
Abstract
Thin films of Cd1−x Zn x S (CZS) were prepared by a novel spin coating/melt method from cadmium ethylxanthato [Cd(C2H5OCS2)2] and zinc ethylxanthato [Zn(C2H5OCS2)2] in x ratios of 0–0.15 and of 1. A solution of the precursor(s) in THF was spin coated onto a glass substrate and then heated at 250 °C for 1 h under N2. The thickness of the film formed can be controlled by varying the solution composition and/or the spin rate of the coating. A total metal precursor solution concentration of 50 mM was used in all cases. The films were characterized by p-XRD, SEM, EDX, ICP-AES, XPS, UV–Vis absorption spectroscopy, Raman spectroscopy and resistivity measurements. The band gaps of the films were between 2.35–2.58 and 3.75 eV (0 ≤ x ≤ 0.15 and at x = 1). The resistivity of Cd1−x Zn x S films was found to vary linearly with zinc contents, and the properties of the films suggest potential application to photovoltaics as window layers. This work is the first study to demonstrate Cd1−x Zn x S thin films by a spin coating/melt method from xanthato precursors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Thin films of Cd1−x Zn x S are interesting because their properties lie between those of ZnS and CdS [1,2,3,4,5]. It is considered a relatively useful transparent metal sulfide, for use as a window in photovoltaic cells. It has a lower absorbance in the UV with band gaps typically in excess of ~ 2.4 eV [1, 5,6,7,8]. There is also a relatively small lattice mismatches to CdS/CdTe or CIGS cells (Supporting Information; Table S6 and Figure S13) [9,10,11,12]. Such materials also establish electric fields at the heterojunction interface and may act as a buffer layer [5, 12]. The resistivity should be high enough to reduce the trap state density at the interface of the buffer and absorber layers, to improve the junction properties [10].
Thin films and nanostructures properties are sensitive to the preparation method used and synthetic route [13,14,15]. Numerous techniques have been used to grow CdS and Cd1−x Zn x S thin films including dip coating [16], electrodeposition [17], chemical vapor deposition (CVD) [18], aerosol-assisted CVD (AA-CVD) [19,20,21], chemical bath deposition (CBD) [2, 8, 22, 23], spray pyrolysis [24, 25], thermal evaporation [26], successive ionic layer and reaction (SILAR) [27], thermolysis [28, 29], electrochemical atomic layer epitaxy (ECALE) [11], sol–gel spin coating, [30, 31] doctor’s blade, [32] and others [14, 21, 33, 34]. Spin coating (SC) is a particularly useful method for the deposition of thin films [35, 36]. A melt of precursors provides a novel low-temperature route to metal chalcogenides; it has good atom efficiency in a high yield of 100% Zn metal as compared to AA-CVD or CBD [37]. In the present work, a metal chalcogenide precursor solution in tetrahydrofuran (THF) is coated onto a glass substrate. The process is repeated until the desired thickness of the chalcogen film has been formed. The final thickness of the film is determined by the precursor used, the concentration of the precursors, the solvent viscosity and the spinning parameters (speed, time). In a related CBD study, Ampong et al. [1] reported a ternary alloy with a hexagonal (wurtzite) structure and a wider band gap than a CdS film [2, 8]. The two most common phases for CdS and ZnS are the well-known hexagonal (greenockite) CdS and the cubic zincblende (sphalerite) [38].
Wurtzite stacking sequence expressed as ABABABAB pattern along the “close-packed” c-axis, whereas the stacking sequence of zincblende expressed as ABCABCABC pattern along the same [111] direction perpendicular to the sulfur planes. Moore et al. [39] determined that the two phases of ZnS are quite similar in structure and can be transformed simply by changing the stacking sequence. Cadmium sulfide is more stable as the hexagonal (wurtzite structure). Singh et al. noted that wurtzite ZnS and CdS structures have cation radii in the ratio of Zn2+/Cd2+ ≈ 0.77 [40].
In this study, we report the thermal decomposition of cadmium ethyl xanthato [Cd(C2H5OCS2)2] (1) and zinc ethylxanthato [Zn(C2H5OCS2)2] (2) as a single molecular precursor (SMPs) for deposition of CdS, ZnS and Cd1−x Zn x S thin films by spin coating. The films, ternary alloys Cd1−x Zn x S, were prepared in the range of mole percentage of Zn from 0 to 15 and at 100%. The optical, electrical and crystalline properties of the deposited films correlate linearly with the mole percentage of zinc content. The CdS and CdZnS films are hexagonal, while the pure ZnS film is a cubic film. The band gap for CdS thin film is 2.35 eV and varies linearly up to 2.55 eV at 15 mol% Zn; the ZnS band gap is 3.75 eV, agreeing well with the literature band gap values of 2.39 and 3.75 eV for CdS and ZnS, respectively, at room temperature. Ampong et al. [1] concluded that the optical band gap mirrors the structural changes in Cd-rich samples, notwithstanding overall the introduction of small quantities of Zn, which leads to a systematic shrinkage of the hexagonal lattice (up to 12% Zn).
