
Abstract
Here we report on the host behaviour of compounds N, N’-bis(9-phenyl-9-xanthenyl)propane-1,3-diamine (H1) and N, N’-bis(9-phenyl-9-xanthenyl)butane-1,4-diamine (H2) in the presence of potential guest species cyclohexanone (CYC) and 2-, 3- and 4-methylcyclohexanone (2MeCYC, 3MeCYC and 4MeCYC). H1 only formed a complex with CYC, whilst all four guest solvents were enclathrated by H2. Thermal analyses in conjunction with SCXRD experiments revealed that more energy was required to remove guest species from the crystals of their complexes when they were housed in discrete cavities compared with guest molecules retained in channels. Only in H1·CYC was identified an intramolecular (host)N‒H···N(host) hydrogen bond, while complexes H2·2(CYC), H2·2(3MeCYC) and H2·4MeCYC all experienced strong (host)N‒H···O(guest) hydrogen bonds which assisted in retention of the guests in the complexes; this interaction type was absent in both H1·CYC and H2·2(2MeCYC). Hirshfeld surface analyses demonstrated that the amounts of (guest)O···H(host) interatomic interactions were comparable and ranged between 11.1 and 13.9%. Guest competition experiments showed that H2 possessed an affinity for, more usually, 3MeCYC, despite the complex H2·2(3MeCYC) being the least thermally stable one. Finally, it was established that H1 and H2 would not be appropriate host compounds for separations of mixed cyclohexanones through supramolecular chemistry strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cyclohexanone (CYC) and its methylated derivatives, 2-methylcyclohexanone (2MeCYC), 3-methylcyclohexanone (3MeCYC) and 4-methylcyclohexanone (4MeCYC) (Scheme 1), may be prepared through the catalytic hydrogenation of cresols, phenols and guaiacol with Pd/C as the catalyst [1,2,3]. Naturally, if the cresol used in this process is not pure (which may often be the case, given the difficulty of purifying the cresols, owing to boiling point similarities), a mixture of these cycloalkanones would result. In many instances, each cyclohexanone has unique chemical functions in synthetic processes. As examples, CYC serves as an important starting material in the synthesis towards caprolactam, which is required to form nylon 6 [4], while CYC and 2MeCYC have been derivatized to form Schiff base complexes that have antifungal activity [5,6,7]; 3MeCYC, on the other hand, has been modified into a cyclohexanone-based chalcone crystal for nonlinear optical applications [8], and 2,6-bis(p-azidobenzylidene)-4-methylcyclohexanone, derived from 4MeCYC, is a useful photosensitizer in photographic processes [9]. Additionally, 4MeCYC (and 4-tert-butylcyclohexanone) have been employed in the production of photoactive linear and hyperbranched polyesters [10].
The isolation of pure MeCYC isomers from their mixtures through fractional distillation processes is not trivial and remains a significant challenge to the chemical industry. This is as a result of their narrow boiling point range (162‒171\(\:\:^\circ\:\)C) [11]. Even unsubstituted CYC has a comparable boiling point to these isomers (156 °C). While chromatographic applications have been reported for the separation of these cyclohexanone mixtures [12], new and improved separation technologies remain extremely attractive.
Alternative separatory protocols for the mixed cyclohexanones may include supramolecular chemistry and, more specifically, the host-guest chemistry field of science. Efficient separations and purifications through this strategy would require that the host compound possesses an enhanced selectivity for one of the cyclohexanones present in the mixture, and this may be affected by the noncovalent interactions present between the host and guest molecules [13,14,15,16]. In fact, some work that focussed on exactly this separation/purification challenge has already been reported in the literature [17,18,19,20]. In those reports, the host compounds were derived from inexpensive tartaric acid or from xanthone and thioxanthone, and demonstrated selectivity when crystallized from such mixtures.
In the present investigation, the host compounds N, N’-bis(9-phenyl-9-xanthenyl)propane-1,3-diamine (H1) and N, N’-bis(9-phenyl-9-xanthenyl)butane-1,4-diamine (H2) were assessed for their host potential for the four cyclohexanones as well as their separatory abilities of mixtures of these through host-guest chemistry principles (Scheme 1). Crystals of any successfully formed complexes with suitable crystal quality were analysed by means of single crystal X-ray diffraction experiments to understand the mode of guest retention as well as thermal analysis in order to ascertain the relative thermal stabilities of each of the complexes. While these two host compounds have been reported to possess selectivity in C8H10 isomers on a prior occasion [21], they have not been presented with cyclohexanone and methylcyclohexanone mixtures before, and we report on all of these findings herein.

Structures of the host systems and guest solvents employed in this investigation; the boiling points of CYC, 2MeCYC, 3MeCYC and 4MeCYC are 156, 162‒163, 169‒170 and 169–171 °C, respectively [11]
Experimental
Materials
All required starting materials/guest solvents were purchased from Merck (South Africa), and these were used without further purification.
1H-NMR spectroscopy
The 1H-NMR experiments were carried using a Bruker Ultrashield Plus 400 MHz spectrometer; CDCl3 was the applicable deuterated solvent. Topspin software was used to analyse the data.
