
Abstract
Enterococcus faecalis, a gram-positive bacterium, is among the most common nosocomial pathogens due to its limited susceptibility to antibiotics and its reservoir of the genes coding for virulence factors. Bacterial enzymes such as kinases and phosphorylases play important roles in diverse functions of a bacterial cell and, thus, are potential antibacterial drug targets. In Gram-positive bacteria, HPr Kinase/Phosphorylase (HPrK/P), a bifunctional enzyme is involved in the regulation of carbon catabolite repression by phosphorylating/dephosphorylating the histidine-containing phosphocarrier protein (HPr) at Ser46 residue. Deficiencies in HPrK/P function leads to severe defects in bacterial growth. This study aimed at identifying novel inhibitors of E. faecalis HPrK/P from a commercial compound library using structure-based virtual screening. The hit molecules were purchased and their effect on enzyme activity and growth of resistant E. faecalis was evaluated in vitro. Furthermore, docking and molecular dynamics simulations were performed to study the interactions of the hit compounds with HPrK/P. Among the identified hit molecules, two compounds inhibited the phosphorylation of HPr as well as significantly reduced the growth of resistant E. faecalis in vitro. These identified potential HPrK/P inhibitors open new research avenues towards the development of novel antimicrobials against resistant Gram-positive bacteria.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Antimicrobial drugs have been used for decades to treat bacterial infections. However, many pathogenic bacteria fail to respond to treatment since these microorganisms have developed strategies to resist the effect of antibiotics [1]. Gram-positive bacteria are involved in more than half of the hospital-acquired infections and Enterococci are among the major pathogens to cause nosocomial bloodstream infections with high morbidity and mortality rate [2, 3]. Approximately, 80–90% of enterococcal infections are caused by Enterococcus faecalis, a commensal bacterium inhabiting the gastrointestinal tracts of humans [1, 4]. E. faecalis has developed resistance to commonly used antibiotics such as vancomycin, aminoglycosides, daptomycin, tetracycline, linezolid, and quinolones [5, 6]. In concert with antibiotic resistance, E. faecalis has also been reported to possess a reservoir of virulence genes and it can transfer these genes to other pathogenic bacteria [7]. Over the past few decades, E. faecalis has emerged as a significant pathogen due to its ability to cause a variety of infections such as urinary tract infection, bacteraemia, sepsis, pelvic infection, abdominal infection and rarely meningitis [8]. In E. faecalis, various mechanisms have been reported to counter the effect of antibiotics such as cell wall and cell membrane modifications, overexpression of efflux pumps, inactivation of antibiotics, and alteration of the drug targets [9]. Now, there is a need to identify new drug targets to reduce the risk of antibiotic resistance development. For instance, bacterial kinases and phosphorylases have been so far relatively overlooked in antimicrobial drug discovery. However, they play an important role in cell growth, intracellular metabolism, gene transcription, translation, signal transduction, and other essential cellular functions [10, 11] and, thus, could serve as suitable drug targets for the discovery of novel antimicrobials.
Bacteria utilize a preferred carbon source from the mixture of different sources through the process of carbon catabolite repression (CCR) that represses the expression of genes involved in the metabolism of secondary carbon sources [12]. CCR is regulated by a bi-functional enzyme that has kinase as well as phosphorylase activity, i.e., HPr Kinase/Phosphorylase (HPrK/P; EC:2.7.11.-, EC:2.7.4.-) [13]. The ATP-dependent HPrK/P phosphorylates Ser46 residue of histidine-containing phosphocarrier protein (HPr) of the phosphotransferase system (PTS) that plays an important role in sugar transport. In addition to kinase activity, it also carries the phosphorylase activity at the same active site. HPrK/P protein is a Ser/Thr protein kinase that shows no sequence similarity to eukaryotic Ser/Thr protein kinases [14,15,16]. The N-terminal (residues 1–134) of HPrK/P has no defined activity [14]. However, the catalytically active C-terminal domain (residues 135–319) contains a characteristic Walker motif A(G/A)xxxxGK(S/T) that forms the phosphate-binding loop (P-loop, residues 155–162), which binds the phosphate moiety of an incoming ATP nucleotide [17]. Residues 203–212 are important for the catalytic mechanism of phosphorylation/dephosphorylation and residues 266–271 (‘K3-loop’ that is connecting βK to α3) for dephosphorylation (UniProtKB; entry O07664, HPRK_ENTFA) [15]. Other important individual residues that have been identified forming the active/catalytic site are His140, Lys161, Asp179 (proton acceptor/donor), Arg245 in the flexible/disordered loop (residues 235–251), and the metal (Mg2+) binding residues Ser162 and Glu204. Crystallographic studies have revealed that HPrK/P is a hexameric enzyme that binds six HPr substrate molecules (Fig. 1a–c) [16]. The substrate-binding area of HPrK/P is exposed when the enzyme adopts an open conformation (~ 1400–2000 Å surface area) and six hydrogen bonds per HPr molecule stabilize the protein–protein interactions. Three molecules of HPr bind on the top and three HPr protein molecules bind on the bottom of the hexameric HPrK/P assembly. Two adjacent HPrK/P enzyme monomers interact with one HPr molecule; one HPrK/P monomer interacts with HPr via its catalytic site and the other monomer through its C-terminal helix [16] (Fig. 1d–f). The most contacts to HPr from one HPrK/P chain are initiated by βA (residues 135–140), the P-loop (connects βC to helix α1) and the βD–βE hairpin (residues 174–185). A conserved residue from the other subunit’s flexible/disordered loop, Arg245 is also important for stabilization of the interaction.

Crystal structure of the hexameric L. casei HprK/P in complex with B. subtilis HPr (PDB ID: 1KKL; [16]); a side view, surface representation; b top view, surface representation; c side view, mixed surface and cartoon representation. Color code: HPrK/P chains in violet blue, brown and light green; HPr protein in pink and light blue (top and bottom respectively). d Cartoon representation of the comparative (homology) model of the homodimeric E. faecalis HPrK/P (without ions). e Cartoon representation of the zoom-in view of HPr binding between two HPrK/P chains (the same structural assembly as in a–c and f panels). Serine 46 of HPr (in elemental ball-and-stick representation and labelled) is projected towards the co-crystallized calcium ion (magenta sphere) and P-loop of HPrK/P (dark pink); K3-loop is in cyan, and the disordered loop in orange. f The same side view as in c panel in cartoon representation
Previous studies have shown that a defect in kinase or phosphorylase activity of HPrK/P severely affects the carbon metabolism, growth, morphology and other cellular functions in Staphylococcus xylosus, Lactobacillus casei, Mycoplasma pneumoniae and Bacillus subtilis [17,18,19,20]. Therefore, bacterial kinases are also gaining pronounced attention as potential targets for antimicrobial drug discovery [11, 21, 22]. In our previous study, we observed an enhanced expression of hprK along with HPr-Ser46 phosphorylation in antimicrobial resistant E. faecalis strains [23]. This suggests that HPrK/P plays an important role in the fitness of antibiotic resistant E. faecalis. Therefore, the present study is aimed at identifying active-site targeted HPrK/P inhibitors against nisin-resistant E. faecalis that showed resistance to antibiotics [5]. Nisin is a class I bacteriocin that is used as a preservative for many food products. Here, we demonstrate that inhibition of bacterial HPr binding to HPrK/P is a strategy that limits the growth of resistant bacteria. To the best of our knowledge, this is the first study reporting HPrK/P inhibitors that have been identified using virtual screening and whose interactions with HPrK/P have been investigated with molecular dynamics (MD) simulations. The results of our study may facilitate the development of a new class of antimicrobials and thus opens up the possibility of combating multi-drug resistant Gram-positive bacteria such as E. faecalis.