Experimental
Materials and instrumentation
All preparations were carried out on a Schlenk line under a dry nitrogen gas stream. All chemicals were used without any purification and were from Sigma-Aldrich or Fisher. The elemental analyses (EA) were carried out with a Flash 2000 thermo-scientific elemental analyzer and using a Thermo iCap 6300 Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES). Thermogravimetric analysis (TGA) measurements were taken using a Seiko SSC/S200 model at a heating rate of 10 °C min−1 from 30 to 600 °C under nitrogen. The optical absorption spectra (UV–Vis) were recorded at room temperature with a Shimadzu double-beam UV-1800 spectrophotometer in the wavelength range of 800–300 nm at 1 nm resolution. Grazing incidence powder X-ray diffraction (p-XRD) patterns were obtained at room temperature using a Bruker D8-Advanced diffractometer with the range of 15°–65°, step size of 0.050° and a dwell time of 8 s, using [Cu Kα radiation source (λ = 1.5418 Å), 40 kV, 40 mA] with X’Pert High Score Plus software. Scanning electron microscopy (SEM) measurements were taken using a Philips XL30. The elemental composition of the samples was determined using an energy-dispersive X-ray spectroscopy (EDX) connected to the scanning electron microscope unit. Before carrying out the SEM and EDX, carbon coated of 11.5 nm thickness was applied by a Quorum Model Q150T-ES precision coating system. The sheet resistivity was determined using a Keithley 2614 type general purpose source meter employing the four-point probe method. X-ray photoelectron spectroscopy (XPS) was carried out to obtain chemical state information using a Kratos Axis Ultra DLD and the data fitted with Gaussian–Lorentzian convolutions using the Casa XPS software (www.casaxps.com).
The synthesis of cadmium ethyl xanthato [Cd(S2COCH2CH3)2] (1)
The cadmium ethylxanthato [Cd(S2COEt)2] was prepared by a method reported in the literature [3, 41]. A solution of potassium ethylxanthato KS2COC2H5 (4.9 g, 31.2 mmol) in 50 ml of distilled water was added dropwise to an aqueous solution of cadmium chloride (2.8 g, 15.6 mmol) dissolved in 25 ml of distilled water, with continuous stirring for 15 min. A creamy-white, yellowish, precipitate formed immediately, which was filtered, then washed with distilled water twice and then dried under vacuum at 25 °C for 6 h. The cadmium ethyl xanthato compound [Cd(S2COEt)2] was purified by recrystallization using ethyl acetate as a solvent with gentle heating. Yield 6.9 g (87.82%) and melting point (mp) 166 °C. EA (%), calculated (found): C, 20.32 (20.31); H, 2.84 (2.79); S, 36.09 (36.31); Cd, 31.72 (31.24). FTIR absorption signals (cm−1): 2985 (m), 2162.45 (n), 1469 (m), 1390.46 (m), 1365.43 (w), 1270.64 (m), 1179.28 (s), 1117.73 (s), 866.51 (m), 817 (m). 1H NMR chemical shift: (400 MHz, DMSO-d6) δ = 1.30 (t, J = 7.08 Hz, 3H), 4.33 (q, J = 7.08 Hz, 2H), (Supporting Information; Figure S14); 13C NMR (DMSO-d6): 14.49 (CH3), 72.76 (CH2) ppm).
The synthesis of zinc ethyl xanthato [Zn(S2COCH2CH3)2] (2)
The zinc ethylxanthato [Zn(S2COEt)2] was prepared according to a modified established method [3, 41]. A solution of potassium ethylxanthato (4.9 g, 31.2 mmol) in 50 ml of distilled water was added dropwise to an aqueous solution of zinc chloride (2.1 g, 15.6 mmol) dissolved in 25 ml of distilled water. A white precipitate formed which was filtered and then washed with distilled water twice and then dried under vacuum at 25 °C for 6 h. The compound was purified by recrystallization using acetonitrile solvent. Yield 5.8 g (81.42%) and melting point 140–143 °C. EA (%), calculated (found): C, 23.42 (23.27); H, 3.28 (3.23); S, 41.61 (41.72); Zn, 21.28 (21.02). FTIR absorption signals (cm−1): 2982 (w), 2251.31 (n), 1462.82 (m), 1386.83 (s), 1364.31 (m), 1287 (s), 1202.66 (s), 1155.21 (w), 1119.11 (s), 1029.68 (s), 999.19 (w), 861.39 (w). 1H NMR chemical shift: (400 MHz, DMSO-d6) δ = 1.26 (t, J = 7.03 Hz, 3H), 4.33 (q, J = 7.28 Hz, 2H), (Supporting Information; Figure S15); 13C NMR (DMSO-d6): 14.48 (CH3), 72.77 (CH2) ppm.
