiu FC (2020) Polyamide 6/poly(vinylidene fluoride) blend-based nanocomposites with enhanced rigidity: Selective localization of carbon nanotube and organoclay. Polymers (Basel) 12:184. https://doi.org/10.3390/polym12010183
Li J, Zhang Y, Cai C et al (2020) Collaborative assembly of doxorubicin and galactosyl diblock glycopolymers for targeted drug delivery of hepatocellular carcinoma. Biomater Sci 8:189–200. https://doi.org/10.1039/c9bm01604j
Pearson S, Scarano W, Stenzel MH (2012) Micelles based on gold-glycopolymer complexes as new chemotherapy drug delivery agents. Chem Commun 48:4695–4697. https://doi.org/10.1039/c2cc30510k
Omurtag Ozgen PS, Atasoy S, Zengin Kurt B et al (2020) Glycopolymer decorated multiwalled carbon nanotubes for dual targeted breast cancer therapy. J Mater Chem B 8:3123–3137. https://doi.org/10.1039/c9tb02711d
Filipová M, Bojarová P, Rodrigues Tavares M et al (2020) Glycopolymers for efficient inhibition of galectin-3: in vitro proof of efficacy using suppression of T lymphocyte apoptosis and tumor cell migration. Biomacromolecules. https://doi.org/10.1021/acs.biomac.0c00515
Song EH, Manganiello MJ, Chow YH et al (2012) In vivo targeting of alveolar macrophages via RAFT-based glycopolymers. Biomaterials 33:6889–6897. https://doi.org/10.1016/j.biomaterials.2012.06.025
Zheng Y, Zhang Y, Zhang T et al (2020) AuNSs@Glycopolymer-ConA hybrid nanoplatform for photothermal therapy of hepatoma cells. Chem Eng J 389:124460. https://doi.org/10.1016/j.cej.2020.124460
Zheng Y, Zhang Y, Wu H et al (2020) Galactose-functionalized GlycoAuNR as a photothermal conversion complex: Its binding to lectin RCA120 and hepatoma-targeting therapy. J Biomater Appl 34:1300–1314. https://doi.org/10.1177/0885328220903950
Jones MW, Otten L, Richards SJ et al (2014) Glycopolymers with secondary binding motifs mimic glycan branching and display bacterial lectin selectivity in addition to affinity. Chem Sci 5:1611–1616. https://doi.org/10.1039/c3sc52982g
Feng K, Peng L, Yu L et al (2020) Universal antifogging and antimicrobial thin coating based on dopamine-containing glycopolymers. ACS Appl Mater Interfaces 12:27632–27639. https://doi.org/10.1021/acsami.0c07949
Theodorou I, Courtin P, Sadovskaya I et al (2020) Three distinct glycosylation pathways are involved in the decoration of Lactococcus lactis cell wall glycopolymers. J Biol Chem 295:5519–5532. https://doi.org/10.1074/jbc.RA119.010844
Smitten KL, Fairbanks SD, Robertson CC et al (2020) Ruthenium based antimicrobial theranostics-using nanoscopy to identify therapeutic targets and resistance mechanisms in: Staphylococcus aureus. Chem Sci 11:70–79. https://doi.org/10.1039/c9sc04710g
Michihata N, Kaneko Y, Kasai Y et al (2013) High-yield total synthesis of (-)-strictinin through intramolecular coupling of gallates. J Org Chem 78:4319–4328. https://doi.org/10.1021/jo4003135
Quagliotto P, Viscardi G, Barolo C et al (2005) Synthesis and properties of new glucocationic surfactants: model structures for marking cationic surfactants with carbohydrates. J Org Chem 70:9857–9866. https://doi.org/10.1021/jo051579s
Song N, Ding M, Pan Z et al (2013) Construction of targeting-clickable and tumor-cleavable polyurethane nanomicelles for multifunctional intracellular drug delivery. Biomacromolecules 14:4407–4419. https://doi.org/10.1021/bm401342t
Li LY, He WD, Li J et al (2010) Shell-cross-linked micelles from PNIPAM- b -(PLL) 2 Y-shaped miktoarm star copolymer as drug carriers. Biomacromolecules 11:1882–1890. https://doi.org/10.1021/bm1004383