SCXRD analysis
Single crystal X-ray diffraction experiments were carried out by means of a Bruker Kappa Apex II diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The X-ray data were collected and analysed by means of program APEXII [22]. Cell refinement and data reduction were conducted using the SAINT program, and numerical absorption corrections were achieved with SADABS. The structure of each crystal was solved with SHELXT-2018/2 [23] and refinement was by means of the least-squares procedure in SHELXL-2018/3 [24], with SHELXLE [25] as the graphical interface. All atoms, excluding hydrogen, were refined anisotropically, while all carbon- and oxygen-bound hydrogen atoms were inserted in idealized geometrical positions in a riding model; nitrogen-bound hydrogen atoms were found on the difference Fourier map and were allowed to refine freely. The crystal structures of complexes H1·CYC, H2·2(CYC), H2·2(2MeCYC), H2·2(3MeCYC) and H2·4MeCYC were deposited at the Cambridge Crystallographic Data Centre (CCDC) and their CCDC numbers are 2,340,021‒2,340,025. Finally, program Mercury [26] was used to prepare all figures pertaining to the SCXRD discussion, such as the unit cells, the host-guest packing motif and the noncovalent interactions present in each of the single solvent complexes. Furthermore, this program facilitated the construction of void diagrams which are also provided in this section: in order to prepare these figures, the guest molecules were deleted from the packing calculations which resulted in voids forming in the molecular packing, and these empty spaces were analysed by means of a probe with a 1.2 Å radius. For the twinned crystal 2,340,021 (H1·CYC), the twin domains were identified and indexed with APEX 5 and scaled with TWINABS version 2021/1. The HKLF4 file was used to solve and refined the data, and then further refinement was carried out on the HKLF5 file. The twin ratio obtained was 0.44382.
Gas chromatography
An Agilent 7890 A gas chromatograph (GC) coupled to an Agilent 5975 C VL mass spectrometer (GC-MS) equipped with an Agilent J&W Cyclosil-B column was employed for GC analyses of the mixed complexes produced in this work. The method involved an initial temperature of 50 °C (with no hold time here), followed by a heating ramp rate of 5 °C∙min‒1 until 60 °C was reached. Subsequently, the instrument was heated to 80 °C at a heating rate of 5 °C∙min‒1, with a hold time of 5 min at this temperature. Finally, a heating rate of 5 °C∙min‒1 was implemented until 105 °C was reached. The split ratio and flow rate were 1:50 and 1.5mL·min‒1, and the carrier gas was nitrogen. Alternatively, a Young Lin YL6500 GC, equipped with the same column, coupled to a flame ionization detector, was used. The method involved an initial temperature of 60 °C with no hold time, followed by heating to 80 °C using a 5 °C·min‒1 heating rate, followed by a 5 min hold time at this temperature. Thereafter, a heating rate of 5 °C∙min‒1 was applied to reach 120 °C (with no hold time). The split ratio and flow rate were 1:50 and 1.5 mL·min‒1, respectively. The carrier gas was, once again, nitrogen.
Thermal analysis
Thermal experiments were conducted on all single solvent complexes prepared in this work by employing a Perkin Elmer STA6000 simultaneous thermal analyser. The complexes were first isolated from their solutions using vacuum filtration and, still under suction, these solids were washed with low boiling petroleum ether (b.p. 40–60 °C) and then padded dry in folded filter paper. Data analysis was by means of Perkin Elmer Pyris 13 thermal analysis software. The purge gas was high purity nitrogen and the samples were heated from approximately 40 to 400 °C at a heating rate of 10 °C·min‒1. Samples were placed in ceramic pans with an empty pan serving as the reference.
Synthesis of host compounds H1 and H2
Host compounds H1 and H2 were synthesized according to a previous report [21].
Host crystallization experiments from each of the cyclohexanones
The host compounds were crystallized independently from each of the cyclohexanones in order to determine whether they possessed host ability for these cyclic ketones. To this end, the host compound (H1 and H2, 0.05 mmol) was dissolved in an excess of the potential guest solvent in glass vials. Mild heat (a hot water bath) was used to ensure complete host dissolution. The vials were left open to the ambient conditions which assisted with the crystallization process. The crystals were collected under suction and washed with petroleum ether (b.p. 40–60 °C) or methanol, also under suction. The solids were then analysed by means of 1H-NMR experiments which revealed whether inclusion had been successful, in which case the host: guest (H: G) ratios were calculated by integrating the areas under applicable host and guest resonance signals.
Host crystallization experiments from equimolar mixed cyclohexanones
Guest solutions were prepared by mixing the cyclohexanone guest solvents in equimolar proportions (7mmol combined amount for H1 and H2) so that every possible guest combination was considered. The host compound (H1 and H2, 0.07 mmol) was then dissolved in these guest solutions, and the vials were closed and stored in a cold room with a temperature setting of 4 °C. These conditions facilitated the crystallization process, and the crystals were isolated and treated in an identical fashion as those emanating from the single solvents. The guest compounds in the mixed complexes that formed in this way were quantified by means of GC analyses, and these experiments were performed in duplicate in order to determine whether the results obtained were replicable. As a consequence, percentage estimated standard deviations (%e.s.d.s) were calculated in each instance and these are also provided here.