Materials and methods
Materials for in vitro assays
Media components, Beef Extract (catalog number: RM002), Peptone (catalog number: CR001), Yeast Extract (catalog number: RM027) and Sodium Chloride (catalog number: TC046) for bacterial culture were purchased from HiMedia Laboratories (Mumbai, India). Anti-phosphoserine antibody was purchased from Sigma-Aldrich Chemical Co., St. Louis, MO (catalog number: SAB5200086-400UL). Inhibitor molecules were purchased from Enamine Ltd., Ukraine. PAGE reagents and buffers were purchased from Biorad and PVDF transfer membrane, 0.2 µm (Catalog number: 88520) was purchased from Thermo Fisher Scientific, Inc. Mini-PROTEAN® Tetra Cell, 2-gel, 10-well combs, 1.0 mm (catalog no. 1658003) from Biorad was used for PAGE and blotting was performed using semi-dry blotting unit (Scie-Plas Ltd., UK). Sensitive and resistant E. faecalis (NisR-147) bacteria were previously isolated from raw buffalo milk in our lab [5, 23]. The nisin-resistant strain used here was found to be resistant to chloramphenicol, ampicillin, ciprofloxacin, rifampicin, vancomycin, carbenicillin, linezolid, oxacillin, and fosfomycin with minimum inhibitory concentrations (MICs) varying between 4 µg/mL (ciprofloxacin) and 512 µg/mL (fosfomycin) [5].
Protein modelling
The amino acid sequence of E. faecalis HPrK/P protein (Accession Number: WP_002357309.1) was retrieved from the NCBI Protein database (https://www.ncbi.nlm.nih.gov/protein/). To find suitable template structures for comparative (homology) modelling, we used the Protein BLAST tool with default parameters against the Protein Data Bank (PDB) [24, 25]. The template crystal structures were then selected from the BLAST output based on the criteria of maximum identity percentage, a high BLAST score and a low E-value (PDB ID 1KO7: Query coverage = 94%, Percent identity = 51.70%, E value = 10–105; PDB ID 1KKL: Query coverage = 58%, Percent identity = 71.04%, E value = 2 × 10–89). The Expect value (E) describes the number of "expected" hits of similarity in a database. It decreases exponentially as the score (S) of the match increases. The crystal structures of the full-length HPrK/P protein of S. xylosus (PDB ID: 1KO7; resolution 1.95 Å) [26] and the C-terminal catalytic domain of L. casei HPrK/P (PDB ID: 1KKM; resolution 2.80 Å, related to 1KKL but containing also the coordinates for the flexible/disordered loop) [16] were retrieved from the PDB and further used to prepare a full-length monomeric model and a homodimeric C-terminal catalytic domain model, respectively. The initial monomeric model based on the single template (1KO7, A chain) was built with the Swiss-Model server (https://swissmodel.expasy.org) [27]. As the template structure lacks the coordinates for the long flexible/disordered loop, the missing region was modelled in by the server. This model was utilized in the virtual screening phase. For the binding mode studies, we built another full-length monomeric model utilizing multiple HPrK/P template structures, some of which contained coordinates also for the disordered loop as well as the complete C-terminus. The following template structures were selected from the BLAST results: A chains of PDB IDs 1KNX [28] (from M. pneumoniae, resolution 2.50 Å), 2QMH [29] (from L. casei, resolution 2.60 Å), 1KO7, 1KKL [16] (from L. casei, resolution 2.80 Å) and 1JB1 [15] (from L. casei, resolution 2.80 Å). Apart from the Swiss-Model-generated monomeric model, the modelling alignments were prepared with the alignment tools (align2d, salign) of Modeller 9.20 [30]. Ten comparative models were built using Modeller 9.20 for both the monomeric and the dimeric forms. The model having the lowest discrete optimized protein energy (DOPE) score was selected for the monomeric form whereas the model with the lowest possible DOPE score and the best stereochemical quality was selected for the dimeric structure [31]. For the molecular dynamics (MD) simulations, two alternative homodimeric models were used: one without ions (Fig. 1d) and the other with an added Mg2+ and a phosphate ion to observe the possible effect of metal coordination on inhibitor binding. The phosphate coordinates were obtained from PDB ID: 1KO7 and Mg2+ coordinates from PDB ID: 1KKL (Mg2+ was replaced for the original Ca2+ since HPrK/P uses Mg2+ for the catalysis). The characteristic P-loop (Fig. 1d, e) offers the binding site for the phosphate ion (normally occupied by the β-phosphate of the bound nucleotide), [26] while Mg2+ stays close to the phosphate ion and facilitates the phosphorylation of Ser46 at HPr. No constraints were needed to keep the ions in their positions during the simulations. The quality assessment of the models was performed using Procheck [32], ProSA [33, 34], and QMEANDisCo scores [35].
Virtual library screening
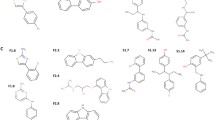
Virtual screening was performed with the INVENTUS Drug Discovery suite v1.1 (Novo Informatics Pvt Ltd). First, the active site was predicted on the Swiss-Model-generated monomeric HPrK/P enzyme model using the PocketDetector™ module of INVENTUS as per the automated active site detection, docking, and scoring (AADS) protocol [36]. The selected active site was formed by the P-loop residues as well as other residues important for the catalytic mechanism of phosphorylation and/or dephosphorylation; residues 158–163, 166–167, 170–171, 206, 232, 267–275, 277. The coordinates of this site were then uploaded to the HitsGen™ module (RASPD protocol) [37] which was used for the virtual high-throughput screening of an inbuilt INVENTUS library that has around four million compounds. HitsGen™ employs a fast-quantitative structure–activity relationship (QSAR)-based methodology to screen for compounds whose physico-chemical properties are complementary to the properties of the target binding site, without actually having to dock the molecules to the target. The ligand binding energy (bio-affinity score) of the screened compounds is estimated with the robust inbuilt QSAR equation that was developed using a representative training set of experimental drug-receptor complexes [37]. Therefore, apart from the structural properties of the given binding site, the following ligand property criteria were used to screen through the compound library: hydrogen bond donors = 0–5, hydrogen bond acceptors = 0–10, LogP (SlogP) = 0–5, molar refractivity = 40–130, number of aromatic atoms = 15–57, length of molecule = 10–100 Å, total number of atoms = 10–100, and binding energy < − 5 kcal/mol. These criteria were set based on a previously described, potent (IC50 17–18 µM at pH 7.0–8.0, respectively), ditopic 2-aminobenzimidazole-type inhibitor “3-B-3” of B. subtilis HPrK/P [10] (Table 1). Three inhibitor candidates from overall 50 hits were selected for in-vitro activity evaluation based on the best (most negative) bio-affinity score.