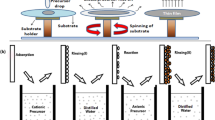
Spin coating method
Samples were prepared by a spin coating method. The precursors (1) and (2) were dissolved in THF with sonication in (0 ≤ x ≤ 0.15 and x = 1) mol ratios; 3.5 cm3 of a solution was used for each deposition with 50 mM at total metal concentration. The clear solution was dropped onto glass substrates by pipette at room temperature. The solution was spin coated on glass substrates at an angular rotation rate of 1500 rpm for 1 min. The samples were then placed in a tube furnace under a nitrogen stream at a flow rate of 20 sccm (cm3/min) at 250 °C for 1 h. All samples were cooled at room temperature for 1.5 h under a nitrogen stream.
Results and discussion
The metal ethylxanthato [Cd(C2H5OCS2)2] (1) and [Zn(C2H5OCS2)2] (2) are both readily soluble in THF. The total precursor solution concentration was 50 mM and was used to form Cd1−x Zn x S films by spin coating, followed by decomposition at 250 °C. The method offers a route to Cd1−x Zn x S films of controlled composition and thickness. This technique is suitable for semiconductor fabrication and uses easily prepared precursors.
The xanthato precursors are expected to decompose by a modified Chugaev elimination mechanism [42]. The Chugaev elimination mechanism (Fig. 1) suggests that the first step of the decomposition of xanthato compounds results in [M(S2COH)2] which leads eventually to MS.

Suggested breakdown of metal ethyl xanthato precursor at 250 °C by a modified Chugaev elimination mechanism [42]
Elemental analysis by EDX and ICP-AES of the dissolved films of Zn, Cd and S both confirmed a consistent stoichiometry. The results are summarized in Fig. 2 and tabulated in the Electronic Supporting Information ESI sheet (Supporting Information, Table S1). The plots in Fig. 2 show linearity over the range 0 ≤ x ≤ 0.125.

Plot of mol% zinc in the precursor mixture against alloy composition as determined by EDX or ICP-AES (Cd1−x Zn x S, 0 ≤ x ≤ 0.125) for thin films produced by spin coating the precursors followed by decomposition at 250 °C
Studies by p-XRD showed that thin films formed at doping ratios between (0 ≤ x ≤ 0.15) were hexagonal. The p-XRD patterns of the deposited films at different compositions are shown in Fig. 3. The peaks of CdS corresponding to the reference pattern (JCPDS, reference code: 00-041-1049) are shifted to higher angles as the zinc content increases. The sequential shifts of the p-XRD patterns confirm that the crystals are Cd1−x Zn x S and not a mixture of ZnS and CdS. The major peaks can be assigned to the (100), (002), (101), (110) and (112) reflections. All patterns were indexed, and unit cell parameters are detailed in the supplementary sheet (Supporting Information; (Equation a), Table S2, and Figures S3 and S8). The deposited films give reasonable diffraction patterning p-XRD. The structure of the ZnS film deposited by this method is cubic, ZnS cubic pattern and standard peaks shown in Fig. 3. Earlier reports have reviewed a nanostructured film with Cd0.9Zn0.1S hexagonal and Cd0.5Zn0.5S polytypic or cubic form existing in the hexagonal phase, while Cd0.1Zn0.9S shows the cubic phase [1, 2, 43].

(p-XRD) patterns of Cd1−x Zn x S films deposited in the composition range (0 ≤ x ≤ 0.15 and x = 1). The p-XRD patterns of a pure CdS; b 7.5 mol% zinc; c 12.5 mol% zinc; d 15 mol% zinc; and e pure ZnS thin films are shown. The intensities and positions of the standard peaks for CdS and ZnS are indicated. The lines capped with triangles correspond to reflections from the cubic phase of ZnS while the others as hexagonal CdS
X-ray photoelectron spectroscopy (XPS) was used to measure the CdS and ZnS films that were annealed at 250 °C. The binding energy (BE) positions of the Cd 3d, Zn 2p and S 2p core level photoelectrons are attributed to CdS and ZnS as expected [44, 45]. The XP spectra showed a single chemical species for Cd with Cd 3d 5/2 at 405.2 eV with a spin–orbit splitting of 6.8 eV as expected for the + 2 Cd state (Supporting Information; Figure S10) [44]. The Zn 2p spectrum shows a single chemical species with a 2p 3/2 peak at 1022.2 eV binding energy with a spin–orbit splitting of 23.2 eV, associated with ZnS (Supporting Information; Figure S11) [45]. The S 2p 3/2 core spectrum was measured with a peak at a binding energy position of 161.6 eV, associated with CdS and ZnS with the same electronegativity (Supporting Information; Figure S12) [44, 45].
None of the diffraction patterns shows evidence for the formation of crystalline CdO, which was also evidenced by the films being well defined, smooth and transparent yellowish in all cases.