Host crystallization experiments from non-equimolar binary mixed cyclohexanones
Binary solutions of the cyclohexanones (Guest A and Guest B, GA and GB) were prepared in which the GA:GB ratios were varied between 20:80 and 80:20 molar amounts. The host compounds H1 and H2 were then crystallized from each solution, and the crystals isolated and treated as described in the equimolar guest experiments. (Host and combined guest molar amounts were also the same.) The guest compounds present in these crystals were, once more, quantified by means of GC analyses. Subsequently, selectivity profiles were constructed by plotting the amount of GA or GB in the crystals (Z) against this amount in the original solution (X), according to the equation by Pivovar and coworkers, K = ZA/ZB × XB/XA; here, XA + XB = 1 [27]. These profiles provide a visual manner in which to observe the host selectivity behaviour in these experimental conditions. When K = 1, the host is not selective for either guest species present, and this is represented by the straight diagonal lines inserted into each of these plots.
Results and discussion
Host crystallization experiments from each of the cyclohexanones
Table 1 summarises the results obtained from 1H-NMR experiments on the solids formed when H1 and H2 were crystallized from each of CYC, 2MeCYC, 3MeCYC and 4MeCYC (Figures S1a‒e in the Supplementary Information (SI)).
From the results contained in Table 1, H1 formed a complex with CYC only (H: G 1:1), while the crystallization experiments involving the MeCYC isomers resulted in the formation of gels, and no crystallization occurred. On the other hand, all of the cyclohexanone solvents were enclathrated by H2, and the H: G ratios were 1:1 for H2·2MeCYC and H2·4MeCYC, and 1:2 for the CYC and 3MeCYC-containing inclusion compounds.
SCXRD analyses
The relevant crystallographic data of the five novel complexes produced in this work after SCXRD analyses are summarized in Table 2. Complex H1·CYC crystallized in the orthorhombic crystal system and space group Pnma, while all of the complexes with H2 crystallized in the triclinic system and space group P‒1. Note that the crystal selected for SCXRD analysis on the 2MeCYC-containing inclusion complex with H2 had a H: G ratio of 1:2 and not 1:1 (as obtained from 1H-NMR analysis).
The guest species in H1·CYC experienced disorder around a mirror plane whilst the nitrogen-bound host hydrogen atoms were also disordered. Furthermore, the guest in H2·2(2MeCYC) was disordered and there were other minor disorder orientations that could not be modelled, and this resulted in a somewhat elevated wR2 value (0.1953, Table 2). Moreover, in the 3MeCYC molecule in H2·2(3MeCYC) was observed disorder between two different inversion conformations; finally, the guest in H2·4MeCYC experienced disorder around an inversion centre whilst the host nitrogen-bound H atoms in this complex were also disordered.
A unique host packing was observed in each of the complexes H1·CYC and H2·4MeCYC, whilst in H2·2(CYC), H2·2(2MeCYC) and H2·2(3MeCYC), this packing was isostructural. These observations were confirmed by deleting the guest molecules from the packing calculations using program Mercury [26], and subsequently generating the powder X-ray diffraction (PXRD) patterns (Supporting Information (SI), Figure S2); these patterns corresponded extremely closely (while that for H2·4MeCYC differed, also in Figure S2).
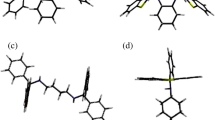
Figure 1a‒c illustrate the unit cell (left) and void (right) diagrams for these five complexes (Fig. 1b for H2·2(CYC) also represents H2·2(2MeCYC) and H2·2(3MeCYC) owing to their shared host packing motifs). Figure 1a and c demonstrate that in H1·CYC and H2·4MeCYC, the guest species were accommodated in discrete cages, while those in H2·2(CYC), H2·2(2MeCYC) and H2·2(3MeCYC) (Fig. 1b) were all housed in channels with varying degrees of constriction.

Unit cell and host-guest packing (left) and void (right) diagrams for (a) H1·CYC [100], (b) H2·2(CYC) [001] (representing also H2·2(2MeCYC) and H2·2(3MeCYC)) and (c) H2·4MeCYC [100]. All host species are represented in stick form while guest molecules are in spacefill representation
The three key features of these tricyclic fused host systems are the geometries of the two xanthenyl moieties with respect to one another, the two free phenyl rings and, finally, the diamino linker. Figure 2 illustrates the planes (which were calculated using program Mercury [26]) between the two xanthenyl moieties in H2·2(2CYC) (left) as well as the two free phenyl rings in H1·CYC (right) (guests have been removed for clarity) as representative examples. These planes were calculated in an identical fashion for each of the five complexes (of H1 and H2), and their relevant features are summarised in Table 3. Remarkably, the relationships between these planes in H2, in its four complexes, was identical: the calculated planes of both xanthenyl moieties as well as the two free phenyl rings were always exactly parallel (180°). In the case of H1·CYC, on the other hand, the planes between the two xanthenyl moieties and the two free phenyl rings were 69.98 and 74.55°, respectively.

