
Abstract
Xerothermic habitats are closely related to continental steppes of Eurasia and contain communities rich in rare and endemic species, especially insects. Considering the dramatic loss of xerothermic habitats due to climatic and anthropogenic changes the evaluation of genetic variation and phylogeographical patterns in xerothermic species is a matter of utmost importance if appropriate conservation measures are to be undertaken. In this paper 3 mitochondrial genes and 3 nuclear markers were used for evaluation of genetic diversity of populations of the weevil Centricnemus leucogrammus from central-east Europe. These data were used for the recognition of conservation units as well as centers of genetic diversity which could be considered as present “warm-stage” refugia for xerothermic beetle assemblages. The most diverse are eastern and central populations from Ukraine and central Poland, however, also isolated groups of populations from the Pannonian Basin and northern Poland possessed a unique genetic signature. This lead to the conclusion that all three regional groups of populations are significantly isolated from each other and represent distinct evolutionary lineages so they should be considered as separate Management Units and Evolutionary Significant Units. C. leucogrammus can be regarded as an excellent representative for typically xerothermophilous, flightless beetles and knowledge about its genetic diversity may be used for the conservation and management of entire xerothermic communities, especially for those with low mobility invertebrate taxa. Further research on the genetic diversity of various organisms with varying levels of mobility should be undertaken to broaden our knowledge on the conservations needs of xerothermic assemblages.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Xerothermic turfs and thickets are threatened environments containing communities rich in rare and endemic species, especially insects. These habitats are closely related to the continental steppes of Eurasia. In central and western Europe they are considered as “warm-stage refugia” of steppe-like habitats (Willis and Van Andel 2004; Ashcroft 2010). However, some probably arose as a result of human land use (mainly deforestation and pasture development) (Mazur 2001; Barańska and Jermaczek 2009; Paul 2010). Due to unfavorable climatic conditions in the temperate zone, xerothermic habitats are presently fragmented and limited to steep, dry and warm slopes of hills and scarps along river valleys.
The fragmentation of natural habitat is generally considered to be a major threat to many species. Population genetic theory predicts that isolation of small populations leads to reduction of genetic diversity (e.g. Frankham et al. 2002). Species with limited dispersal ability are particularly affected by isolation, which may produce substantial divergence among populations (e.g. Conner and Hartl 2004). Consequently, the evaluation of the extent of isolation of the existing populations and their genetic diversity are of major concern in assessing the risk of local extinction of threatened taxa. The rarity of xerothermic habitats has resulted in their protection in reserves and the Natura 2000 network of protected sites according to the Habitats Directive of the European Union. However, such protected areas are scarce in Europe; most have a very limited area and are isolated. Almost nothing is known about the actual level of isolation of particular xerothermic patches, in continental, regional and local scales. Xerothermic habitats are characterized by their specialized vegetation and fauna, with many species having their main distributions in this habitat type. Considering the dramatic loss of xerothermic habitats as a consequence of changes in land use taking place particularly in central and western Europe (Michalik and Zarzycki 1995; Mazur and Kubisz 2000; Varga 2001), but also in eastern Europe (Cremene et al. 2005), the evaluation of genetic variation and phylogeographical patterns in xerothermic species is of immediate importance if appropriate conservation measures are to be undertaken. Although knowledge of the genetic diversity, phylogeography and population genetics of steppe plants (Franzke et al. 2004; Szczepaniak and Cieślak 2006; Szczepaniak et al. 2007; Schlee et al. 2007; Wróblewska 2008) and vertebrates, mainly rodents (Biedrzycka and Konopiński 2008; Banaszek et al. 2009; Kryštufek et al. 2009), has increased significantly in the last years, information concerning invertebrate populations are scarce and limited mostly to semi-xerothermophilous butterflies (Schmitt and Hewitt 2004; Gratton et al. 2008) and snails (Pfenninger and Posada 2002).
Among xerothermic habitats, turfs are particularly interesting due to their limited distribution and the high number of rare taxa they sustain. The best known xerothermic communities of insects in central Europe include Lepidoptera (butterflies) (Kostrowicki 1953; Cremene et al. 2005; Goloborod’ko and Fedenko 2008); Orthoptera (grasshoppers and crickets) (Liana 1987; Varga 2001), and Cucujiformia (leaf beetles and weevils) (Mazur 2001; Wąsowska 2006).
The most numerous group of xerothermic insects are weevils (Curculionidae) (Mazur 2001). Previous studies on xerothermic weevils were focused mainly on their biogeography and ecology (Mazur 2001). Only populations of two species from central Europe have been studied genetically: Polydrosus inustus (Kajtoch et al. 2009; Kajtoch and Lachowska-Cierlik 2009) and Centricnemus leucogrammus (Kajtoch et al. 2009). The latter species is flightless and ecologically restricted to dry xerothermic turfs, therefore its mobility is very limited. C. leucogrammus is polyphagous so its distribution does not depend on a particular food (plant) source. These ecological and biological characters partially explain its present distribution. The main range of this beetle is located in the steppe zone of southern Russia and Ukraine, however, it occurs there very sparsely. According to Winkler (1932) the range of this species is restricted to Europe, it is also known from only the European part of Russia (http://www.zin.ru/ANIMALIA/COLEOPTERA/eng/entimru2.htm). The last information about its existence in central Asia was probably presented by Von Heyden (1880–1881) so it is uncertain if this species actually occurs there, or if this data concerned another taxon. The western limits of its range are in the highlands of central Poland. C. leucogrammus also occupies steppe-like habitats in the Pannonian Basin, along the Danube valley and in the uplands of Germany. Isolated populations were recently found in northern Poland in the lower Vistula and lower Oder river valleys. In central Europe this species does not have a continuous range, but rather consists of a network of small populations, partially or fully isolated from each other. However, the sizes of these populations are quite large, enabling and facilitating the collection of samples of reasonable size. Recent studies on C. leucogrammus mitochondrial diversity (Kajtoch et al. 2009) showed that its populations consist of at least six phylogenetic lineages, partially overlapping in central Poland and Ukraine. Populations of C. leucogrammus have probably existed for the last 400 000 years in central Europe (in steppes, in front of the ice sheet). This weevil, in common with other steppe and xerothermic species, was probably more common during Pleistocene glaciations when climatic and environmental conditions favored dry grasslands (Adams 1997; Lindner et al. 2006; Magyari et al. 2010). Its present distribution is limited to “warm-stage refugia” (Willis and Van Andel 2004; Ashcroft 2010). However, C. leucogrammus is not protected by law and is not consider as a threatened species, despite a shrinking range in the last few decades, especially at the western edge of the species range (e.g. in Polish Silesia) but also inside its continuous distribution (Kajtoch Ł., unpublished data).