Cubic zincblende “sphalerite” is the more stable phase at lower temperatures [14, 19, 33, 46]. Ramasamy et al. reported that the structure of ZnS is temperature dependent; the deposited films were cubic ZnS at 300 and 350 °C, while at 400 and 450 °C hexagonal ZnS with granular crystallites predominant [19]. Zhao et al. found that synthesized ZnS by colloid chemistry usually gives a stable phase of a cubic sphalerite structure at low temperatures. The wurtzitic phase typically forms at a temperature greater than 1023 °C (1296 K) [47]. Ramasamy et al. reported that the XRD pattern of ZnS films displays a cubic-to-hexagonal phase conversion above 350 °C [46]. The p-XRD of ZnS structure (Fig. 3) shows broad peaks corresponding to sphalerite. Barnes et al. [48] first suggested sulfur fugacity (fS2) is a key in determining the phase form. Low sulfur fugacity decreases the transition temperature to ca. 500 °C, while for a higher fugacity of sulfur the transition temperature moves to above 1000 °C. Wold et al. [49] determined that the growth of hexagonal ZnS thin films generally occurs at higher temperatures. The cubic ZnS film can form at high temperatures on closely lattice-matched substrates.
The surface morphology of thin films is important for applicability and has a strong influence on the optical properties of the films. Abdelhady et al. [29] found a major influence on surface morphology that happens by the precursor concentration, growth temperature and reaction time, while the optical properties of the particles are strongly dependent on the ratio of the ZnS to CdS in the feed solution. The SEM images confirm that the effect of low Zn doping content changes the microstructure of Cd1−x Zn x S thin films.
A smooth surface leads to transparency and enhanced light transmission, whereas irregular films scatter light. Significant research has been conducted, including Lee et al. [50] where it was demonstrated that a non-smooth surface could cause light scattering. SEM images of the as-deposited films reveal the presence of compact films and smooth grains, and therefore, an enhanced transmission efficiency is accomplished (Fig. 4). Kamuruzzaman et al. [51] demonstrated that increasing the annealing temperature leads to an improved surface homogeneity, and the crystallinity increases due to Cd diffusion over the surface which reduces defects and surface roughness. Increasing the annealing temperature of the films leads to the amorphous phase diminishing, since more energy is supplied for crystallite growth. Increasing the zinc content of the films improves the surface morphology and microstructure. SEM confirms that even low zinc content changes the microstructure of Cd1−x Zn x S films. Binary and ternary alloys often obey Vegard’s law, with a linear relationship between the lattice constants and composition. This is often valid for substituted solid solutions such as CdZnS [1, 52, 53]. Peter et al. found that a gradual decrease in lattice parameters is subject to the Zn content increases and that increasing Cd concentration in the film leads to the grain size increasing [53].

Secondary electron microscopy SEM images (10 kV) of Cd1−x Zn x S films deposited in the composite range (0 ≤ x ≤ 0.15 and x = 1); the images: a pure CdS; b 7.5 mol% zinc; c 10 mol% zinc; d 12.5 mol% zinc; e 15 mol% zinc; and f pure ZnS thin films. (All magnification scales are 500 nm.) EDX spectrum mapping of the elemental distributions (20 kV) of Zn, Cd and S (colored images); g Cd Lα1, h Zn Kα1 and i S Kα1 emission at 12.5 mol% zinc-doped CdS films. (All EDX scale bar corresponds to 1 μm.)
The optical properties of the films were measured over the wavelength range 300–800 nm at room temperature. The optical density at the absorption edge did not exceed 0.55 in all cases. A plot of (αhν)2 versus hν was plotted for different zinc compositions in order to estimate the optical band gap and transition type of the films from the Stern relation for near edge absorption (Supporting Information; Equation b) [54]. The band gap energy was obtained by extrapolating the linear portion of (αhν)2/n versus hν to the energy axis. A straight line at higher energies indicates a direct optical transition for the majority of II–VI compounds, and hence, the n value of 1 is assumed (Supporting Information; Equation b).
Alloying Cd1−x Zn x S allows a systematic variation in the band gap of Cd1−x Zn x S with composition [55]. The behavior is in accordance with the Burstein/Moss shift [56], which describes the shift in doped samples to higher energies of the band gap due to an increase in the carrier concentration. The band gap of CdS film is 2.35 eV. The alloy in the range (2.5–15) mol% of Zn content allows for the band gap to be tuned between 2.38 and 2.55 eV with doping (Supporting Information; Figure S16). Figure 5 shows a plot of (αhν)2 versus hν for Cd1−x Zn x S thin film at zinc concentration of 12.5 mol% Zn with a linear variation in the band gap.

A typical Tauc plot showing extrapolation of the absorption spectrum to obtain the optical band gap for 12.5 mol% zinc film deposited from (1) and (2) spin coated on a glass substrate (250 °C for 1 h) under N2 gas stream (Supporting Information; Table S4)
Band gap defects are often electrically active and introduce energy levels within the band gap. The origin of the defect states is related to cadmium and zinc vacancies, which are shallow donor levels [7, 57, 58]. Defect states change the electrical properties of the films, observed as a sharp increase in the band gap, and a single optical gap was observed. A band gap for zinc sulfide of 3.75 eV was measured as expected for bulk ZnS (Supporting Information; Figure S17) [1].