Calculated planes of the two xanthenyl moieties in H2·2(2CYC) (left) and the two free phenyl rings in H1·CYC (right)
The geometrical relationship between the two nitrogen atoms of the diamino linker and the geometry of the linker itself were subsequently explored. This was achieved by superimposing the CH2 carbon atoms directly bonded to each nitrogen atom in the linker. When sighting along this view, the nitrogen atoms in H1·CYC were “syn-periplanar” with respect to one another. Furthermore, both the free aromatic moieties were on the same side of the molecule. For the complexes with H2, the nitrogen atoms were all “anti-periplanar” with respect to one another. Stereoviews of these geometries were prepared using X-Seed [28] and POV-Ray [29], and these are provided in Fig. 3 (H1·CYC) and 4a‒d (for each of the complexes of H2). Visually, the geometry of the diamino linker of the host molecule (H2) in all four complexes (H2·2(CYC), H2·2(2MeCYC), H2·2(3MeCYC) and H2·4MeCYC) is consistent (Fig. 4a‒d), where the N‒C‒C‒C‒C‒N unit is in a zig-zag orientation. Moreover, the free aromatic moieties are positioned on either side of the molecule in these complexes. This linker in H1·CYC adopts a more folded conformation (Fig. 3).

Stereoview demonstrating the geometry of H1 in H1·CYC; for clarity, the guest molecules were deleted; the host compound is displayed in stick form

Stereoviews showing the geometry of H2 in (a) H2·2(CYC), (b) H2·2(2MeCYC), (c) H2·2(3MeCYC) and (d) H2·4MeCYC; again, the guest species were deleted for clarity, and the host molecules are represented in stick form
We subsequently considered the noncovalent interactions present in these complexes.
Several (guest)C–H···π(host) contacts were observed in the complexes with H1 and H2. In the case of H1·CYC, two interactions of this type were identified, occurring between the hydrogen atoms of one of the disorder guest components and the free aromatic ring of the host molecule. Their C···π distances were 3.581(19) Å and 3.752(18) Å, respectively. In the complex H2·2(2MeCYC), a pair of interactions of this type was also identified but involving a methyl hydrogen atom of both guest disorder components and an aromatic ring of the xanthenyl moiety; these interaction distances (C···π) were 3.847(8) and 3.506(13) Å (Fig. 5, left, only one guest disorder component is shown). A single contact of this type was also identified in H2·2(3MeCYC), but which involved a guest cyclohexyl hydrogen atom and an aromatic ring of the host xanthenyl moiety (3.781(4) Å, Fig. 5, right). Finally, complexes H2·2(CYC) and H2·4MeCYC were not involved in interactions of this kind ((guest)C\(\:-\)H···π(host)).

Intermolecular (guest)C–H···π(host) interactions present in H2·2(2MeCYC) (left, only one guest disorder component is shown) and H2·2(3MeCYC) (right); both host and guest species are displayed in capped stick form
The complexes of both H1 and H2 experienced both classical and non-classical hydrogen bonding interactions (Table 4). In H1·CYC, the nitrogen atoms of the diamino linker were orientated in such a manner so as to facilitate a classical intramolecular (host)N\(\:-\)H···N(host) hydrogen bonding contact (the host molecule was thus not involved in a classical hydrogen bond with the CYC guest species). The interaction parameters were 2.38 Å (H···N) and 2.975(2) Å (N···N), with a small 123° (N\(\:-\)H···N) bond angle. Furthermore, a non-classical intermolecular (host)C\(\:-\)H···O(host) interaction was also detected in this complex that measured 3.387(3) Å (C···O). These interactions were unique to H1·CYC. In the complexes with H2 were identified strong classical intermolecular (host)N\(\:-\)H···O(guest) hydrogen bonding, with an exception noted in H2·2(2MeCYC) (in this complex, the distance between the involved atoms was too long to be considered significant [30,31,32]). Their H···O distances ranged between 2.38(2) and 2.48(2) Å with associated angles between 170.5(17) and 175.0(15)°. The N···O distances measured between 3.272(11) Å and 3.387(14) Å. Illustrations of these hydrogen bonds are presented in Fig. 6 for H2·2(CYC) (left) and H2·2(3MeCYC) (right) where, in both cases, two guest and four host species occupied the unit cell. Both disorder guest components in H2·2(3MeCYC) experienced this contact type. All of these complexes, additionally, interacted by means of a non-classical (host)C\(\:-\)H···N(host) hydrogen bond, which was intramolecular in nature. The C···N distances ranged between 2.776(2) and 2.819(3) Å. Furthermore, non-classical (host)C\(\:-\)H···O(guest) interactions were also identified in H1·CYC, H2·2(CYC) and H2·2(2MeCYC) (3.305‒3.580 Å (C···O), Table 4).