It is essential to know the genetic structure of species throughout their entire ranges in order to develop wide-scale conservation strategies and plan management actions according to contemporary genetic differentiation (Avise et al. 1987; Moritz 1994; Haig 1998). Genetic analyses have thus become an important tool in many studies of threatened or endangered species (Moritz 1994; Haig 1998), and by using genetic markers the evolutionary history of a group can be investigated to determine whether smaller management units may exist below the species level (Moritz 1994; Knapen et al. 2003). It has recently been suggested that focusing on species as the unit of conservation concern may not be the appropriate scale at which diversity should be measured (Hughes et al. 1997; Luck et al. 2003). If gene flow among populations within a species is restricted, then traits important to a species interaction in an ecosystem may become geographically structured. Therefore, local populations will only represent a subset of the range of ecosystem services exhibited within the entire species (Hughes et al. 1997). The implication of this finding is that simply conserving species without regard to population structure may fail to conserve the full spectrum of functions provided by that species. Consequently, it becomes critical to understand the nature of population structure of species.
It is probable that in the fragmented range of C. leucogrammus, conservation units such as “Evolutionary Significant Units” (ESUs) (Ryder 1986; Waples 1991; Moritz 1994) and “Management Units” (MUs) (Moritz 1994) can be identified. It is also likely that within the range of C. leucogrammus areas harboring high genetic diversity may be identified. These would probably constitute important warm-stage refugia for this, and also other xerothermic species.
To determine the genetic diversity and phylogeographic structure of central European populations and their level of isolation I used partial sequences of three mitochondrial genes: cytochrome oxidase II (COII), cytochrome B (CytB) and nicotinamide adenine dinucleotide dehydrogenase 1 (ND1) along with the tRNALeu gene and a small portion of 16S RNA gene, as well as partial sequences of three nuclear markers: internal transcribed spacer 2 (ITS2) of rDNA, the gene encoding the Elongation factor-α (EF-α) and anonymous sequences (AS1). These data were used for recognition of conservation units in central European populations of C. leucogrammus. C. leucogrammus may be regarded as an excellent representative for typically xerothermophilous, flightless beetles and knowledge about its genetic diversity may be used for the conservation and management of entire xerothermic communities, particularly of low mobility invertebrate taxa.
Methods
Material
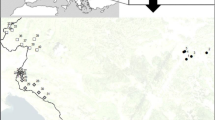
Specimens were collected from most of the C. leucogrammus range in central Europe: (1) main eastern range in Ukraine (4 populations, numbers: 4, 10, 11, 12), (2) part of the species range in central Poland (4 populations, numbers: 1, 2, 3, 9), (3) Pannonian Basin and Danube valley (Moravia in the Czech Republic and Slovakia) (3 populations, numbers: 6, 13, 14), and (4) from isolated localities in northern Poland (lower part of the Oder and Vistula river valleys) (3 populations, numbers: 5, 7, 8). The locations of sampled populations have been presented previously (Kajtoch et al. 2009), with the exception of one additional population from the Slovakian White Carpathians (number 14 in Fig. 1). Adult beetles were collected during several expeditions in 2005–2008. Samples were first preserved in 99% ethanol and then stored at −22°C. For DNA analyses, 5 individuals per population were taken (total 70 specimens from 14 populations).

C. leucogrammus simplified range in central-eastern Europe (grey shaded) and location of sampled populations (black squares and their numbers—see “Material”) (†, Populations extinct in XX-century; ?, probably extinct populations)
Laboratory techniques
DNA isolation techniques, primers for mtDNA markers, PCR conditions, DNA purification and sequencing are described in Kajtoch et al. (2009). For amplification of ITS2 sequences primers ITS3 and ITS4 (White et al. 1990) were used, for EF-α primers ef1F and ef1R (Hughes and Vogler 2004) and for AS1, primers Cl-AS1F ATCTCAGTCGAATTGAAACG and Cl-AS1R AGTAGTCGATGGCGATGTTA. The anonymous sequence was found via 454-sequencing (Roche 454 GS-FLX System Using Titanium Chemistry) of C. leucogrammus genomic DNA. The two longest sequences (more than 400 bp) among about 1700 obtained sequences were used for primer design (Primer3 http://frodo.wi.mit.edu/primer3/) and tested for their level of polymorphism. Only one showed polymorphism and was therefore chosen as a new marker. BLAST search did not reveal any matching sequences so it is probably a non-coding nuclear sequence. All sequences were deposited in GenBank. Accession numbers for mitochondrial DNA FJ442860–FJ442930 and GU565634–GU565642, for ITS2 GU565643–GU565645, for EF-α GU565646–GU565656 and for AS1 HM214632-HM214635.