ICP-AES analysis showed that zinc content increased over the doping and that the molar changes in the zinc percentage are reflected in the changes in structural and optical properties which linearly mirror each other over the range (0 ≤ x ≤ 0.125) (Fig. 6).

Mol% zinc in precursor mixture against optical band gap and d-exp (002) (Cd1−x ZnxS, 0 ≤ x ≤ 0.15) of thin films produced by spin coating precursors (1) and (2) followed by decomposition at 250 °C for 1 h. The literature d-spacing (002) value of the CdS 2H-wurtzite structure 3.360 Å [2]
As the zinc contents increase (2.5 to 15%), the c/2 (002) reflection decreases, as may be expected due to the lattice contraction along the [001] direction (Fig. 6). Wurtzite CdS usually grows along the [001] direction (c-axis) [1]; the c-axis is energetically preferred for the growth/relaxation of the wurtzite lattice [59]. At mol% of Zn greater than 12%, the lattice contraction becomes uncorrelated [1]. The systematic variations in the lattice constants (a and c) (Supporting Information; Table S2 and Figure S3) are consistent with the substitution of tetrahedrally coordinated Zn2+ (ionic radius = 0.74 Å) into the hexagonal CdS (rCd+2 = 0.97 Å) [60, 61]. A gradual linear decrease in lattice parameters (a and c) with increasing zinc composition was observed in the range up to 12.5 mol%. Figure 7 shows the variations of unit cell volume (Å3) that are linear with the Zn content, associated with defects in the lattice [62].

Mol% zinc in precursor mixture against unit cell volume (Cd1−x Zn x S, 0 ≤ x ≤ 0.15) of thin films produced by spin coating, followed by decomposition at 250 °C for 1 h, lattice parameters were measured by p-XRD. Literature value of crystalline wurtzite-2H CdS is 99.53 Å3, Yeh et al. [38]
The structure is hexagonal with a smooth variation of the lattice parameters up to 15 mol% of Zn [1]. Linearity ceases at this level of doping and is followed by an abrupt change to cubic ZnS at Zn > 75%. The yellow color of the films lightens as the zinc content increases. The zinc-containing films are smoother and less light scattering.
A wider band gap window material is useful for applications in heterojunctions of photovoltaic cells, and a low resistivity less than 102 Ω cm is generally required [63], and here the sheet resistance is often linearly dependent on the zinc content [64, 65]. The electrical resistivity of CdS films decreases with film thickness [66] and decreases to 105 Ω cm as the temperature is increased (200–400 °C) [67]. The electrical resistivity and surface defects are strongly dependent on the deposition method [66]. In this study, the resistivity measurement was taken using the four-probe technique; the current was transferred through the outer ends and the likely drop measured across the other two inner ends. The resistivity of the films was found to increase linearly in the range of 0 ≤ x ≤ 0.15, (Fig. 8) (Supporting Information; Table S5 and Figure S9). The resistivity can be reduced considerably by increasing the film thickness, which can be attributing to the effect of crystallite size on carrier mobility. Al Kuhaimi [66] referred to the reversible correlation between the resistivity and film thickness and that this was related to the stoichiometry of the film, the effect of the crystallite size, the degree of preferred orientation and internal microstrain. Further work is therefore required to make these films applicable to photovoltaics; now the film deposition method has been well characterized.

Sheet resistance is a one-dimensional entity; as with the band gap, it is linearly dependent on zinc doping. The electrical resistivity of Cd1−x Zn x S films in CdTe devices increases in the range 1–1010 Ω cm with the zinc content (0–1) [10].
Conclusion
Thin films of CdS, Cd1−x Zn x S and ZnS have been deposited from xanthato complexes by spin coating the precursor(s) onto glass substrates, followed by thermal decomposition. Zinc-doped CdS thin films were deposited at different molar ratios of the precursors. At lower doping levels x ≤ 0.125, a linear variation in structural, optical and electrical properties is observed. The thin films had a range of morphologies, measured by SEM. The films become smoother and less scattering and are more transparent as the zinc content is increased, and the band gap increases from 2.35 eV (CdS) to 2.55 eV at 15 mol% of Zn. The lattice of CdZnS mismatch with common semiconductors lies in the range 0.03–9.5 %, (Supporting Information Table S6 and Figure S13). The method is potentially useful for window layers in CdS/CdTe and CIGS photovoltaic cells.