Strong classical (host)N–H···O(guest) hydrogen bonds in complexes H2·2(CYC) (left) and H2·2(3MeCYC) (right). In the latter case, both guest disorder components experienced this interaction type. For clarity, hydrogen atoms not involved in this interaction have been deleted. Host and guest species are provided in stick and ball-and-stick representation, respectively
Finally, we compared the remaining significant interactions in the five complexes where these contact distances were less than the sum of the van der Waals radii of the atoms involved. While no further stabilizing interactions could be identified in H1·CYC, H2·2(CYC) and H2·4MeCYC, both H2·2(2MeCYC) and H2·2(3MeCYC) experienced a single short interaction between a carbon atom of the xanthenyl moiety and a hydrogen atom of the guest molecule (2.51 Å, 141° and 2.86 Å, 142°).
Hirshfeld surface analyses were also considered in order to quantify the various guest···host atom···atom interactions using program Crystal Explorer [33, 34]. Three-dimensional surfaces were created around the guest molecule, and these were converted into the two-dimensional (2D) fingerprint plots as provided in Fig. 7. These 2D plots illustrate the distance between the nearest host atom outside the surface (de) and guest atom inside this surface (di). Where guest disorder was present, each disorder component was considered independently by deleting each one in turn. However, owing to the nature of the disorder in complexes H1·CYC and H2·4MeCYC, the surfaces of the guest species could not be computed in these two instances. The O···H interatomic interactions are represented by the ‘spikes’ (1) whilst the ‘wings’ (2 and 3) refer to the C···H and H···H interactions, respectively, in Fig. 7. (Figure S3 in the SI is a bar graph comparing, quantitatively, the various guest···host interatomic interactions present in these complexes.) These analyses demonstrated that the amounts of (guest)O···H(host) interatomic interactions were comparable, and ranged between 11.1 and 13.9% (Figure S3, SI).

Calculated three-dimensional surfaces around the guest species (left) and the 2D plots showing all of the interatomic guest···host interactions (right) in (a) H2·2(CYC), (b) H2·2(2MeCYC) (disorder guest component 1), (c) H2·2(2MeCYC) (disorder guest component 2), (d) H2·(3MeCYC) (disorder guest component 1) and (e) H2·(3MeCYC) (disorder guest component 2)
Thermal analysis
In order to determine the relative thermal stabilities of these complexes, thermal analyses were carried out on each one. A stack plot of the thermogravimetric (TG) curves of the five host-guest complexes produced in this work are presented in Fig. 8, and these were obtained by heating the samples from approximately 40 to 400 °C at a heating rate of 10 °C·min‒ 1. Table 5 contains a summary of the temperatures at which the guest release event commenced (Ton, which serves as a measure of the relative thermal stabilities of these complexes), the experimental mass loss, the expected (theoretical) mass loss and the nature of the guest accommodation (obtained from the SCXRD experiments).
It is evident from Table 5 that the type of guest accommodation correlated with the thermal stability of each complex: inclusion compounds H1·CYC and H2·4MeCYC, in which the guest species were housed in discrete cavities, possessed the greater thermal stabilities (Ton 92.8 and 105.0 °C, respectively), while these stabilities of H2·2(CYC), H2·2(2MeCYC) and H2·2(3MeCYC), where guests were located in unilateral channels, were significantly lower; Ton values were 68.5 and 49.8 °C for the first two of these, while H2·2(3MeCYC) was unstable at ambient conditions (the measured and expected mass losses did not correlate (13.0, 27.2%, Table 5), possibly as a result of early guest release during sample preparation). Interestingly, when considering the complexes involving H2 and where classical host···guest hydrogen bonding was present, greater Ton values were observed (H2·2(CYC), 68.5 °C and H2·4MeCYC, 105.0 °C) compared with H2·2(2MeCYC) (49.8 °C), where no classical hydrogen bonding of this type could be identified (the exception is the complex H2·2(3MeCYC) which was unstable at ambient conditions despite the classical hydrogen bond between the host and guest species). All remaining experimental and expected mass losses were in close agreement (for H1·CYC, H2·2(CYC), H2·2(2MeCYC) and H2·4MeCYC, the measured mass losses were 14.4, 22.6, 19.7 and 16.4%, whilst those expected were 14.3, 24.6, 21.9 and 16.0%). With the exception of H1·CYC and H2·2(2MeCYC) (where the guest release process was more convoluted, all guest species were released in a single step (Figure S4a‒e). Finally, the thermal stabilities of the complexes involving H2 may therefore be written as in the order H2·4MeCYC > H2·2(CYC) > H2·2(2MeCYC) > H2·2(3MeCYC) when considering their Tonvalues.

Overlaid thermogravimetric curves for complexes with H1 and H2 and the cyclohexanone guests. For clarity, DSC and dTG curves have been omitted. No Ton is indicated for complex H2·2(3MeCYC) due to potential premature guest loss
Equimolar guest competition experiments
In order to assess the preferential behaviour of H1 and H2 for the cyclohexanones, these host compounds were crystallized from all combinations of equimolar guest mixtures (binary, ternary and quaternary). H1 was very soluble in these equimolar mixtures, but (or as a consequence of) the rate of crystal growth was very slow, often requiring several months for crystals to form. Therefore, H1 was disregarded in any further crystallization experiments from mixed guests since this observation implied that separation procedures on an industrial scale using H1 would not be practicable. However, H2 crystallized from such mixed guests significantly faster, and so these experiments were indeed considered. Table 6 contains a summary of the guest ratios in the so-formed mixed complexes (these results were obtained by means of GC analyses). To ensure that the results were reproducible, all of these crystallization experiments were conducted in duplicate and, therefore, the percentage estimated standard deviations (%e.s.d.s) are also provided in this table. Furthermore, the preferred guest in each mixture is presented in bold red text for facile comparison purposes.