Sequence analysis
Sequences were checked and aligned using BioEdit v.7.0.5.2 (Hall 1999) and ClustalX (Thompson et al. 1997). No indels (i.e. insertions or deletions) or stop codons were observed in the mitochondrial sequences. Also no indels were found in nuclear sequences except for two short polymorphic microsatellites in the ITS2 sequences. Identical sequences were combined into unique haplotypes. All ITS2 and EF-α sequences were homozygous as judged from the lack of double peaks in chromatograms in from both directions; the only exceptions were microsatellites inside ITS2. The number of microsatellite motifs repeats was determined for heterozygous specimens on the basis of forward and reverse sequence comparison. In AS1 sequences some individuals were heterozygous in the 154 and 155 nucleotide positions (most had “T” in both positions, some “C” and four “Y”). BLAST search showed that between 347 and 432 bp of the obtained EF-α sequence is an intron (85 bp). The three fragments of mtDNA genes obtained (COII, CytB, and ND1-tRNALeu) were subjected to a partition homogeneity test (Farris et al. 1995) using PAUP* 4.10b (Swofford 2002), which showed that further analyses could be performed on combined sequences. Nuclear sequences were analyzed separately for each marker. Haplotypes were identified and standard genetic indices such as haplotype diversity (h), nucleotide diversity (π), number of segregating sites (S) and frequency of private haplotypes (Np) for populations were computed using the program DnaSP v.5 (Librado and Rozas 2009).
In most analyses C. leucogrammus populations were a priori clustered into four groups representing the main geographic parts of the species range (1) eastern (Ukraine), (2) central (central Poland), (3) southern (Moravia and Slovakia) and (4) northern (northern Poland). This grouping of samples is in accordance with the genetic distinctiveness of other steppe-like species. I tested if this pattern is also characteristic for C. leucogrammus.
Mitochondrial sequences were used for phylogenetic tree reconstruction in previous work (Kajtoch et al. 2009). As polymorphism of nuclear sequences was low, standard phylogenetic methods for identification of phylogenetic clusters were not applied. Instead, haplotype networks were constructed using the statistical parsimony method (SP) (Templeton et al. 1992) and the TCS 1.21 program (Clement et al. 2000). Circles representing haplotypes were drawn according to the four main groups of populations. This graphical representation was made to show how the relationships among haplotypes correspond to their geographic distribution.
Nucleotide pairwise distances were calculated only for mitochondrial sequences using MEGA v.4 (Tamura et al. 2007). F ST indices were calculated in ARLEQUIN 3.1 using 1.000 permutations to test for statistical significance (Schneider et al. 2000). A Mantel test (Mantel 1967) was performed in ARLEQUIN 3.1 to check if the genetic structure of the populations fits an isolation by distance model (IBD) (Slatkin 1993), using pairwise F ST values and straight-line geographic distances in kilometers. To test for geographical structuring of populations, an analysis of molecular variance (AMOVA) was conducted using ARLEQUIN 3.1. Clustering was based on the four main groups described above. The level of migration between populations was estimated using the frequency of private haplotypes (Np) (Slatkin 1985)) and Nm indices (Wright 1951). Nm indices were calculated according to formulas: Nm ≈ (1 − F ST )/4 F ST (for nuclear sequences) and Nm ≈ (1 − F ST )/2 F ST (for mtDNA). Values of Nm > 1 indicate a high level of gene flow (Cockerham and Weir 1993). Migration rates (M) were also estimated in program the Lamarc v.2.1.3 (Kuhner 2006) which takes advantage of Likelihood Analysis with Metropolis Algorithm using Random Coalescence (Kuhner and Smith 2007). Bayesian analyses were used separately for mtDNA (joined sequences) and nuDNA (sequences of all three markers were treated as separate regions). Four short chains (10,000 generations, sampling every 20) and two long chains (1,000,000 generations, sampling every 100) were run, 2,000 initial samples were regarded as burn-in. Analyses were repeated twice. For calculation of F ST indices, Mantel test, AMOVA and M, all nuclear markers were concatenated. Mitochondrial sequences were also combined. The two microsatellites in the ITS2 sequences were excluded from the above-mentioned analyses. The polymorphism exhibited by these microsatellites was only described in the results.
Results
Genetic diversity
I obtained a 1,632 bp fragment of mitochondrial DNA (661 bp of COII, 435 bp of CytB and 536 bp of ND1-tRNALeu-16S), 553–564 bp of ITS2, 658 bp of EF-α and 333 bp of AS1. Among the 70 studied specimens of C. leucogrammus 45 mitochondrial, 3 ITS2 (excluding microsatellites), 11 EF-α and 4 AS1 haplotypes were identified.
A total of 47 segregating sites were detected in the mitochondrial DNA. Mean haplotype diversity (h) was 0.98 ± 0.01 and mean nucleotide diversity (π) was 0.57% (Table 1). No population was monomorphic. Except for one pair of populations from central Poland and one pair of populations from Slovakia, no haplotypes were shared between populations, however, neighboring populations often had similar haplotypes (differing by only one to a few mutations).