In-vitro evaluation
Effect of inhibitors on growth and morphology of resistant E. faecalis
To evaluate the effect of the three identified candidate inhibitors on bacterial growth, Colony-Forming Units (CFU) were counted. Resistant E. faecalis was grown in the presence of 1 mM inhibitors (100 mM stock dissolved in DMSO) and without any inhibitor as a control from 0 to 8 h and plated on nutrient agar Petri dish. Then, CFU mL−1 was calculated. To visualize the effects of inhibitors on bacterial morphology, Gram staining of the bacteria was done and examined under a microscope (×1000).
Detection of HPr-Ser-phosphorylation using western blotting
To determine the effect of the candidate inhibitors on the activity of HPrK/P, western blotting was performed. Nisin resistant E. faecalis strain was grown in nutrient broth till the exponential phase in the presence of 1 mM inhibitors and without any inhibitor as a control, harvested and washed with ice-cold 10 mM Tris–HCl buffer. Cells were lysed using bead beater (FastPrep®-24, Bio101/Savant, Farmingdale, NY), followed by centrifugation at 10,000 rpm at 4 °C for 20 min. The crude sample was loaded on 12% SDS-PAGE without heating. Anti-phosphoserine antibody was used to detect the serine phosphorylation of the HPr protein as described in [23]. The experiment was performed in triplicate and relative density was analysed using ImageJ (1.51v 9). Statistical analysis was performed using GraphPad Prism 8.0.0.
Computational binding mode analysis of the hit compounds
Molecular docking
Since our virtual screening method utilized only the properties of the reference ligand and the selected pocket in a monomeric subunit, we did not get any realistic view of the possible binding modes of the ligands at the hexameric enzyme. Thus, the binding mode of the three candidate compounds at HPrK/P was investigated by molecular docking using the Glide docking tool [38, 39] of Schrödinger’s Maestro Molecular Modeling Suite (Schrödinger Release 2022-1: Maestro, Schrödinger, LLC, New York, NY). The protein structures were also visualised using the Pymol Molecular Graphics System (version 2.3, Schrödinger, LLC). The structures of the reference and hit compounds were prepared with the LigPrep tool of Maestro. The missing hydrogen atoms were added, and alternative protonation/ionization states (if any) were generated at pH 7 ± 2 using Epik [40]. Finally, the structures were optimised using the OPLS4 force field [41] to get low-energy 3D conformers of the ligands. The reference and hit compounds’ binding was investigated at all the three Modeller-built comparative models of HPrK/P (i.e., the multi-template-based monomeric model and the dimeric model with and without the Mg2+ and phosphate ions). All three models were processed with the Protein Preparation Wizard [42] of Maestro: the missing hydrogen atoms were added, and the hydrogen bond network was optimized with PROPKA at pH 7.0 [43, 44]. The protein models were energy-minimized using the OPLS3e force field [41] with the convergence criteria of the root-mean-square deviation (RMSD) 0.3 Å for all heavy atoms. Due to the absence of a co-crystallized ligand, we identified the docking sites using Maestro’s SiteMap tool [45, 46], which also detected the same binding pocket whose properties had been used for the virtual screening protocol. However, that site was not the best-ranked among the pockets analysed. Moreover, in the virtual screening phase we had not considered the multimeric form of the enzyme and observed that the initial site detected in the monomeric model would not even exist in the hexameric molecule as the K3-loop of an adjacent subunit would be located at that site (thus, we named the site the ‘K3-loop site’). The best-ranked pocket by SiteMap was the substrate-binding pocket on the phosphate-binding side of the P-loop, next to the flexible/disordered loop. Since the reference inhibitor has also been suggested to act on the HPrK/P substrate site [10], we chose both the initial K3-loop site in the monomeric model and the substrate binding site in the dimeric models for the binding mode studies. With Maestro’s Receptor Grid Generation tool, the docking site for the monomer model was defined using the same residues as at the virtual screening stage. The size of the inner grid box was kept at the default size (10 × 10 × 10 Å3) whereas the outer box size was set to 30 × 30 × 30 Å3. For the dimeric models (with and without ions), the centre of the docking grid was defined by the residues forming the substrate-binding site (Arg245, Thr294, Leu301, Ile305 and His140′, Ser157′, Asp179′, Arg180′, Ile198′, Leu199′, Glu204′; a prime (′) denotes the residues from the other subunit). The inner and outer box sizes were set to 15 × 15 × 15 Å3 and 35 × 35 × 35 Å3, respectively. The most suitable grid box size in each case was selected based on the test docking results of the reference inhibitor. The maximum length of the ligands to be docked was set to 20 Å for all three models. The docking was carried out using the Glide extra precision (XP) [47] mode with flexible ligand sampling. The Epik [40] state penalties for different ionization states/tautomers of the compounds were added to the final docking scores. This can be crucial when ranking compounds with more than one ionization state/tautomer. For each ligand, ten poses were taken for post-docking minimization and a minimum of one pose was generated per ligand.
Molecular dynamics simulations
Molecular dynamics (MD) simulations of the docked ligand-enzyme complexes were carried out for 100 ns with Desmond (Schrödinger Release 2022-1: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2022. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2022) [48] using the OPLS4 force field [49]. Three replicate simulations for each system were performed. The simulation systems were prepared with the System Builder tool of Desmond. An orthorhombic simulation box with Periodic Boundary Conditions (PBC) was filled with single point charge (SPC) water [50] and a 10-Å buffer space was left between the solute (ligand-enzyme complex) and the box edge. In addition, the system was neutralized using an appropriate number of chloride ions for each model. The simulation system was initially relaxed with and without restraints on the solute heavy atoms using the following stepwise relaxation protocol: 12 ps of Brownian dynamics in NVT ensemble at 10 K using a Berendsen thermostat and a 1-fs time step with restraints; 12 ps of Langevin dynamics in NPT ensemble at 300 K and 1 atm pressure using a Berendsen thermostat and barostat with restraints and 24 ps without restrains. The actual production simulations were performed in NPT ensemble for 100 ns with a 2-fs time step. The temperature was set at 300 K with the Nosé-Hoover chain thermostat [51, 52] and pressure at 1.01325 bar with the Martyna-Tobias-Klein barostat [53] using isotropic coupling and relaxation time of 1 ps and 2 ps, respectively. For handling the short-range Coulombic interactions a cut-off radius of 9.0 Å was used whereas the u-series decomposition of the Coulomb potential was used for the long-range electrostatic interactions [54]. The simulation trajectories were analysed using the Maestro in-built Simulation Interactions Diagram tool after which the interaction analysis data generated was processed with Microsoft Excel360 to prepare illustrating graphs to aid the analysis.