References
Ampong FK, Awudza JAM, Nkum RK, Boakye F, Thomas PJ, O’Brien P (2015) Ternary cadmium zinc sulphide films with high charge mobilities. Solid State Sci 40:50–54
Boyle DS, O’Brien P, Otway DJ, Robbe O (1999) Novel approach to the deposition of CdS by chemical bath deposition: the deposition of crystalline thin films of CdS from acidic baths. J Mater Chem 9:725–729
Nair PS, Radhakrishnan T, Revaprasadu N, Kolawole G, O’Brien P (2002) Cadmium ethylxanthate: a novel single-source precursor for the preparation of CdS nanoparticles. J Mater Chem 12:2722–2725
Antohe S, Ion L, Antohe VA (2003) The effect of the electron irradiation on the structural and electrical properties of A II-B VI thin polycrystalline films. Optoelectron Adv Mater 5:801–816
Mosiori C (2014) Inorganic ternary thin films; analysis of optical properties, 1st edn. Anchor Academic Publishing, Hamburg
Jana S, Maity R, Das S, Mitra MK, Chattopadhyay KK (2007) Synthesis, structural and optical characterization of nanocrystalline ternary thin films by chemical process. Phys E Low-dimens Syst Nanostruct 39:109–114
Khefacha Z (2004) Electrical and optical properties of Cd1−x Zn x S (0 ≤ x≤0.18) grown by chemical bath deposition. J Cryst Growth 260:400–409
Boyle DS, Robbe O, Halliday DP, Heinrich MR, Bayer A, O’Brien P, Otway DJ, Potter MDG (2000) A novel method for the synthesis of the ternary thin film semiconductor cadmium zinc sulfide from acidic chemical baths. J Mater Chem 10:2439–2441
Han J, Fu G, Krishnakumar V, Liao C, Jaegermann W, Besland MP (2013) Preparation and characterization of ZnS/CdS bi-layer for CdTe solar cell application. J Phys Chem Solids 74:1879–1883
Kumar SG, Rao KSRK (2014) Physics and chemistry of CdTe/CdS thin film heterojunction photovoltaic devices: fundamental and critical aspects. Energy Environ Sci 7:45–102
Colletti LP, Flowers BH, Stickney JL (1998) Formation of thin films of CdTe, CdSe, and CdS by electrochemical atomic layer epitaxy. J Electrochem Soc 145:1442–1449
Jackson P, Hariskos D, Wuerz R, Wischmann W, Powalla M (2014) Compositional investigation of potassium doped Cu(In, Ga)Se2 solar cells with efficiencies up to 20.8%. Phys Status Solidi Rapid Res Lett 8:219–222
Afzaal M, Malik MA, O’Brien P (2010) Chemical routes to chalcogenide materials as thin films or particles with critical dimensions with the order of nanometres. J Mater Chem 20:4031–4040
Ramasamy K, Malik MA, Revaprasadu N, O’Brien P (2013) Routes to nanostructured inorganic materials with potential for solar energy applications. Chem Mater 25:3551–3569
Matthews PD, McNaughter PD, Lewis DJ, O’Brien P (2017) Shining a light on transition metal chalcogenides for sustainable photovoltaics. Chem Sci 8:4177–4187
Karanjai MK, Dasgupta D (1987) Preparation and study of sulphide thin films deposited by the dip technique. Thin Solid Films 155:309–315
Mammadov MN, Sh Aliyev A, Elrouby M (2012) Electrodeposition of cadmium sulfide. Int J Thin Film Sci Technol 1:43–53
Barreca D, Gasparotto A, Maragno C, Seraglia R, Tondello E, Venzo A, Krishnan V, Bertagnolli H (2005) Cadmium O-alkylxanthates as CVD precursors of CdS: a chemical characterization. Appl Organomet Chem 19:59–67
Ramasamy K, Malik MA, Helliwell M, Raftery J, O’Brien P (2011) Thio-and dithio-biuret precursors for zinc sulfide, cadmium sulfide, and zinc cadmium sulfide thin films. Chem Mater 23:1471–1481
Wattoo MHS, Tirmizi SA, Quddos A, Khan MB, Wattoo FH, Wadood A, Ghangro AB (2011) Aerosol-assisted chemical vapor deposition of thin films of cadmium sulfide and zinc sulfide prepared from bis(dibutyldithiocarbamato)metal complexes. Arab J Sci Eng 36:565–571
Memon AA, Dilshad M, Revaprasadu N, Malik MA, Raftery J, Akhtar J (2015) Deposition of cadmium sulfide and zinc sulfide thin films by aerosol-assisted chemical vapors from molecular precursors. Turk J Chem 39:169–178
Alexander JN, Higashiya S, Caskey D, Efstathiadis H, Haldar P (2014) Deposition and characterization of cadmium sulfide (CdS) by chemical bath deposition using an alternative chemistry cadmium precursor. Sol Energy Mater Sol Cells 125:47–53
Ouachtari F (2011) Influence of bath temperature, deposition time and S/Cd ratio on the structure, surface morphology, chemical composition and optical properties of CdS thin films elaborated by chemical bath deposition. J Mod Phys 2:1073–1082