When H2 was crystallized from the equimolar binary cycloalkanone mixtures, a marked preference towards 3MeCYC was observed when this guest was present, with the highest selectivity being 75.6% (from the 3MeCYC/4MeCYC experiment) (Table 6). In the CYC/4MeCYC mixture, the host selectivity was then for CYC (76.6%), while experiments involving CYC/2MeCYC and 2MeCYC/4MeCYC afforded mixed complexes with 53.1 and 66.2% 2MeCYC and 4MeCYC, respectively. The host affinity behaviour in the ternary solutions was more ambivalent, but neither CYC nor 4MeCYC was ever preferentially selected. In fact, the amounts of 4MeCYC enclathrated in these ternary experiments were always extremely low (11.7‒17.9%). Broadly speaking, from a consideration of all of these equimolar mixed guest crystallization experiments, it may be concluded that 2MeCYC and 3MeCYC were the more preferred guest compounds of H2, while CYC and 4MeCYC were guests less favoured. These observations are remarkable when considering the results from the thermal experiments: in that investigation, CYC and 4MeCYC formed more stable complexes with H2 (Ton 68.5 and 105.0 °C), and the complexes containing 2MeCYC and 3MeCYC were lower in thermal stability (Ton 49.8 °C in the former, while the latter was unstable at ambient conditions). Clearly, in this work, the relative thermal stabilities of the complexes are not related to the selectivity behaviour of the host compound.
Non-equimolar binary guest competition experiments
Selectivity profiles could not be constructed when the host compound was H1 due to the slow rate of crystallization from these non-equimolar binary guest solutions, as was the case in the equimolar crystallization experiments. However, these were, once again, possible in the case of H2 and, after plotting Z (the molar ratio of GA or GB in the mixed complex) against X (this molar ratio in the original solution), Fig. 9a‒f were obtained.

Selectivity profiles of H2 when crystallized from the binary mixtures (a) 2MeCYC/CYC, (b) 3MeCYC/CYC, (c) CYC/4MeCYC, (d) 3MeCYC/2MeCYC, (e) 4MeCYC/2MeCYC and (f) 3MeCYC/4MeCYC
When H2 was crystallized from the various CYC/2MeCYC solutions (Fig. 9a), the experimentally-obtained data points, for all intents and purposes, were along the line of no selectivity (K = 1), and H2 did not possess the ability to discern between CYC and 2MeCYC. However, in 3MeCYC/CYC mixtures (Fig. 9b), the host demonstrated an affinity towards 3MeCYC when the molar concentration for this guest compound exceeded 40%. K values were low (1.1‒2.2 for experiments in favour of 3MeCYC), and separations of these two guest compounds would not be feasible using supramolecular chemistry strategies and H2, according to Nassimbeni and coworkers (K should be 10 or greater for these separations to be feasible) [35]. In Fig. 9c (CYC/4MeCYC) and 9f (3MeCYC/4MeCYC), these profiles were S-shaped demonstrating that the preference of the host compound depended on the amounts of the two guest species present. In both of the 80:20 and 60:40 mixtures involving CYC/4MeCYC (Fig. 9c), the complex that was isolated was significantly enriched with CYC, 93.9% (K = 3.8) and 86.7% (K = 4.3), respectively. In the case of 3MeCYC/4MeCYC (Fig. 9f), molar concentrations of 3MeCYC that contained 40% or more of this guest species resulted in crystals that were significantly enriched with 3MeCYC, and K ranged between 2.9 and 3.5. However, the 20:80 and 30:70 mixtures of 3MeCYC/4MeCYC resulted in H2 favouring 4MeCYC (89.5 (K = 2.1) and 76.7% (K = 1.4)). In the binary 3MeCYC/2MeCYC and 4MeCYC/2MeCYC solutions (Fig. 9d and e), H2 consistently selected 3MeCYC and 4MeCYC across the concentration range, correspondingly; however, K values were, once more, too low (1.6‒1.9 and 1.7‒2.4) to allow for effective separations. These results largely concur with observations made in the equimolar mixed guest experiments, where 3MeCYC was oftentimes the selected guest solvent of H2. As was the case there, this was not entirely expected given that the thermal analyses demonstrated the 3MeCYC-containing complex to be the least stable one, decomposing even at ambient conditions.
In summary, H1 and H2 were demonstrated to not have the ability to separate these cyclohexanone mixtures using host-guest chemistry strategies.