Zhang L, Murata H, Amitai G et al (2020) Catalytic detoxification of organophosphorus nerve agents by butyrylcholinesterase-polymer-oxime bioscavengers. Biomacromol 21:3867–3877. https://doi.org/10.1021/acs.biomac.0c00959
Koontz JL, Liggans GL, Redan BW (2020) Temperature and pH affect copper release kinetics from copper metal foil and commercial copperware to food simulants. Food Addit Contam—Part A Chem Anal Control Expo Risk Assess 37:465–477. https://doi.org/10.1080/19440049.2019.1704447
Bielas R, Mielańczyk A, Skonieczna M et al (2019) Choline supported poly(ionic liquid) graft copolymers as novel delivery systems of anionic pharmaceuticals for anti-inflammatory and anti-coagulant therapy. Sci Rep 9:14410
Neugebauer D, Sumerlin BS, Matyjaszewski K et al (2004) How dense are cylyndrical brushes grafted from a multifunctional macroinitiator? Polymer (Guildf) 45:8173–8179
He H, Zhong M, Adzima B et al (2013) A simple and universal gel permeation chromatography technique for precise molecular weight characterization of well-defined poly(ionic liquid)s. J Am Chem Soc 135:4227–4230. https://doi.org/10.1021/ja4012645
Kobatake S, Yamada B (1996) Sterically hindered elementary reactions in radical polymerization of α-ethylacrylic esters as studied by ESR spectroscopy. Polym J 28:535–542. https://doi.org/10.1295/polymj.28.535
Maksym P, Tarnacka M, Dzienia A et al (2019) Efficient metal-free strategies for polymerization of a sterically hindered ionic monomer through the application of hard confinement and high pressure. RSC Adv 9:6396–6408. https://doi.org/10.1039/C8RA09242G
Neugebauer D, Odrobińska J, Bielas R, Mielańczyk A (2016) Design of systems based on 4-armed star-shaped polyacids for indomethacin delivery. New J Chem 40:10002–10011. https://doi.org/10.1039/c6nj02346k
Theodorakis PE, Paul W, Binder K (2010) Interplay between chain collapse and microphase separation in bottle-brush polymers with two types of side chains. Macromolecules 43:5137–5148. https://doi.org/10.1021/ma100414u
Leibler L (1980) Theory of microphase separation in block copolymers. Macromolecules 13:1602–1617. https://doi.org/10.1021/ma60078a047
Thomas EL, Alward DB, Kinning DJ et al (1986) Ordered bicontinuous double-diamond structure of star block copolymers: a new equilibrium microdomain morphology. Macromolecules 19:2197–2202. https://doi.org/10.1021/ma00162a016
Yao Y, Liu L, Li H et al (1994) Glass transition and crystallization of poly(vinyl alcohol)-g-poly(methyl methacrylate) graft copolymers. Polymer (Guildf) 35:3122–3126
Zhao Q, Evans CM (2021) Effect of molecular weight on viscosity scaling and ion transport in linear polymerized ionic liquids. Macromolecules 54:3395–3404. https://doi.org/10.1021/acs.macromol.0c02801
Löwe R, Hanemann T, Zinkevich T, Hofmann A (2021) Structure–property relationship of polymerized ionic liquids for solid-state electrolyte membranes. Polymers (Basel) 13:1–12. https://doi.org/10.3390/polym13050792
Maksym P, Tarnacka M, Bernat R et al (2020) Pressure-assisted strategy for the synthesis of vinyl pyrrolidone-based macro-star photoiniferters. A route to star block copolymers. J Polym Sci 58:1393–1399. https://doi.org/10.1002/pol.20200037
Pakula T, Vlassopoulos D, Fytas G, Roovers J (1998) Structure and dynamics of melts of multiarm polymer stars. Macromolecules 31:8931–8940. https://doi.org/10.1021/ma981043r
Klein J (1986) Dynamics of entangled linear, branched, and cyclic polymers. Macromolecules 19:105–118. https://doi.org/10.1021/ma00155a018