Binding free energy analysis
Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) method implemented in Prime [55, 56] (Schrödinger Release 2022-1: Prime, Schrödinger, LLC, New York, NY, 2022) of Maestro was used to predict the binding free energy (ΔGbind) of the docked ligands at the HPrK/P substrate binding site. The ΔGbind was calculated using the following equation:
where, Gcomplex is the free energy of the ligand–protein complex, Greceptor is the free energy of the protein and Gligand is the free energy of the ligand. These energy values are calculated as
where, GMM is the calculated molecular mechanics (MM) energy for the force field applied, and GSolv is the solvation energy in the generalized Born approximation. Prime/MM-GBSA analysis was carried for both the initial docking poses and the final simulated poses after MD using the ‘thermal_mmgbsa.py’ script from Schrödinger with the variable-dielectric generalized Born model (VSGB 2.1 solvation model) [57] and the OPLS4 force field [49]. The Prime/MM-GBSA energies were calculated for the complete 100-ns trajectories by evaluating the snapshot structures of the complexes at every 100 ps. The results from the replicate simulations were averaged.
Results and discussion
Structural analysis and quality of the HPrK/P comparative models
To illustrate the sequence identities and locate the key C-terminal domain loops and some of the active site residues, a multiple sequence alignment of the E. faecalis HPrK/P sequence with the PDB-retrieved sequences of the S. xylosus and L. casei HPrK/P crystal structures is shown in Fig. 2.

Multiple sequence alignment of E. faecalis HPr kinase/phosphorylase (HPrK/P) sequence (WP_002357309.1/UniProtKB entry: O07664) with the Protein Data Bank-retrieved sequences of selected template crystal structures: both the structure of S. xylosus HPrK/P (PDB ID: 1KO7, A chain) and the structure of L. casei HPrK/P (PDB ID: 1KKL, A chain) were among the templates used for the full-length monomeric model. The three important loops; P-loop (residues 155–162), flexible/disordered loop (residues 235–251) and K3-loop (residues 267–272) are highlighted with red, cyan and pink boxes, respectively. A red and a cyan color arrow point to the catalytic Asp-156 and Arg-245 residues at the P-loop and the disordered loop, respectively. The multiple sequence alignment was created with the Clustal Omega tool [58] and visualized with Mview [59] at EMBL-EBI [60]; cov—sequence coverage; pid—percent identity; identical amino acids are colored according to their physicochemical properties. The coordinates for the disordered loop residues W233-L245 and the C-terminal residues 299–314 are missing from the S. xylosus HPrK/P crystal structure (1KO7, A chain), whereas the N-terminal residues up till E135, the disordered loop residues K241-G249, and the C-terminal residues T311-K319 are missing from the L. casei structure (1KKL, A chain)
The Modeller modelling alignments for the monomeric and dimeric models are shown in Supplementary Information (p. S2, S3). The best comparative models of E. faecalis HPrK/P of each type (monomer/dimer) were selected based on the DOPE score (− 32,637.54/− 39,614.91 for the monomeric/dimeric models, respectively) and the stereochemical quality of the models. The stereochemical quality of the generated models was assessed using the Ramachandran plots generated by PROCHECK [32]. The Ramachandran plot of the monomeric model showed 92.0% of the residues in the most favoured regions, 6.9% in additionally allowed regions, 0.7% in generously allowed regions and only 0.4% (Asp179) in a disallowed region (see Supplementary Information, Fig. S1). On the other hand, the Ramachandran plot of the homodimeric protein model showed 94.2% of the residues in the most favoured regions, 5.1% in additionally allowed regions, 0.0% in generously allowed regions and only 0.6% (Asp179 in both chains; Asp46 and Asp224 according to the dimeric model’s residue numbering: 1st chain 1–178, 2nd chain 179–356) in disallowed regions (Fig. S1). The residue found in the disallowed region has been identified as the proton donor/acceptor in the dephosphorylation/phosphorylation reaction, respectively (UniProtKB entry O07664). It is located at the tip of the βD–βE hairpin loop and its phi/psi angles differ from the usual values in the template structures (e.g. 1KKM and 1KO7), explaining why it is in the disallowed region also in the models. The RMSD of Cα atoms of the full-length monomeric model was found to be 0.747 Å when superimposed on one of the templates (PDB ID: 1KO7, A chain) and the RMSD of the C-terminal catalytic domain models of the homodimeric HPrK/P was found to be 0.272 Å when superimposed on the template (PDB ID: 1KKM, A and B chains) using PyMol. The overall model quality and degree of nativeness for both the models were evaluated using the ProSA web server. The Z-score was found to be − 6.92 for the monomer model and − 5.67 for the homodimeric protein model, confirming that the model structures are within the Z-score range that is typically found for native proteins of similar size (Fig. S1). The Qualitative Model Energy Analysis (QMEAN) was done using the QMEANDisCo scoring function. It includes a term for estimating the local per-residue quality based on the similarity of pairwise residue-residue distances in the model compared to the sets of distance constraints obtained from homologous structures [35]. The Global Score is an average of the per-residue lDDT (the local Distance Difference Test) score that ranges from 0 to 1 (1 is good) and was found to be 0.73 ± 0.05 for the monomeric protein model and 0.78 ± 0.05 for the homodimeric protein model (more details are provided in Supplementary Information, Fig. S1). We also compared our models with the artificial intelligence-based AlphaFold [61] model of E. faecalis HPrK/P (alphafold.ebi.ac.uk). When aligned with the AlphaFold full-length monomeric model, the RMSD of Cα atoms for our monomeric models was 0.994 Å (initial model) and 0.852 Å (multi-template model) whereas the RMSD was only 0.601 Å for the C-terminal catalytic domains in our dimeric models. The disordered loop conformation and the C-terminal helix orientation in the dimeric models was very similar to the AlphaFold structure (more similar than in the monomer models). On the other hand, the AlphaFold model was very similar (RMSD of the Cα atoms: 0.585 Å) to the template that was used for building the dimeric model (PDB ID: 1KKM, A and B chains).