There were only 2 polymorphic segregating sites in the ITS2 sequences. Mean haplotype diversity (h) was 0.27 ± 0.62 and mean nucleotide diversity (π) was 0.05% (Table 1). Moreover within the ITS2 sequences I found two neighboring microsatellites, a GTC repeat between sites 417 and 431–440 and a GTA repeat between sites 434–443 and 459–462. The GTC microsatellite consisted of 5–8 repeats whereas the GTA microsatellite had 5 or 6 repeats [(GTC)5–8CAA(GTA)5–6]. Most specimens (38) were homozygous in both microsatellites [(GTC)7CAA(GTA)5]. Three specimens from one of the northern populations had 5 GTC repeats (1 homozygous and 2 heterozygous), six specimens from Ukraine populations had 6 GTC repeats (1 and 5, respectively), ten specimens from Slovakia and Moravia had 8 GTC repeats (4 and 4, respectively) and four specimens from Slovakia and Moravia had 6 repeats of GTA (3 and 3, respectively). Data on the polymorphism of microsatellites should be taken with caution. The number of repeats was identified on the basis of a comparison of forward and reverse sequences what were not always reliable. Also the structure of ITS2 sequences, which are members of tandemly repeated motifs of ribosomal DNA, might cause problems with genotype identification.
There were 10 segregating sites in the EF-α sequences. Mean haplotype diversity (h) was 0.53 ± 0.07 and mean nucleotide diversity (π) was 0.11% (Table 1). Most variable populations were found in Ukraine. The non-coding short fragment of the EF-α gene was similarly conservative as the coding fragments. The AS1 marker had 4 segregating sites. Mean haplotype diversity (h) was 0.64 ± 0.04 and mean nucleotide diversity (π) was 0.03% (Table 1). Most variable populations were found in central Poland. All nuclear sequences (1,535 bp) contained 16 segregating sites and 19 haplotypes. Mean haplotype diversity (h) was 0.858 ± 0.034 and mean nucleotide diversity (π) was 0.15%.
Geographical structure of populations
Mitochondrial haplotypes found in C. leucogrammus were grouped into six clades (Fig. 2). Populations from Ukraine harbored three clades (M1, M2, M3). Two of these were found only in Ukraine. Haplotypes belonging to the third clade (M3) were closely related to some of the haplotypes from central Poland. About half of the specimens from central Poland had haplotypes from clade M3, the rest had haplotypes from clade M4. All weevils collected in Slovakia and Moravia belong to clade M5. Beetles from northern, isolated populations had haplotypes from two clades: M4 (one population from the lower Oder valley) or M6 (two populations from the lower Vistula valley). The mean pairwise distance between mitochondrial clades of C. leucogrammus was between 0.3 and 0.6%.

Networks of C. leucogrammus haplotypes: mitochondrial DNA (mtDNA), ITS2 fragment, part of EF-α gene, AS1 sequences and combined of all studied nuclear sequences (nucDNA). Marked parts of species range on map indicate areas from where studied weevils were sampled (black colour Ukraine, darker grey colour central Poland, lighter grey colour northern Poland, white colour Moravia and Slovakia)
In the ITS2 network, the most common haplotype (I1) was shared among almost all populations (Fig. 2). A second haplotype (I2) grouped three out of four of the weevil populations from central Poland together. Another haplotype was found only in a single individual from central Poland. Haplotypes in this network were separated by only 1–2 mutational steps.
Among nuclear markers, the EF-α network was most diverse (Fig. 2). More than half of the individuals from most populations had a single, most common haplotype (E1). Six haplotypes (E5–E11) were found in single individuals (from Ukraine). One haplotype (E2) was found in two individuals from Moravia, another (E3) in one individual from Ukraine and one from Poland. All weevils from two populations from the lower Vistula valley in northern Poland had a common haplotype (E4). Haplotypes in this network were separated by only 1–3 mutational steps.
The AS1 network was also simple. Most specimens from all regional groups of populations had the same, main haplotype M1. About half of the specimens from Ukraine possessed haplotype M2. Most specimens from central Poland belong to a separate cluster (M3) consisting of the two haplotypes. Among them were specimens from one of the northern Polish populations (from lower Vistula valley).
A network based on all nuclear sequences reveals three main groups. The most common nuclear haplotype and its satellites was found in specimens mainly from Moravia–Slovakia but also from Ukraine and northern Poland (Oder valley). Haplotypes from the N2 cluster were detected only in Ukrainian populations, those from N3 were from central and northern Poland populations.
Results of the AMOVA analyses for C. leucogrammus populations (conducted separately for the nuclear and mtDNA data sets) indicate that about 20% (mtDNA) or 40% (nucDNA) of the genetic variation is partitioned among geographical groups of populations (Table 2). According to the mitochondrial markers, about 50% of the variance is due to among population variation within groups and about 30% within populations. AMOVA for nuclear markers showed that about 40% of variation is partitioned among populations within groups and about 20% within populations (Table 2). These results indicate moderate structure between four geographical groups and among populations at a regional scale and that individuals are rather uniform within groups of populations.
Isolation and migrations
Mitochondrial and nuclear Nm indices for most pairs of C. leucogrammus regional groups of populations are c. 1.0 or lower, indicating that these populations are isolated from each other (Table 3). Nm values over 1.0 were found between populations from central Poland and Ukraine (mtDNA) and northern Poland and Slovakia–Moravia (mtDNA). In these regions migration events may have been recent.
The results of the Lamarc analyses (Table 4) showed that mitochondrial migration rates are highest between populations from central and northern Poland (in both directions), slightly lower between populations from Ukraine and central Poland (but higher from Ukraine to Poland than the reverse) and much lower from/into southern populations (Slovakia and Moravia) which seemed to be most isolated from any others. On the other hand, M values obtained from nuclear sequences were similar indicating moderate gene flow between regional groups of populations. It is worth noticing that the obtained M values have very wide confidence intervals so these data and conclusions should be treated only as approximations.
Values of both indices of migration, Nm and M, could be violated by the low level of sequence polymorphism, especially in respect to nucDNA.