Yadav AA, Barote MA, Masumdar EU (2010) Studies on nanocrystalline cadmium sulphide (CdS) thin films deposited by spray pyrolysis. Solid State Sci 12:1173–1177
Yadav AA, Masumdar EU (2011) Photoelectrochemical investigations of cadmium sulphide (CdS) thin film electrodes prepared by spray pyrolysis. J Alloys Compd 509:5394–5399
Sahay PP, Nath RK, Tewari S (2007) Optical properties of thermally evaporated CdS thin films. Cryst Res Technol 42:275–280
Pathan HM, Lokhande CD (2004) Deposition of metal chalcogenide thin films by successive ionic layer adsorption and reaction (SILAR) method. Bull Mater Sci 27:85–111
Osakada K, Yamamoto T (1987) Formation of ZnS and CdS. J Chem Soc 1117:1117–1118
Abdelhady AL, Malik MA, O’Brien P (2014) CdS and Zn × Cd1−x S Nanoparticles from zinc and cadmium thiobiuret complexes colloidal synthesis of ZnS. J Inorg Organomet Polym Mater 24:226–240
Lahewil ASZ, Al-Douri Y, Hashim U, Ahmed NM (2013) Structural, analysis and optical studies of cadmium sulfide nanostructured. Procedia Eng 53:217–224
Lahewil ASZ, Al-Douri Y, Hashim U, Ahmed NM (2013) Structural and morphological studies of cadmium sulfide nanostructures. Adv Mater Res 795:228–232
De Kergommeaux A, Fiore A, Faure-Vincent J, Pron A, Reiss P (2013) Colloidal CuInSe2 nanocrystals thin films of low surface roughness. Adv Nat Sci Nanosci Nanotechnol 4:1–6
Ramasamy K, Malik MA, O’Brien P, Raftery J (2010) Metal complexes of thiobiurets and dithiobiurets: novel single source precursors for metal sulfide thin film nanostructures. Dalt Trans 39:1460–1463
Thomas PJ, Stansfield GL, Komba N et al (2015) Growth of nanocrystalline thin films of metal sulfides [CdS, ZnS, CuS and PbS] at the water–oil interface. RSC Adv 5:62291–62299
Hall DB, Underhill P, Torkelson JM (1998) Spin coating of thin and ultrathin polymer films. Polym Eng Sci 38:2039–2045
Lewis EA, McNaughter PD, Yin Z, Chen Y, Brent JR, Saah SA, Raftery J, Awudza JAM, Malik MA, O’Brien P (2015) In situ synthesis of PbS nanocrystals in polymer thin films from lead (II) xanthate and dithiocarbamate complexes: evidence for size and morphology control. Chem Mater 27:2127–2136
McNaughter PD, Saah SA, Akhtar M, Abdulwahab K, Malik MA, Raftery J, Awudza JAM, O’Brien P (2016) The effect of alkyl chain length on the structure of lead (II) xanthates and their decomposition to PbS in melt reactions. Dalton Trans 45:16345–16353
Yeh CY, Lu ZW, Froyen S, Zunger A (1992) Zinc-blendewurtzite polytypism in semiconductors. Phys Rev B 46:10086–10097
Moore D, Wang ZL (2006) Growth of anisotropic one-dimensional ZnS nanostructures. J Mater Chem 16:3898–3905
Singh VP, Singh S (1976) Some physical properties of ZnCdS solid solutions. Czechoslov J Phys 26:1161–1166
Rohovec J, Toušková J, Toušek J, Schauer F, Kuvritka I (2011) New cadmium sulfide nanomaterial for heterogeneous organic photovoltaic cells. In: World Renewable Energy Congress, Sweden, pp 2815–2822
Velez E, Quijano J, Notario R, Murillo J, Ramirez JF (2008) Computational study of the mechanism of thermal decomposition of xanthates in the gas phase (the Chugaev reaction). J Phys Org Chem 21:797–807
Garaje SN, Apte SK, Naik SD, Ambekar JD, Sonawane RS, Kulkarni MV, Vinu A, Kale BB (2013) Template-Free Synthesis of Nanostructured Cd × Zn1−x S with Tunable Band Structure for H2 production and organic dye degradation using solar light. Environ Sci Technol 47:6664–6672
Marychurch M, Morris GC (1985) X-ray photoelectron spectra of crystal and thin film cadmium sulphide. Surf Sci 154:251–254
Laajalehto K, Kartio I, Nowak P (1994) XPS study of clean metal sulfide surfaces. Appl Surf Sci 81:11–15
Ramasamy K, Malik MA, O’Brien P, Raftery J (2008) ZnS, CdS and Zn x Cd1−x S thin films from Zn (II) and Cd (II) complexes of 1, 1, 5, 5-tetramethyl-2-4-dithiobiuret as single molecular precursors. Mater Res Soc Proc 4:1145–1151
Zhao Y, Zhang Y, Zhu H, Hadjipanayis GC, Xiao JQ (2004) Low-temperature synthesis of hexagonal (wurtzite) ZnS nanocrystals. J Am Chem Soc 126:6874–6875
Scott SD, Barnes HL (1972) Sphalerite-wurtzite equilibria and stoichiometry. Geochim Cosmochim Acta 36:1275–1295