Drug screening and docking
The virtual screening of putative HPrK/P inhibitors was performed with the HitsGen™ module of the INVENTUS software suite by (i) scanning the inbuilt compound library based on physicochemical and structural properties of a known HPrK/P inhibitor [10] and the selected binding site (K3-loop site) and (ii) ranking the hit molecules with the bio-affinity score. Three hit compounds with the best score were selected for further investigations (Table 1).
The binding mode of the hit compounds at HPrK/P was further investigated by docking them both to the K3-loop site in the monomeric model and the substrate-binding area of the dimeric HPrK/P models. The initial assessment of the docked poses revealed that the compounds had been successfully docked at these sites; in the K3-loop site between the P-loop (residues 155–162) and the K3-loop (residues 266–271) and in the substrate-binding area, close to the flexible loop region that contains the catalytically critical Arg245. The initial docking scores (Table 2) are seemingly poor for all three hit compounds at both sites, although slightly better at the substrate binding site, and there might be several reasons to this. Either the compounds do not bind to the particular site, or they bind with only low affinity, or the comparative models of HPrK/P are not accurate, especially regarding the conformation of the side chains and flexible regions. The binding site conformation crucially affects the ligand docking poses and possible ligand–protein interactions. Since the flexible/disordered loop containing Arg245 is part of the substrate binding site, the site is likely to be very dynamic and is affected by the binding partner’s size and other properties. Therefore, we further investigated the docked poses using molecular dynamics (MD) simulations to see whether the docked complexes were stable and if we could see any improvement in the estimated binding free energies (ΔGbind, kcal/mol) during the MD simulations (Table 2). Of note, the reference inhibitor 3-B-3 most likely binds to the substrate-binding site and its ditopic structure is beneficial for the inhibitory activity [10]. This is consistent with the predicted binding affinities that are much better for the substrate-binding site than for the K3-loop site. The docked pose of NITSKI5508 in all three HPrK/P models before the MD simulation is shown in Fig. 3 (the poses of the other hits are presented in Supplementary Information, Figs. S2, S3).

The virtual screening hit compound NITSKI5508 docked in the E. faecalis HPrK/P monomeric and substrate-binding site. The docking site interactions are shown at all the three HPrK/P models in 3D (left) and 2D (right). Top: full-length monomeric HPrK/P model; middle: homodimeric model of the HPrK/P C-terminal catalytic domain; bottom: homodimeric model of the HPrK/P C-terminal catalytic domain with Mg2+ and PO43− ions. Left panel: one monomer chain of HPrK/P is shown in gray color cartoon representation while the other one is in yellow. The docked ligand is shown in ball-and-stick representation (cyan carbon atoms) and the binding site residues are in sticks (green carbon atoms); Mg2+ ion is shown as a pink sphere and the phosphate ion in ball-and-stick representation; oxygen atoms are shown in red, nitrogen atoms in blue, phosphorus in dark pink, hydrogen atoms in white. Yellow dashed lines indicate hydrogen bond interactions. Residue numbering in the dimeric models: 1st monomer chain 1–178, 2nd monomer chain 179–356 (corresponding to residues 134–311 in the monomer model)
MD simulation analysis
Stability of the complexes
The stability of the ligand-HPrK/P (monomer and homodimer models with and without ions) interactions was assessed by observing the changes in the protein backbone RMSD and backbone root-mean-square fluctuation (RMSF) per residue as well as the ligand RMSD with respect to the protein during the MD simulation (Supplementary Information, Figs. S4–S10). In general, the protein RMSD converged for all models with all ligands. However, the RMSD values were significantly higher for the monomeric model. The RMSF plot reveals that it is mainly the N-terminal domain residues of the monomeric model and the C-terminus of all models that are fluctuating significantly, although the flexible/disordered loop at the substrate-binding site also shows a visible peak in the fluctuation graphs. Ligand fluctuations with respect to the protein revealed that all hit compounds found a relatively stable pose in the monomeric model towards the end of the simulation although the initial fluctuations were large especially for NITSKI152 and NITSKI5508. On the other hand, positions of NITSKI8583 and NITSKI5508 fluctuated more in the dimeric models than the position of NITSKI152 (Fig. S10).
Interaction analysis
Detailed interaction data from the MD simulations of the ligands with the HPrK/P models are presented in the Supplementary Material (Table S1, Figs. S11–S16). At the substrate-binding site, many ligand–protein interactions were observed with the highly disordered loop (residues 235–251) and its surrounding region. This region plays a significant role in the protein–protein interaction of HPrK/P and its substrate HPr [62]. Therefore, the binding of inhibitors in this region can interfere with the binding of HPr and, thus, will impede the catalytic reactions. The impairment will further abolish the carbon catabolism, required for the essential functioning of a bacterial cell [17]. For example, in the simulations of the dimeric models, Arg245 and Leu246 of the flexible loop frequently formed hydrogen bonding or hydrophobic interactions with the ligands, respectively. On the other hand, in the simulations of the monomeric model, especially residues Ile159 (P-loop region) and Val273 (K3-loop region) formed polar (via the backbone atoms) or hydrophobic interactions (via the side chains) with the ligands at the K3-loop site. Furthermore, it has been shown that residues from the C-terminal α-helix (α3) of HPrK/P also take part in binding the substrate [16]. In the simulations of the dimeric models, Phe297 and Leu301 of the C-terminal helix form aromatic or hydrophobic interactions with the ligands. In addition to the direct hydrogen bonding interactions, many water-mediated interactions also contribute to the binding affinities of the ligands (Figs. S11–S16).
Binding free energy calculations
During the MD simulations, the Prime/MM-GBSA binding free energies were calculated for all the three hit compounds at the three different HPrK/P protein models at 100-ps intervals (Table 2 and Supplementary Information, Fig. S17). In general, the binding free energies of the hit compounds improved somewhat during the MD simulations as they found a more favorable pose compared to the initial docking pose. The reference compound 3-B-3 shows the best binding free energies throughout the simulations at both sites, but the energies are much better at the substrate binding site. On the other hand, NITSKI152 exhibits the best binding free energies of the hit compounds at both sites, and comparable energies at the K3-loop site with the reference compound. The presence of the added ions in the dimer model had a positive effect on the Glide XP docking scores especially for the hit compounds, but this effect was not seen in the Prime/MM-GBSA energies before or during the MD simulations (Table 2). The effect of the pose refinement by MD simulations was evidently beneficial for all the other compounds but not NITSKI5508. However, the improvement in the average binding energy was not significant for the reference compound at the K3-loop site, which would support the fact that it rather binds at the substrate-binding site. Despite the seemingly unfavorable binding energies, a crude decoy docking test suggests that it was not mere serendipity that the three hit compounds were discovered using the K3-loop site (Supplementary Information p. S19, Fig. S18). In case the enzyme is an obligate hexamer to be functional, these compounds may disrupt the multimerization process once the protein chains have folded and are assembled into the hexameric form. For example, human thymidylate synthase (EC 2.1.1.45) is an obligate dimer and indeed, it has been shown experimentally that the dimeric enzyme can be disrupted by small molecules, shifting the equilibrium to monomers that are then degraded easily [63].