Populations of C. leucogrammus fit the IBD model on the basis of their mitochondrial sequences (Mantel test r = 0.293, P = 0.014) (Fig. 3). Generally populations more genetically distinct are more distant in space, however, this correlation is rather weak (Fig. 2). There is no such correlation for nuclear sequences (Mantel test r = 0.000058, P = 0.460) (Fig. 3).

The relationship between genetic (F ST ) and geographic distance (km) for studied populations of C. leucogrammus (Mantel tests) obtained for mitochondrial (a) and nuclear (b) sequences
Discussion
Genetic diversity
The presented analyses have revealed a moderate degree of genetic differentiation among populations. All sampled localities possessed unique mtDNA haplotypes from single or two–three haplogroups leading to a highly structured relationship between genetic diversity and geography. The network obtained from mitochondrial sequence data suggests very little historic interconnectivity between populations in geographic proximity. Consequently, most populations are effectively isolated from each other. The only exceptions are a single pair of neighboring populations from central Poland and closely located populations in Slovakia, with shared single haplotypes indicative of recent or presently restricted gene flow. Related haplotypes identified in Ukraine and central Poland probably represent past connection between these two regions during the Pleistocene glaciations, when dry grasslands were more common (Adams 1997; Lindner et al. 2006; Magyari et al. 2010). Also one of most northern populations (from the lower Oder river valley), containing haplotypes very closely related to some haplotypes from central Poland, may be the result of species expansion from the south and subsequent extinction of intermediate populations (Kajtoch et al. 2009). Indeed a low level of migration between these regional groups of populations (Ukraine and central Poland, central and northern Poland, central Poland and Moravia–Slovakia) was detected (Tables 3, 4).
On the other hand the extensive nuclear DNA haplotype sharing among populations either results from gene flow or ancestral polymorphism with an absence of substitution since population isolation. This pattern may result from a combination of slower rates of evolution and the longer time necessary for lineage sorting for the nuclear polymorphism compared to variation observed in the mtDNA genes (Maddison 1997; Carstens and Knowles 2007), rather than any recent gene flow. Differences in migration rates calculated from mitochondrial and nuclear sequences may also be the result of female philopatry and male dispersal. This explanation is unlikely in the light of knowledge on C. leucogrammus ecology and biology, particularly dispersal abilities, which are very poor (Mazur 2001). Both sexes of this species are flightless so probably males are not more migratory than females. Geographically distant populations that display highly divergent mtDNA lineages, like southern and northern populations, share part of their nuclear haplotypes. However, in some instances, especially when nuclear sequences are analyzed jointly, the nuclear DNA data reinforces the notion of ongoing differentiation among populations of four geographic groups: Ukraine, central-Poland, northern-Poland (at least lower Vistula valley) and Slovakia–Moravia, supporting further the general observation of moderate population structure derived from mtDNA. The use of more rapidly evolving DNA markers such as microsatellites may help resolve relationships among populations and show present migration routes and barriers among them, both in regional and local scales.
The highest genetic diversity was found in populations from Ukraine where three out of six mitochondrial clades exist (M1, M2, M3). Nuclear markers revealed significant diversity of weevils from these eastern populations. Different microsatellite alleles from the ITS2 sequences were found, both in homo- and heterozygotes, a private AS1 haplotype (A2) and the highest haplotype and nucleotide diversity of EF-α (nine of eleven haplotypes, except E2 and E4) and joined nuclear DNA. Populations from central Poland also exhibit a high level of genetic diversity. These populations contain two mitochondrial clades (M3 and M4), one of which (M4) is also central for the whole mtDNA network. Most weevils from this part of the species range have a more distinct ITS2 haplotype (I2) than the common one (I1) found in other populations, leading to high haplotype diversity, a private AS1 haplotype (from the A3 cluster). They also belong to a separate cluster in the nuclear DNA network. The variability of sequences from southern populations (Slovakia and Moravia) is lower but still significant. All these populations belong to one mitochondrial clade (M5), which is quite diversified (very high haplotype diversity) and also have different alleles in the ITS2 microsatellites. Surprisingly, populations from the northern part of the C. leucogrammus range, where this species could have existed for no more than 10,000 years due to unfavourable environmental conditions (ice-sheet) (Adams 1997; Lindner et al. 2006; Magyari et al. 2010), are also quite differentiated genetically. These populations belong to two mitochondrial clades (M4 and M6), but only those from the lower Vistula river possessed haplotypes (clustered in M6 clade) not closely related to haplotypes from central Poland (clade M4). These populations also have a single private EF1-α haplotype (E4) and joined nuclear haplotypes (from cluster N3) and a private number of repeats in the microsatellites. The data presented above additionally supports the hypothesis of the long-term survival of C. leucogrammus populations in situ in central Europe (Mazur 2001; Kajtoch et al. 2009). The highest genetic diversity in Ukraine showed that this part of the species range is probably the most important present (warm-stage) refuge, at least in central-east Europe. This finding is congruent with other studies that have found high genetic diversity and refugia of other steppe animals (e.g. Spotted suslik Spermophilus suslicus Biedrzycka and Konopiński 2008; Biedrzycka and Radwan 2008; Common hamster Cricetus cricetus Banaszek et al. 2009) and plants (e.g. Oxytropis pilosa Schlee et al. 2007; Melica ciliata and M. transsilvanica Szczepaniak and Cieślak 2006; Elymus hispidus Szczepaniak et al. 2007; Iris aphylla Wróblewska 2008). In Ukraine large areas of steppe-like and similar dry habitats still exist, which could sustain vital populations of steppe organisms. Also the high genetic diversity of populations from fragmented parts of C. leucogrammus show that the central Poland highlands, Pannonian Basin and even xerothermic “islands” south of the Baltic Sea are important for C. leucogrammus.