Stetefeld J, McKenna SA, Patel TR (2016) Dynamic light scattering: a practical guide and applications in biomedical sciences. Biophys Rev 8:409–427. https://doi.org/10.1007/s12551-016-0218-6
Pranantyo D, Xu LQ, Hou Z et al (2017) Increasing bacterial affinity and cytocompatibility with four-arm star glycopolymers and antimicrobial α-polylysine. Polym Chem 8:3364–3373. https://doi.org/10.1039/c7py00441a
Muthukumar M (2017) 50th Anniversary perspective: a perspective on polyelectrolyte solutions. Macromolecules 50:9528–9560. https://doi.org/10.1021/acs.macromol.7b01929
Yang C, Gao L, Shao M et al (2020) End-functionalised glycopolymers as glycosaminoglycan mimetics inhibit HeLa cell proliferation. Polym Chem 11:4714–4722. https://doi.org/10.1039/d0py00384k
Filipová M, Bojarová P, Rodrigues Tavares M et al (2020) Glycopolymers for efficient inhibition of Galectin-3. In vitro proof of efficacy using suppression of T lymphocyte apoptosis and tumor cell migration. Biomacromolecules 21:3122–3133. https://doi.org/10.1021/acs.biomac.0c00515
Rawlinson LAB, O’Brien PJ, Brayden DJ (2010) High content analysis of cytotoxic effects of pDMAEMA on human intestinal epithelial and monocyte cultures. J Control Release 146:84–92. https://doi.org/10.1016/j.jconrel.2010.05.002
Jones RA, Poniris MH, Wilson MR (2004) PDMAEMA is internalised by endocytosis but does not physically disrupt endosomes. J Control Release 96:379–391. https://doi.org/10.1016/j.jconrel.2004.02.011
Kim ES (2016) Chemotherapy resistance in lung cancer. In: Lung cancer and personalized medicine. Advances in experimental medicine and biology. Springer International Publishing, pp 189–209
Mielańczyk A, Skonieczna M, Mielańczyk Ł, Neugebauer D (2016) In vitro evaluation of doxorubicin conjugates based on sugar core nonlinear polymethacrylates toward anticancer drug delivery. Bioconjug Chem 27:893–904. https://doi.org/10.1021/acs.bioconjchem.5b00671
Bhattacharjee S, Ershov D, Van Der GJ et al (2013) Surface charge-specific cytotoxicity and cellular uptake of tri-block copolymer nanoparticles. Nanotoxicology 7:71–84. https://doi.org/10.3109/17435390.2011.633714
Sun J, Sheng R, Luo T et al (2016) Synthesis of diblock/statistical cationic glycopolymers with pendant galactose and lysine moieties: Gene delivery application and intracellular behaviors. J Mater Chem B 4:4696–4706. https://doi.org/10.1039/c6tb00969g
Kizjakina K, Bryson JM, Grandinetti G, Reineke TM (2012) Cationic glycopolymers for the delivery of pDNA to human dermal fibroblasts and rat mesenchymal stem cells. Biomaterials 33:1851–1862. https://doi.org/10.1016/j.biomaterials.2011.10.031
Acknowledgements
R. B. and K. K. are grateful for the financial support from SONATA-BIS 2015/18/E/ST4/00320 from National Science Centre (Poland).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Handling Editor: Chris Cornelius.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The graphical abstract was created with BioRender.com.
Supplementary Information
Below is the link to the electronic supplementary material.
10853_2021_6755_MOESM1_ESM.docx
SEC traces, DSC thermograms, intensity and number DLS histograms, FTIR spectra of polymers, examples of NMR spectra of polymers and higher resolution images of linear polymer (DOCX 29983 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bielas, R., Maksym, P., Erfurt, K. et al. Sugar decorated star-shaped (co)polymers with resveratrol-based core – physicochemical and biological properties. J Mater Sci 57, 2257–2276 (2022). https://doi.org/10.1007/s10853-021-06755-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-021-06755-8