Effect of inhibitors on morphology and growth of resistant strain
Microscopy results showed that resistant E. faecalis formed clumps and a web-like structure by joining the chains (Fig. 4a). However, in the presence of inhibitors, NITSKI8583 and NITSKI5508, the clump formation and webbing were observed to be significantly reduced (Fig. 4c, d). The growth of the resistant strain was also found to be significantly decreased when grown in the presence of these inhibitors (Fig. 5). These results well corroborate with the previous reports that showed that deficiency of HPrK/P protein leads to a pleiotropic effect on bacterial physiology [64] and inactivation of HPrK/P activities results in the deleterious effect on the bacterial growth [20]. HPrK/P deficient strains are unable to grow on the phosphoenolpyruvate:glycose phosphotransferase system (PTS) and on most non-PTS carbohydrates [65].

Gram staining of E. faecalis examined under a light microscope (magnification ×1000). Resistant E. faecalis strain-control (a), effect of NITSKI152 (b), NITSKI8583 (c), and NITSKI5508 (d) on resistant E. faecalis

Growth curve of resistant E. faecalis. Resistant strain grown in the absence (control) and in the presence of NITSKI152, NITSKI8583 and NITSKI5508 compounds. Data points are presented as mean (n = 3) ± SD
Western blot analysis of HPr-Ser-phosphorylation
When resistant E. faecalis was grown in the presence of inhibitors, the phosphorylation of HPr-Ser46 residue was found to be decreased significantly. Inhibitors NITSKI8583 and NITSKI5508 were found to be the most potent to reduce the activity of HPrK/P, which was in concordance with the decreased HPr-Ser phosphorylation (Fig. 6). In our previous study, we found that with the progression of nisin resistance in E. faecalis, the phosphorylation of the Ser46 residue of HPr protein was enhanced [23]. Researchers [64] also reported that inactivation of hprK gene results in the loss of kinase and phosphorylase activity, which severely reduced the growth of bacteria.

Western blot analysis of HPr-Ser phosphorylation of E. faecalis: HPr-Ser46 phosphorylation in a sensitive and a resistant strain. Sensitive: nisin sensitive E. faecalis; Resistant: nisin resistant E. faecalis; Resistant + N: nisin resistant E. faecalis grown in the presence of nisin; N: Nisin; A: NITSKI152; B: NITSKI8583; C: NITSKI5508. Data represent relative density mean (n = 3) ± SD. Statistical analysis was performed using One Way ANOVA Tukey test. *P < 0.05, **P < 0.01and **P < 0.001
Conclusions
Enterococci are Gram-positive bacteria and are considered the third most common nosocomial pathogens. In Gram-positive bacteria, HPrK/P regulates the transport of carbon source required for growth and other essential functions. It also regulates the phosphorylation of HPr protein, implicated in the virulence processes of pathogenic bacteria. Most importantly, deficiency of HPrK/P reduces the growth of bacteria. Therefore, a search for inhibitors of HPrK/P is of clinical interest. The present study was conducted to identify drug-like compounds able to inhibit the activity of E. faecalis HPrK/P. A commercial compound library was computationally screened to identify compounds with high predicted binding energy at an identified site between the P-loop and K3-loop of HPrK/P (named as the ‘K3-loop site'), utilizing a comparative model of E. faecalis HprK/P monomer and the structure of a known benzimidazole inhibitor. To evaluate the efficacy of the screened inhibitors, in-vitro assays were carried out. Out of the three hit compounds, two (NITSKI8583 and NITSKI5508) potently reduced the activity of HPrK/P and the growth of multi-drug resistant E. faecalis in vitro. Putative binding modes of the hit compounds were predicted by molecular docking both at the K3-loop site and at the substrate-binding site of HPrK/P. Subsequent MD simulations of the ligand–protein complexes were carried out to study the stability of the binding interactions, suggesting that the compounds are able to form favorable interactions at the studied sites. However, the predicted binding energies suggest a relatively low affinity of the hit compounds for both the sites, whereas the reference compound clearly prefers the substrate-binding site, consistent with previous research. To be able to confirm the real binding mode and site of the hit compounds, experimental research is required. In conclusion, we have reported potential HPrK/P inhibitor candidates (NITSKI8583 and NITSKI5508) against E. faecalis. Further studies are required to verify the compounds’ efficacy on other Gram-positive bacteria to evaluate their usefulness as valuable antimicrobial agents.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding authors.