Conservation units
The recognition of conservation units below the species level is a crucial task if we are to avoid the loss of genetic diversity in species inhabiting threatened environments such as xerothermic turfs and thickets. One widely used framework to distinguish units for conservation has been that of evolutionary significant units (ESU), originally proposed by Ryder (1986) but further developed by others (e.g. Waples 1991; Avise 1994; Vogler and DeSalle 1994; Fraser and Bernatchez 2001). The results here show the existence of six discrete genetic units within C. leucogrammus at a relatively small geographical scale in central-east Europe, which could be considered as ESUs. I am aware of sampling limitations in defining ESUs based exclusively on maternally inherited mtDNA, which does not reflect male-mediated gene flow (Moritz 1994; Avise and Hamrick 1995). The nuclear sequences which I used were much less polymorphic but they support the recognition of two or three ESUs. The ESU of mitochondrial clade M6, restricted only to the lower Vistula valley, is also supported by a distinct EF-α (E4) haplotype and the presence of a 5-repeat GTC motif in the microsatellite. Populations from Slovakia and Moravia belong to the second ESU (mitochondrial clade M5) and some of the specimens from this region also have a distinct number of microsatellite repeats (8 GTC and 6 GTA), found only in these populations. In Moravia a distinct EF-α haplotype (E2) was also found. Most weevils from central Poland had a different ITS2 haplotype (I2) than the common one found in other populations (I1) and also belong to a separate nucDNA cluster, together with populations from northern Poland. However these weevils belonged to two mitochondrial clades, so they do not form a monophyletic group and cannot be considered as an ESU.
Fraser and Bernatchez (2001) integrated the ESU into a more general and flexible concept—Adaptive Evolutionary Conservation (AEC). In general, any criteria providing evidence of lineage sorting through highly reduced gene flow are potentially useful for conservation initiatives under the ESU definition of AEC. The mtDNA data presented here suggest significant geographical structuring of genetic variation across the range of C. leucogrammus, and the phylogeographic analyses point to long-term historical isolation among at least the three geographical areas where this species occurs: (1 and 2) from Ukraine to central Poland, (3) southern (Pannonian, represented here by populations from Slovakia and Moravia) and (4) northern Poland (only lower Vistula valley). Because of the existence of large areas of habitat discontinuity between these three regions, their genetic singularity and the inferred history of the group it seems unlikely that genetic exchange occurs. These three groups of populations can be considered as ESUs under the AEC.
The four regional (geographic) groups mentioned above (1) “eastern” Ukrainian, (2) “central” central Poland, (3) “southern” Slovakia–Moravia and (4) “northern” northern Poland, at least from the lower Vistula valley, could be considered as independent management units (MUs), defined by Moritz (1994). Such a division of the C. leucogrammus range (at least in central Europe) is also clearly supported by the distribution of many xerothermic and steppe-like taxa. Ranges of these species in central Europe are mainly limited to the steppes of Ukraine (Cremene et al. 2005) and/or Pannonian Basin (Varga 2001), highlands of Poland (Mazur 2001) and Germany and also sometimes to isolated localities in larger river valleys south of the Baltic Sea (Mazur 2001). The definition of MUs within C. leucogrammus corresponding to the main steppe/xerothermic areas of central Europe is also in good correspondence with differences of conservation status. Anthropogenic habitat transformation is not equal in the four areas where the populations of the species are found. Land-use changes are less intense in the eastern part of the species range (Ukraine, Russia) where large steppe-like habitats still exist. However, they have recently been considered as threatened (Cremene et al. 2005). On the other hand, xerothermic habitats in central and western Europe, inhabited by this weevil among others, are much more threatened. Despite protection of these environments in many nature reserves and the Natura 2000 network of the European Union, xerothermic patches are scarce, fragmented and isolated from each other. Moreover, they are often devastated because of agricultural conversion or through forestation. These sites are also susceptible to nitrification which changes their specific vegetation (used to poor, dry and warm soil conditions) into more dense herbaceous areas or shrubland. The current practice of strict xerothermic turf protection is not working. Many such habitats in present climatic and environmental conditions, modified by human activity, must be protected actively by grazing, bush-cutting and even periodically prescribed burning, but these actions are expensive, particularly for large habitat patches.
C. leucogrammus can be used as a model organism to study population genetics of species with restricted dispersal ability in xerothermic and steppe habitats. Knowledge about genetic differentiation of the extant and mostly isolated populations of this weevil species with a fragmented distribution is extremely important for the success of future conservation and restoration actions of xerothermic patches. Further research on the genetic diversity of various organisms with different mobility patterns should be undertaken to broaden our knowledge on the conservations needs of entire xerothermic assemblages.