Conclusions
From the single solvent crystallization experiments, H1 only formed a complex with CYC, while H2 included all four cycloalkanones, and H: G ratios were 1:1 or 1:2. Thermal and SCXRD analyses on the single solvent complexes demonstrated that the thermally more stable complexes housed their guest molecules in discrete cages and inclusion compounds with guests in endless channels possessed lower thermal stabilities. Furthermore, the SCXRD analyses revealed that strong classical host···guest hydrogen bonding assisted in retaining the guest species in the complexes H2·2(CYC), H2·2(3MeCYC) and H2·4MeCYC; these interactions were not present in H1·CYC and H2·2(2MeCYC). Hirshfeld surface analyses demonstrated that the quantities of (guest)O···H(host) were comparable in each of the five complexes, ranging between 11.1 and 13.9%. Furthermore, the selectivity behaviour of H2 in guest mixtures were found to be unrelated to the relative thermal stabilities of these complexes: equimolar and binary non-equimolar guest experiments showed that H2 usually preferred 3MeCYC, despite its complex being the least stable one. Due to a very slow rate of crystal growth when employing H1 in these guest mixtures, its selectivity could not be ascertained in analogous mixed guest experiments. Finally, it was established that neither host compound possessed the ability to separate any of the mixed solutions employed in this work.
Data availability
No datasets were generated or analysed during the current study.
References
Cheng, H., Liu, R., Wang, Q., Wu, C., Yu, Y., Zhao, F.: Selective Reduction of Phenol Derivatives to Cyclohexanones in Water under Microwave Irradiation. New. J. Chem. 36, 1085–1090 (2012)
Shafaghat, H., Sirous Rezaei, P., Daud, W.M.A.W.: Catalytic Hydrogenation of Phenol, Cresol and Guaiacol over physically mixed catalysts of Pd/C and zeolite solid acids. RSC Adv. 5, 33990–33998 (2015)
Fraga, G., Yin, Y., Konarova, M., Hasan, M.D., Laycock, B., Yuan, Q., Batalha, N., Pratt, S.: Hydrocarbon Hydrogen Carriers for Catalytic Transfer Hydrogenation of Guaiacol. Int. J. Hydrogen Energy. 45(51), 27381–27391 (2020)
Lewis, R.J., Ueura, K., Fukuta, Y., Davies, T.E., Morgan, D.J., Paris, C.B., Singleton, J., Edwards, J.K., Freakley, S.J., Yamamoto, Y., Hutchings, G.J.: Cyclohexanone Ammoximation via in situ H2O2 production using TS-1 supported catalysts. Green. Chem. 24, 9496–9507 (2022)
Synthesis: Spectral Studies of Some New Chalcone and Schiff Base Derivatives Derived from Cyclohexanone and Molecular Docking and Biological Activity Studies. pnr 13 (SO3). (2022)
Garba, H., Bashir Yakasai, J., Waziri, I., Bisiriyu, O.: Transition Metal Complexes of Schiff Base Ligand derived from Trimethoprim with Cyclohexanone: Synthesis, characterization, Antimicrobial and Computational studies. Act. Scie Pharma. 4(5), 36–45 (2020)
Kumar, U., Chandra, S., Synthesis: Spectroscopic characterization of some Schiff Base complexes derived from 2-Methylcyclohexanone and their activity against some Fungi. J. Saudi Chem. Soc. 15, 19–24 (2011)
Jebapriya, J.C., Jonathan, D.R., Kirupavathy, S.S., Ragu, R., Prasana, J.C.: Growth and characterization of a Cyclohexanone based Chalcone Crystal 2(E)-(4-N,N-Dimethylaminobenzylidene)-5-methylcyclohexanone for nonlinear optical applications. Opt. Mater. 107, 110035 (2020)
Ritchie, K., Malkiewicz, C.: Thermal analysis of the Photosensitizer 2,6-Bis(p-azidobenzylidene)-4-methylcyclohexanone. Thermochim Acta. 1, 173–179 (1970)
Aly, K.I., Abdel-Rahman, M.A., Qutai, M.M.: Photoactive Linear and Hyperbranched Polyesters based on 4-Methylcyclohexanone and 4-Tert-butylcyclohexanone moieties in the Main Chain: Synthetic methodology, characterization and cytotoxicity. J. Polym. Res. 25 (8). (2018)
Bingham, E., Cohrssen, B. (eds.): Patty’s Toxicology: 6 Volume Set, 6th edn. Wiley- Blackwell, Hoboken, NJ (2012)
Dvořák, B., Buryan, P., Pašek, J.: Chromatographic analysis of a mixture of Isomeric methylcyclohexanones and Methylcyclohexanols. J. Chromatogr. A. 124, 1–7 (1976)
Steed, J.W., Atwood, J.L.: Supramolecular Chemistry. John Wiley & Sons, Ltd. United States of America (2022)
Lehn, J.-M.: Supramolecular Chemistry. Proc. Indian Acad. Sci. (Chem Sci). 106, 915–922 (1994)
Atwood, J.L., Steed, J.W.: Encyclopedia of Supramolecular Chemistry, vol. 1. Marcel Dekker, United States of America (2004)
Wagner, B.D.: Host-Guest Chemistry: Supramolecular Inclusion in Solution. De Gruyter, Berlin, Germany (2020)