References
Jeong K-W, Lee J-Y, Kang D-I et al (2009) Screening of flavonoids as candidate antibiotics against Enterococcus faecalis. J Nat Prod 72:719–724. https://doi.org/10.1021/np800698d
Rice LB (2006) Antimicrobial resistance in gram-positive bacteria. Am J Med. https://doi.org/10.1016/j.amjmed.2006.03.012
Zhang Y, Du M, Chang Y et al (2017) Incidence, clinical characteristics, and outcomes of nosocomial Enterococcus spp. bloodstream infections in a tertiary-care hospital in Beijing, China: a four-year retrospective study. Antimicrob Resist Infect Control 6:73. https://doi.org/10.1186/s13756-017-0231-y
Barnes AMT, Dale JL, Chen Y et al (2017) Enterococcus faecalis readily colonizes the entire gastrointestinal tract and forms biofilms in a germ-free mouse model. Virulence 8:282–296. https://doi.org/10.1080/21505594.2016.1208890
Kumar S, Devi S, Sood SK et al (2019) Antibiotic resistance and virulence genes in nisin-resistant Enterococcus faecalis isolated from raw buffalo milk modulate the innate functions of rat macrophages. J Appl Microbiol 127:897–910. https://doi.org/10.1111/jam.14343
Agudelo HN, Huycke MM (2014) Enterococcal Disease, Epidemiology, and Implications for Treatment. In: Gilmore MS, Clewell DB, Ike Y, Shankar N (eds) Enterococci: From Commensals to Leading Causes of Drug Resistant Infection. Massachusetts Eye and Ear Infirmary, Boston
Nallapareddy SR, Wenxiang H, Weinstock GM, Murray BE (2005) Molecular characterization of a widespread, pathogenic, and antibiotic resistance-receptive Enterococcus faecalis lineage and dissemination of its putative pathogenicity island. J Bacteriol 187:5709–5718. https://doi.org/10.1128/JB.187.16.5709-5718.2005
Moellering RC (1992) Emergence of enterococcus as a significant pathogen. Clin Infect Dis 14:1173–1176. https://doi.org/10.1093/clinids/14.6.1173
Miller WR, Munita JM, Arias CA (2014) Mechanisms of antibiotic resistance in enterococci. Expert Rev Anti Infect Ther 12:1221–1236
Bunyapaiboonsri T, Ramström H, Ramström O et al (2003) J Med Chem 46:5803–5811. https://doi.org/10.1021/jm030917j
Schaenzer AJ, Wlodarchak N, Drewry DH et al (2017) A screen for kinase inhibitors identifies antimicrobial imidazopyridine aminofurazans as specific inhibitors of the Listeria monocytogenes PASTA kinase PrkA. J Biol Chem 292:17037–17045. https://doi.org/10.1074/jbc.M117.808600
Görke B, Stülke J (2008) Carbon catabolite repression in bacteria: Many ways to make the most out of nutrients. Nat Rev Microbiol 6:613–624
Kravanja M, Engelmann R, Dossonnet V et al (1999) The hprK gene of Enterococcus faecalis encodes a novel bifunctional enzyme: the HPr kinase/phosphatase. Mol Microbiol 31:59–66. https://doi.org/10.1046/j.1365-2958.1999.01146.x
Nessler S (2005) The bacterial HPr kinase/phosphorylase: a new type of Ser/Thr kinase as antimicrobial target. Biochim Biophys Acta - Proteins Proteom 1754:126–131
Fieulaine S, Morera S, Poncet S et al (2001) X-ray structure of HPr kinase: a bacterial protein kinase with a P-loop nucleotide-binding domain. EMBO J 20:3917–3927. https://doi.org/10.1093/emboj/20.15.3917
Fieulaine S, Morera S, Poncet S et al (2002) X-ray structure of a bifunctional protein kinase in complex with its protein substrate HPr. Proc Natl Acad Sci USA 99:13437–13441. https://doi.org/10.1073/pnas.192368699
Poncet S, Mijakovic I, Nessler S et al (2004) HPr kinase/phosphorylase, a Walker motif A-containing bifunctional sensor enzyme controlling catabolite repression in Gram-positive bacteria. Biochim Biophys Acta - Proteins Proteom 1697:123–135
Steinhauer K, Jepp T, Hillen W, Stülke J (2002) A novel mode of control of Mycoplasma pneumoniae HPr kinase/phosphatase activity reflects its parasitic lifestyle. Microbiology (N Y) 148:3277–3284. https://doi.org/10.1099/00221287-148-10-3277
Dossonnet V, Monedero V, Zagorec M et al (2000) Phosphorylation of HPr by the bifunctional HPr kinase/P-Ser-HPr phosphatase from Lactobacillus casei controls catabolite repression and inducer exclusion but not inducer expulsion. J Bacteriol 182:2582–2590. https://doi.org/10.1128/JB.182.9.2582-2590.2000
Hanson KG, Reizer J, Steinhauer K et al (2002) HPr kinase/phosphatase of Bacillus subtilis: expression of the gene and effects of mutations on enzyme activity, growth and carbon catabolite repression. Microbiology (N Y) 148:1805–1811. https://doi.org/10.1099/00221287-148-6-1805
Pensinger DA, Aliota MT, Schaenzer AJ et al (2014) Selective pharmacologic inhibition of a PASTA kinase increases Listeria monocytogenes susceptibility to β-lactam antibiotics. Antimicrob Agents Chemother 58:4486–4494. https://doi.org/10.1128/AAC.02396-14
Lougheed KEA, Osborne SA, Saxty B et al (2011) Effective inhibitors of the essential kinase PknB and their potential as anti-mycobacterial agents. Tuberculosis 91:277–286. https://doi.org/10.1016/j.tube.2011.03.005
Kumar S, Narayan KS, Shandilya S et al (2019) Role of non-PTS dependent glucose permease (GlcU) in maintaining the fitness cost during acquisition of nisin resistance by Enterococcus faecalis. FEMS Microbiol Lett. https://doi.org/10.1093/femsle/fnz230
Altschul SF, Gish W, Miller W et al (1990) Basic local alignment search tool. J Mol Biol 215:403–410. https://doi.org/10.1016/S0022-2836(05)80360-2
Berman HM (2000) The protein data bank/biopython. Presentation 28:235–242. https://doi.org/10.1093/nar/28.1.235
Márquez JA, Hasenbein S, Koch B et al (2002) Structure of the full-length HPr kinase/phosphatase from Staphylococcus xylosus at 1.95 Å resolution: mimicking the product/substrate of the phospho transfer reactions. Proc Natl Acad Sci USA 99:3458–3463. https://doi.org/10.1073/pnas.052461499
Biasini M, Bienert S, Waterhouse A et al (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–W258. https://doi.org/10.1093/NAR/GKU340
Allen GS, Steinhauer K, Hillen W et al (2003) Crystal structure of HPr kinase/phosphatase from Mycoplasma pneumoniae. J Mol Biol 326:1203–1217. https://doi.org/10.1016/S0022-2836(02)01378-5
Chaptal V, Vincent F, Gueguen-Chaignon V et al (2007) Structural analysis of the bacterial HPr kinase/phosphorylase V267F mutant gives insights into the allosteric regulation mechanism of this bifunctional enzyme. J Biol Chem 282:34952–34957. https://doi.org/10.1074/JBC.M705979200
Webb B, Sali A (2016) Comparative protein structure modeling using MODELLER. Curr Protoc Bioinform 54:5.6.1-5.6.37. https://doi.org/10.1002/CPBI.3
Shen M, Sali A (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci 15:2507–2524. https://doi.org/10.1110/PS.062416606
Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26:283–291. https://doi.org/10.1107/s0021889892009944
Sippl MJ (1993) Recognition of errors in three-dimensional structures of proteins. Proteins: Struct Funct Bioinform 17:355–362. https://doi.org/10.1002/PROT.340170404
Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35:407–410
Studer G, Rempfer C, Waterhouse AM et al (2020) QMEANDisCo—distance constraints applied on model quality estimation. Bioinformatics 36:1765–1771. https://doi.org/10.1093/bioinformatics/btz828