References
Adams JM (1997) Global land environments since the last interglacial. Oak Ridge National Laboratory. http://www.esd.ornl.gov/ern/qen/nerc.html
Ashcroft MB (2010) Identifying refugia from climate change. J Biogeogr 37:1407–1413
Avise JC (1994) Molecular markers, natural history and evolution. Chapman and Hall, New York
Avise JC, Hamrick JL (eds) (1995) Conservation genetics: case histories from nature. Chapman and Hall, New York
Avise JC, Arnold J, Ball RM, Bermingham E, Lamb T, Neigel JE, Reeb CA, Saunders NC (1987) Intraspecific phylogeography: the mitochondrial DNA bridge between population genetics and systematics. Annu Rev Ecol Syst 18:489–522
Banaszek A, Jadwiszczak KA, Ratkiewicz M, Ziomek J (2009) Low genetic diversity and significant structuring of the common hamster populations Cricetus cricetus in Poland revealed by the mtDNA control region sequence variation. Acta Theriol 54:289–295
Barańska K, Jermaczek P (2009) Poradnik utrzymania i ochrony siedliska przyrodniczego 6210 murawy kserotermiczne. [Guidebook for preservation and conservation of xerothermic habitats 6210]. Wydawnictwo Klubu Przyrodników, Świebodzin
Biedrzycka A, Konopiński MK (2008) Genetic variability and the effect of habitat fragmentation in spotted suslik Spermophilus suslicus populations from two different regions. Conserv Genet 9:1211–1221
Biedrzycka A, Radwan J (2008) Population fragmentation and major histocompatibility complex variation in the spotted suslik, Spermophilus suslicus. Mol Ecol 17:4801–4811
Carstens BC, Knowles LL (2007) Estimating phylogeny from gene tree probabilities in Melanoplus grasshoppers despite incomplete lineage sorting. Syst Biol 56:400–411
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Cockerham CC, Weir BS (1993) Estimation of gene flow from F-statistics. Evolution 47:855–863
Conner JK, Hartl DL (2004) A primer of ecological genetics. Sinauer Associates, Inc, Sunderland
Cremene C, Groza G, Rakosy L, Schileyko A, Baur A, Erhardt A, Baur B (2005) Alterations of steppe-like grasslands in Eastern Europe: a threat to regional biodiversity hotspots. Conserv Biol 19:1606–1618
Farris JS, Kallersjo M, Kluge AG, Bult C (1995) Constructing a significance test for incongruence. Syst Biol 44:570–572
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to Conservation Genetics. Cambridge University Press, Cambridge
Franzke A, Hurka H, Janssen D, Neuffer B, Friesen N, Markov M, Mummenhoff K (2004) Molecular signals for Late Tertiary Early Quaternary range splits of an Eurasian steppe plant: Clausia aprica (Brassicaceae). Mol Ecol 13:2789–2795
Fraser DJ, Bernatchez L (2001) Adaptive evolutionary conservation: towards a unified concept for defining conservation units. Mol Ecol 10:2741–2752
Goloborod’ko KK, Fedenko VS (2008) Parameters of the wing coloration in the butterfly genus Colias Fabr. (Lepidoptera, Pieridae) of the steppe zone of eastern and Central Europe. Entomol Rev 87:1109–1114
Gratton P, Konopinski MK, Sbordoni SV (2008) Pleistocene evolutionary history of the clouded Apollo (Parnassius mnemosyne): genetic signatures of climate cycles and a “time-dependent” mitochondrial substitution rate. Mol Ecol 17:4248–4262
Haig SM (1998) Molecular contributions to conservation. Ecology 79:413–425
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser (Lond) 41:95–98
Hughes J, Vogler AP (2004) The phylogeny of acorn weevils (genus Curculio) from mitochondrial and nuclear DNA sequences: the problem of incomplete data. Mol Phylogenet Evol 32:601–615
Hughes J, Daily BGC, Ehrlich PR (1997) Population diversity: its extent and extinction. Science 278:689–692
Kajtoch Ł, Lachowska-Cierlik D (2009) Genetic constitution of parthenogenetic form of Polydrusus inustus (Coleoptera: Curculionidae)—hints of hybrid origin and recombinations. Folia Biol-Krakow 57:149–156
Kajtoch Ł, Lachowska-Cierlik D, Mazur M (2009) Genetic diversity of xerothermic weevils Polydrusus inustus and Centricnemus leucogrammus (Coleoptera: Curculionidae) in central Europe. Eur J Entomology 106:325–334
Knapen D, Knaepkens G, Bervoets L, Taylor MI, Eens M, Verheyen E (2003) Conservation unis based on mitochondrial and nuclear DNA variation among European bullhead populations (Cottus gobio L., 1758) from Flanders, Belgium. Conserv Genet 4:129–140
Kostrowicki AS (1953) Studia nad fauną motyli wzgórz kserotermicznych nad dolną Nidą. [Studies on butterflies fauna of xerothermic hills in vicinity of Nida valley]. Fragm faun Mus Zool Polon Warszawa 6:263–447
Kryštufek B, Bryja J, Bužan EV (2009) Mitochondrial phylogeography of the European ground squirrel, Spermophilus citellus, yields evidence on refugia for steppic taxa in the southern BalkansMitochondrial phylogeography of the ground squirrel. Heredity 103:129–135
Kuhner MK (2006) LAMARC 2.0: maximum likelihood and Bayesian estimation of population parameters. Bioinformatics 22:768–770
Kuhner MK, Smith LP (2007) Comparing likelihood and Bayesian coalescent estimation of population parameters. Genetics 175:155–165
Liana A (1987) Orthoptera of xerothermic habitats in Poland and their origin. In: Baccetti B (ed) Evolutionary biology of orthopteroid insects. Ellis Horwood, Chichester, pp 342–346
Librado P, Rozas J (2009) DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Lindner L, Bogutsky A, Gozhik P, Marks L, Łanczont M, Wojnatowicz J (2006) Correlation of Pleistocene deposits in the area between the Baltic and Black Sea, Central Europe. Geological Quarterly 50:195–210
Luck GW, Daily GC, Ehrlich PR (2003) Population diversity and ecosystem services. TREE 18:331–336
Maddison WP (1997) Gene trees in species trees. Syst Biol 46:523–536