Barton, B., Caira, M.R., Hosten, E.C., McCleland, C.W., Weitz, S.: Clathrates of TETROL: Selective inclusion of Methylcyclohexanones in their energetically unfavorable Axial Methyl conformations. Chem. Commun. 50, 13353–13355 (2014)
Barton, B., Caira, M.R., Hosten, E.C., McCleland, C.W., Weitz, S.: Clathrates of TETROL: Further aspects of the selective inclusion of Methylcyclohexanones in their energetically unfavorable Axial Methyl conformations. J. Org. Chem. 80, 7184–7192 (2015)
Barton, B., Dorfling, S.-L., Hosten, E.C.: Cyclohexanone-driven discriminatory Behaviour change of Host Compound (+)-(2R,3R)-TETROL for the Isomeric Methylcyclohexanone guests. Cryst. Growth Des. 17, 6725–6732 (2017)
Barton, B., de Jager, L., Senekal, U., Hosten, E.C.: Comparing the effect of selected substituent changes on host ability and selectivity in four xanthenyl-type host compounds in the Presence of Cyclohexanone and Methylcyclohexanone isomers. J. Incl. Phenom. Macrocycl. Chem. 95, 259–271 (2019)
Barnardo, B., Barton, B., Caira, M.R., Hosten, E.C.: Evaluation of the Behavior of two tricyclic-fused Host Systems in the Presence of single and mixed isomers of the C8H10 aromatic crude oil fraction. Cryst. Growth Des. (2024)
Bruker, A.: APEX2, SADABS and SAINT. Madison (WI), USA,, AXS Inc. (2010). Bruker
Sheldrick, G.M.: SHELXT – Integrated Space-Group and Crystal structure determination. Acta Cryst. A71, 3–8 (2015)
Sheldrick, G.M.: Crystal structure refinement with SHELXL. Acta Cryst. C71, 3–8 (2015)
Hübschle, C.B., Sheldrick, G.M., Dittrich, B., ShelXle: A qt graphical user interface for SHELXL. J. Appl. Crystallogr. 44, 1281–1284 (2011)
Macrae, C.F., Sovago, I., Cottrell, S.J., Galek, P.T.A., McCabe, P., Pidcock, E., Platings, M., Shields, G.P., Stevens, J.S., Towler, M., Wood, P.A.: Mercury 4.0: From visualization to analysis. Design and prediction. J. Appl. Crystallogr. 53, 226–235 (2020)
Pivovar, A.M., Holman, K.T., Ward, M.D.: Shape-selective separation of Molecular isomers with Tunable Hydrogen-Bonded host frameworks. Chem. Mater. 13, 3018–3031 (2001)
Barbour, L.J.: X-Seed – A software tool for supramolecular crystallography. J. Supramol Chem. 1, 189–191 (2001)
POV-Ray for: Windows, Version 3.1e, The persistence of vision team, ©1991–1999
Grabowski, S.J.: In: Grabowski, S. (ed.) Hydrogen Bonding: New Insights. Springer, New York, NY (2006)
Grabowski, S.J.: Understanding Hydrogen Bonds: Theoretical and Experimental Views. Royal Society of Chemistry, Cambridge, England (2020)
Gilli, G., Gilli, P.: The Nature of the Hydrogen Bond: Outline of a Comprehensive Hydrogen Bond Theory. OUP Oxford (2009)
Spackman, M.A., Jayatilaka, D.: Hirshfeld Surf. Anal. CrystEngComm. 11, 19–32 (2009)
Spackman, P.R., Turner, M.J., McKinnon, J.J., Wolff, S.K., Grimwood, D.J., Jayatilaka, D., Spackman, M.A.: CrystalExplorer: A Program for Hirshfeld Surface Analysis, Visualization and Quantitative Analysis of Molecular Crystals. J. Appl. Crystallogr. 54 (3), 1006–1011. (2021)
Sykes, N.M., Su, H., Weber, E., Bourne, S.A., Nassimbeni, L.R.: Selective enclathration of Methyl- and Dimethylpiperidines by Fluorenol Hosts. Cryst. Growth Des. 17, 819–826 (2017)
Acknowledgements
Financial support is acknowledged from the Nelson Mandela University and the National Research Foundation (South Africa).
Funding
Open access funding provided by Nelson Mandela University.
Author information
Authors and Affiliations
Contributions
B. Barnardo: Investigation; Methodology; Validation; Writing the original draft. B. Barton: Conceptualization; Funding acquisition; Methodology; Project administration; Resources; Supervision; Visualization; Assistance with editing the original draft. E.C.H.: Data curation; Formal analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10847_2024_1256_MOESM1_ESM.docx
Supplementary Material 1: The Supporting Information (SI) contains 1H-NMR spectra of the single solvent complexes (Figure S1), calculated stacked PXRD patterns (Figure S2), quantitative representations of the guest···host interatomic interactions present in the complexes (Figure S3) and the DSC, TG and dTG traces from thermal analysis (Figure S4). Table S1 contains a summary of the calculated K values applicable to this work.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barnardo, B., Barton, B. & Hosten, E.C. The host behaviour of 9-phenyl-9 H-xanthene derivatives in mixtures of cyclohexanone and the methylcyclohexanone isomers. J Incl Phenom Macrocycl Chem (2024). https://doi.org/10.1007/s10847-024-01256-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10847-024-01256-y