Singh T, Biswas D, Jayaram B (2011) AADS—an automated active site identification, docking, and scoring protocol for protein targets based on physicochemical descriptors. J Chem Inf Model 51:2515–2527. https://doi.org/10.1021/ci200193z
Mukherjee G, Jayaram B (2013) A rapid identification of hit molecules for target proteins via physico-chemical descriptors. Phys Chem Chem Phys 15:9107–9116. https://doi.org/10.1039/c3cp44697b
Friesner RA, Banks JL, Murphy RB et al (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47:1739–1749. https://doi.org/10.1021/jm0306430
Halgren TA, Murphy RB, Friesner RA et al (2004) Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 47:1750–1759. https://doi.org/10.1021/jm030644s
Shelley JC, Cholleti A, Frye LL et al (2007) Epik: a software program for pKa prediction and protonation state generation for drug-like molecules. J Comput Aided Mol Des 21:681–691. https://doi.org/10.1007/s10822-007-9133-z
Lu C, Wu C, Ghoreishi D et al (2021) OPLS4: improving force field accuracy on challenging regimes of chemical space. J Chem Theory Comput. https://doi.org/10.1021/acs.jctc.1c00302
Madhavi Sastry G, Adzhigirey M, Day T et al (2013) Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 27:221–234. https://doi.org/10.1007/s10822-013-9644-8
Søndergaard CR, Olsson MHM, Rostkowski M, Jensen JH (2011) Improved treatment of ligands and coupling effects in empirical calculation and rationalization of p K a values. J Chem Theory Comput 7:2284–2295. https://doi.org/10.1021/CT200133Y/SUPPL_FILE/CT200133Y_SI_001.PDF
Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH (2011) PROPKA3: consistent treatment of internal and surface residues in empirical p K a predictions. J Chem Theory Comput 7:525–537. https://doi.org/10.1021/CT100578Z/SUPPL_FILE/CT100578Z_SI_001.PDF
Halgren T (2007) New method for fast and accurate binding-site identification and analysis. Chem Biol Drug Des 69:146–148. https://doi.org/10.1111/j.1747-0285.2007.00483.x
Halgren TA (2009) Identifying and characterizing binding sites and assessing druggability. J Chem Inf Model 49:377–389. https://doi.org/10.1021/ci800324m
Friesner RA, Murphy RB, Repasky MP et al (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49:6177–6196. https://doi.org/10.1021/jm051256o
Bowers KJ, Chow DE, Xu H et al (2007) Scalable algorithms for molecular dynamics simulations on commodity clusters. Institute of Electrical and Electronics Engineers (IEEE), pp 43–43
Roos K, Wu C, Damm W et al (2019) OPLS3e: extending force field coverage for drug-like small molecules. J Chem Theory Comput 15:1863–1874. https://doi.org/10.1021/acs.jctc.8b01026
Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J (1981) Interaction models for water in relation to protein hydration. Springer, Dordrecht, pp 331–342
NosÉ S (2002) A molecular dynamics method for simulations in the canonical ensemble. Mol Phys 100:191–198. https://doi.org/10.1080/00268970110089108
Nosé S (1998) A unified formulation of the constant temperature molecular dynamics methods. J Chem Phys 81:511. https://doi.org/10.1063/1.447334
Martyna GJ, Tobias DJ, Klein ML (1994) Constant pressure molecular dynamics algorithms. J Chem Phys 101:4177. https://doi.org/10.1063/1.467468
Predescu C, Lerer AK, Lippert RA et al (2020) The u-series: a separable decomposition for electrostatics computation with improved accuracy. J Chem Phys 152:084113. https://doi.org/10.1063/1.5129393
Jacobson MP, Pincus DL, Rapp CS et al (2004) A hierarchical approach to all-atom protein loop prediction. Proteins: Struct Funct Genet 55:351–367. https://doi.org/10.1002/prot.10613
Jacobson MP, Friesner RA, Xiang Z, Honig B (2002) On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol 320:597–608. https://doi.org/10.1016/S0022-2836(02)00470-9
Li J, Abel R, Zhu K et al (2011) The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins: Struct Funct Bioinform 79:2794–2812. https://doi.org/10.1002/prot.23106
Sievers F, Wilm A, Dineen D et al (2011) Fast scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. https://doi.org/10.1038/msb.2011.75
Brown NP, Leroy C, Sander C (1998) MView: a web-compatible database search or multiple alignment viewer. Bioinformatics 14:380–381. https://doi.org/10.1093/BIOINFORMATICS/14.4.380
Madeira F, Park YM, Lee J et al (2019) The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res 47:W636–W641. https://doi.org/10.1093/NAR/GKZ268
Jumper J, Evans R, Pritzel A et al (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596(7873):583–589. https://doi.org/10.1038/s41586-021-03819-2
Maurer T, Meier S, Kachel N et al (2004) High-resolution structure of the histidine-containing phosphocarrier protein (HPr) from Staphylococcus aureus and characterization of its interaction with the bifunctional HPr kinase/phosphorylase. J Bacteriol 186:5906–5918. https://doi.org/10.1128/JB.186.17.5906-5918.2004
Luca C, Ferrari S, Santucci M et al (2021) Disrupters of the thymidylate synthase homodimer accelerate its proteasomal degradation and inhibit cancer growth. ChemRxiv. https://doi.org/10.26434/CHEMRXIV.14156918.V1
Huynh PL, Jankovic I, Schnell NF, Brückner R (2000) Characterization of an HPr kinase mutant of Staphylococcus xylosus. J Bacteriol 182:1895–1902. https://doi.org/10.1128/JB.182.7.1895-1902.2000
Monedero V, Poncet S, Mijakovic I et al (2001) Mutations lowering the phosphatase activity of HPr kinase/phosphatase switch off carbon metabolism. EMBO J 20:3928–3937. https://doi.org/10.1093/emboj/20.15.3928
Acknowledgements
This research was supported by funds from the ICAR-National Dairy Research Institute and a fellowship awarded by the Council of Scientific & Industrial Research, India (Suman Kapila and Sandeep Kumar) and by the Åbo Akademi University research mobility programme within the research profiling area “Drug Development and Diagnostics” (R.B.). The Sigrid Jusélius Foundation, Biocenter Finland Bioinformatics and Drug Discovery and Chemical Biology networks, CSC IT Center for Science, Joe, Pentti and Tor Borg Memorial Fund and Prof. Mark Johnson and Dr. Jukka Lehtonen are gratefully acknowledged for the excellent computational infrastructure at the Åbo Akademi University. This work contributes also to the activities within the strategic research profiling area Solutions for Health at Åbo Akademi University (Academy of Finland, # 336355).
Funding
Open access funding provided by Åbo Akademi University (ABO).
Author information
Authors and Affiliations
Contributions
All authors were involved in study design. Sandeep Kumar and SS performed in-vitro experiments and supervised by Suman Kapila; RB conducted the molecular dynamics analysis; Sandeep Kumar and RB participated in writing the manuscript; OMHS-A supervised the MD analysis and critically reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
Authors have no conflict of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kumar, S., Bhadane, R., Shandilya, S. et al. Identification of HPr kinase/phosphorylase inhibitors: novel antimicrobials against resistant Enterococcus faecalis. J Comput Aided Mol Des 36, 507–520 (2022). https://doi.org/10.1007/s10822-022-00461-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-022-00461-6