Magyari EK, Chapman JC, Passmore DG, Allen JRM, Huntley JP, Huntley B (2010) Holocene persistence of wooded steppe in the Great Hungarian Plan. J Biogeogr 37:915–935
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Mazur M (2001) Ryjkowce kserotermiczne Polski (Curculionoidea: Nemonychidae, Attelabidae, Apionidae, Curculionidae). Studium zoogeograficzne [Xerothermic weevils of Poland (Curculionoidea: Nemonychidae, Attelabidae, Apionidae, Curculionidae). Zoogeographic studies]. Monografie Fauny Polski 22:1–378
Mazur M, Kubisz D (2000) Ochrona owadów siedlisk kserotermicznych Polski (Insect conservation in xerothermic habitats). (In:) Ochrona owadów w Polsce u progu integracji z Unią Europejską (Insect conservation in Poland in the beginning of integration with European Union). Wiadomości Entomologiczne 2:129–137
Michalik S, Zarzycki K (1995) Management of xerothermic grasslands in Poland: botanical approach. Colloques Phytosociologiques 24:881–895
Moritz C (1994) Defining “Evolutionarily significant units” for conservation. Tree 9:373–375
Paul W (2010) Szlaki holoceńskiej migracji roślin kserotermicznych na ziemie Polski—przegląd ustaleń i hipotez oraz perspektywy badań [Migration routes of xerothermic plants into Poland—review of knowledge and hypothesis and research perspectives] [in:] Ratyńska H., Walodn B. 2010. Ciepłolubne murawy w Polsce. [Thermophilous turfs of Poland]. Bydgoszcz
Pfenninger M, Posada D (2002) Phylogeographic history of the land snail Candidula unifasciata (Poiret, 1801) (Helicellinae, Stylommatophora): fragmentation, corridor migration and secondary contact. Evolution 56:1776–1788
Ryder OA (1986) Species conservation and systematics: the dilemma of the subspecies. Tree 1:9–10
Schlee M, Grimm G, Göker M, Sauer W, Hemleben V (2007) Molecular and phytosociological insight into postglacial relicts Lathyrus pannonicus and Oxytropis pilosa. In: Phylogeography and conservation of postglacial relicts. Symposium Abstrakt Book. National Museum of Natural History (Luxembourg), pp 31–32. http://www.symposium.lu/relicts/docs/Abstractbook.pdf
Schmitt T, Hewitt GM (2004) The genetic pattern of population threat and loss: a case study of butterflies. Mol Ecol 13:21–31
Schneider S, Roessli D, Excoffier L (2000) ARLEQUIN: a software for population genetics data analysis. University of Geneva, Geneva
Slatkin M (1985) Rare alleles as indicators of gene flow. Evolution 39:53–65
Slatkin M (1993) Isolation by distance in equilibrium and nonequilibrium populations. Evolution 47:264–279
Swofford DL (2002) PAUP*. Phylogenetic analysis using parsimony (*and other methods). Sinauer Associates, Sunderland
Szczepaniak M, Cieślak E (2006) Genetic variation and structure in natural populations of Melica ciliata and M. trassilvanica (Poaceae) as indicated by AFLP markers. Biodiv Res Conserv 3–4:39–43
Szczepaniak M, Cieślak E, Bednarek PT (2007) Natural hybridization between Elymus repens and E. hispidus assessed by AFLP analysis. Acta Soc Bot Pol 76:225–234
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular evolutionary genetics analysis (mega) software version 4.0. Mol Biol Evol 24:1596–1599
Templeton AR, Crandall KA, Sing CF (1992) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data III. Cladogram estimation. Genetics 132:619–633
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl Acids Res 24:4876–4882
Varga Z (2001) Post-glacial dispersal strategies of Orthoptera and Lepidoptera in Europe and in the Carpathian basin. In: Proc. 13th Int. Coll. EIS, September 2001, pp 93–105
Vogler A, Desalle R (1994) Diagnosing units of conservation management. Conserv Biol 8:354–363
Von Heyden L (1880–1881) Catalog der Coleopteren von Sibirien, mit Einschluss derjenigen der Turanischen Länder, Turkestans und der chinesischen Grenzgebiete. (The Catalogue of Coleoptera of Siberia with adjacent areas of Turan Land, Turkestan and borderline of China). der Deutschen Entomologischen Gesellschaft. Berlin
Waples RS (1991) Pacific salmon, Oncorhynchus spp., and the definition of “species” under the Endangered Species Act. US Natl Mar Fish Serv. Mar Fish Rev 53:11–22
Wąsowska M (2006) Chrysomelid communities (Chrysomelidae, Coleoptera) of xerothermic grasslands (Inuletum ensifoliae) in the Wyzyna Miechowska Uplands (Central Poland). Biologia 61:565–572
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Shinsky JJ, White TJ (eds) PCR Protocols: a guide to methods and applications. Academic Press, San Diego, pp 315–322
Willis KJ, Van Andel TA (2004) Trees or no trees? The environments of central and eastern Europe during the Last Glaciation. Quat Sci Rev 23:2369–2387
Winkler A (1932) Catalogus Coleopterorum regionis palaearcticae. Pars 12. Wien, pp 1393–1520
Wright S (1951) The genetic structure of populations. Ann Eugen 15:323–354
Wróblewska A (2008) From the center to the margins of geographical range: molecular history of steppe plant Iris aphylla L. in Europe. Plant Syst Evol 272:49–65
Acknowledgments
I would like to greatly thank M. Mazur, M. Holecova, D. Kubisz and D. Lachowska-Cierlik for collecting of some of studied specimens. W. Babik, K. Nadachowska and D. Lachowska-Cierlik helped in different steps of laboratory or analytical work. I am also grateful to W. Babik for helpful comments to first draft of manuscript. This research was partially supported by the Polish Ministry of Science and Higher Education, grant no N N303 612238.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kajtoch, Ł. Conservation genetics of xerothermic beetles in Europe: the case of Centricnemus leucogrammus . J Insect Conserv 15, 787–797 (2011). https://doi.org/10.1007/s10841-011-9377-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10841-011-9377-8