Pike RD, Cui H, Kershaw R, Dwight K, Wold A, Blanton TN, Wernberg AA, Gysling HJ (1993) Preparation of zinc sulfide thin films by ultrasonic spray pyrolsis from bis (diethyldithiocarbamato) zinc (II). Thin Solid Films 224:221–226
Lee K-T, Guo LJ, Park HJ (2016) Neutral- and multi-colored semitransparent perovskite solar cells. Molecules 21:475–496
Kamuruzzaman M (2014) Synthesis and characterization of Zn1−x Cd x S thin films prepared by the spray pyrolysis technique. Asian J Appl Sci 7:607–620
Hotje U, Rose C, Binnewies M (2003) Lattice constants and molar volume in the system ZnS, ZnSe, CdS, CdSe. Solid State Sci 5:1259–1262
Peter AJ, Lee CW (2012) Electronic and optical properties of CdS/CdZnS nanocrystals. Chin Phys B 21:87302
Kassim A, WeeTee T (2010) Preparation and characterization of iron sulphide thin films by chemical bath deposition method. Indo J Chem 10:8–11
Dzhafarov TD, Ongul F, Karabay I (2006) Formation of CdZnS thin films by Zn diffusion. J Phys D Appl Phys 39:3221–3225
Sarkar A, Ghosh S, Chaudhuri S, Pal AK (1991) Studies on electron transport properties and the Burstein-Moss shift in indium-doped ZnO films. Thin Solid Films 204:255–264
Chavhan SD, Senthilarasu S, Lee S-H (2008) Annealing effect on the structural and optical properties of a Cd1−x Zn x S thin film for photovoltaic applications. Appl Surf Sci 254:4539–4545
Sebastian PJ, Narvaez J (1996) Photosensitive ZnCdS nanoparticles in a CdS matrix formed by high temperature sintering of ZnS and CdCl2 in argon. Thin Solid Films 287:130–133
Lee Y, Kim S, Kim HS et al (2017) Highly luminescent blue-emitting CdZnS/ZnS nanorods having electric-field-induced fluorescence switching properties. J Mater Chem C 5:2098–2106
Shannon RD (1976) Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr Sect A 32:751–767
Xing C, Zhang Y, Yan W, Guo L (2006) Band structure-controlled solid solution of Cd1−x Zn x S photocatalyst for hydrogen production by water splitting. Int J Hydrog Energy 31:2018–2024
Verma KC, Kotnala RK (2016) Realizing ferromagnetic ordering in SnO2 and ZnO nanostructures with Fe Co, Ce ions. Phys Chem Chem Phys 18:17565–17574
Chynoweth TA, Bube RH (1980) Electrical transport in ZnCdS films deposited by spray pyrolysis. J Appl Phys 51:1844–1846
Al Kuhaimi SA, Tulbah Z (2000) Structural, compositional, optical, and electrical properties of solution-grown Zn x Cd1−x S films. J Electrochem Soc 147:214–218
Rajathi S, Subramanian NS, Ramanathan K, Senthamizhselvi M (2013) Influence of deposition temperature on the structural and optoelectronic properties of CdZnS thin films. Adv Mater Res 699:606–611
Al Kuhaimi SA (1998) Influence of preparation technique on the structural, optical and electrical properties of polycrystalline CdS films. Vacuum 51:349–355
Tepantlan CS (2008) Structural, optical and electrical properties of CdS thin films obtained by spray pyrolysis. Rev Mex Fis 54:112–117
Acknowledgements
The authors grateful to the Ministry of Higher Education and Scientific Research of Iraq (MOHESR-IQ) for funding AB, Grant # MO/2708, the Iraqi Culture Attaché-London for the financial support A. Bakly (S1743) also thanks the College of Materials Engineering CME at the University of Babylon UOB, Iraq for granting leave for study at the University of Manchester, UK. Some of the specialist equipment used in this study was provided by the Engineering and Physical Sciences Research Council UK (Core Capability in Chemistry, EPSRC Grant # EP/K039547/1).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
10853_2017_1872_MOESM1_ESM.docx
Characterization of CZS thin films produced by spin coating at 250 °C, specifics to chemical synthesis, experimental technique, and more analytical data (EDX, ICP-AES, p-XRD), band gap, lattice parameters, d-space, Raman, and resistivity) (DOCX 1553 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bakly, A.A.K., Spencer, B.F. & O’Brien, P. The deposition of thin films of cadmium zinc sulfide Cd1−x Zn x S at 250 °C from spin-coated xanthato complexes: a potential route to window layers for photovoltaic cells. J Mater Sci 53, 4360–4370 (2018). https://doi.org/10.1007/s10853-017-1872-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-017-1872-1