
Abstract
Purpose
Advances in structural biology, genetics, bioinformatics, etc. resulted in the availability of an enormous pool of information enabling the analysis of the ancestry of pro- and eukaryotic genes and proteins.
Methods
This review summarizes findings of structural and/or functional homologies of pro- and eukaryotic enzymes catalysing analogous biological reactions because of their highly conserved active centres so that non-antibiotics interacted with bacterial targets.
Results
Protease inhibitors such as staurosporine or camostat inhibited bacterial serine/threonine or serine/tyrosine protein kinases, serine/threonine phosphatases, and serine/threonine kinases, to which penicillin-binding-proteins are linked, so that these drugs synergized with β-lactams, reverted aminoglycoside-resistance and attenuated bacterial virulence. Calcium antagonists such as nitrendipine or verapamil blocked not only prokaryotic ion channels but interacted with negatively charged bacterial cell membranes thus disrupting membrane energetics and inducing membrane stress response resulting in inhibition of P-glycoprotein such as bacterial pumps thus improving anti-mycobacterial activities of rifampicin, tetracycline, fluoroquinolones, bedaquilin and imipenem-activity against Acinetobacter spp. Ciclosporine and tacrolimus attenuated bacterial virulence. ACE-inhibitors like captopril interacted with metallo-β-lactamases thus reverting carbapenem-resistance; prokaryotic carbonic anhydrases were inhibited as well resulting in growth impairment. In general, non-antibiotics exerted weak antibacterial activities on their own but synergized with antibiotics, and/or reverted resistance and/or attenuated virulence.
Conclusions
Data summarized in this review support the theory that prokaryotic proteins represent targets for non-antibiotics because of a common evolutionary origin of bacterial- and mammalian targets resulting in highly conserved active centres of both, pro- and eukaryotic proteins with which the non-antibiotics interact and exert antibacterial actions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Two lines of evidence have demonstrated that non-antibiotics exert antibacterial activities. First, antibacterial effects of non-antibiotic drugs are well documented [1,2,3,4,5,6,7,8,9]. Agents discussed in comprehensive reviews will not be alluded to once again in this manuscript. Second, commonly used medications were found to have a significant impact on the faecal microbiome [9,10,11]. Proton pump inhibitors (PPIs), metformin used for the treatment of type II diabetes, laxatives, and psychotropic drugs had the biggest impact on the faecal flora; opioids, serotonine re-uptake inhibitors, tricyclic antidepressants, antihistamines, and even local anesthetics affected the faecal flora, too, but had a much smaller effect. The antibacterial effect of proton pump inhibitors is due to their proton dependent conversion into sulfen-amide derivatives with sulfides constituting the main end products of degradation. PPIs exhibit a non-selective antibacterial activity at an acidic environment but inhibit selectively Helicobacter spp. and Campylobacter spp. at a neutral pH. PPIs exert no specific antibacterial action since binding to a particular target was not observed. PPIs bind to a broad range of intracellular proteins in Helicobacter spp.; binding was enhanced in an acidic environment. The lipophilic nature of PPIs may also result in an unspecific interaction with cell membrane constituents [12,13,14,15,16]. The concentration- and time-dependent antihyperglycaemic effects of metformin are due to AMP-activated protein kinase (AMPK) dependent and independent effects including their downstream effects. Furthermore, metformin inhibited mitochondrial respiration and probably also mitochondrial glycerophosphate dehydrogenase [17, 18]. Copper complexation may also contribute to the antihyperglycaemic action of metformin [19, 20]. Metformin exerted effects not only on the gut microbiome but also against Legionella pneumophila [21] and H. pylori [22,23,24] and several other bacterial- and viral species [25] in vitro, in vivo and in the clinical setting. Proposed mechanisms of antibacterial action encompass those effects attributed to antihyperglycaemic effects of metformin, inhibition of electron transport, and also AMPK independent effects such as immunomodulation, or production of mitochondrial reactive oxygen species thus enhancing bactericidal activities of macrophages [25, 26]. Laxatives constitute a diverse group of agents inhibiting multiple targets with a wide range of biological effects including anti-bacterial-, -viral-, -fungal-, -inflammatory-, and -oxidant activities [27]. Other laxatives bind irreversibly to nucleophilic amino acids, thus inactivating unspecifically the corresponding proteins [28, 29]. Psychotropic drugs interacted with bacterial membranes and transport systems [5, 9]. These data demonstrate that those non-antibiotic drugs exerting most frequently pronounced antibacterial activities affect bacteria unspecifically and target multiple functions.
However, non-antibiotics may hypothetically interact specifically with prokaryotic targets because of structural and/or functional homologies of eukaryotic- and prokaryotic proteins. Evidence has been provided in a companion paper [30] that antibiotics interact with eukaryotic targets because of evolutionarily conserved functions. This review summarizes data describing antibacterial activities of non-antibiotics with processes considered to be essential in pro—as well as eukaryotes. These essential reactions may be evolutionary highly conserved thus offering the chance for inhibitory activities of drugs beyond the borders of the domains eukarya, archae and bacteria. Selected examples for evolutionarily conserved processes are first, phosphorylation and dephosphorylation as a frequently employed mechanism of signalling; second, initiation and propagation of electrical signalling by voltage-gated ion channels; third, metalloenzymes with zinc as a key ion for catalytic functions and as an essential structural element.
Phosphorylation and dephosphorylation
Reversible phosphorylation and dephosphorylation of proteins are essential for the regulation of protein activity and signalling in pro- and eukaryotes. Protein phosphorylation triggers essential processes like cell wall biosynthesis in bacteria and neurologic- or immune responses, endocrine actions, etc. in humans [31,32,33]. Therefore, any interaction with protein phosphorylation or dephosphorylation has far-reaching ramifications.
Inhibition of bacterial serine/threonine/tyrosine protein kinases
Posttranscriptional modification of eukaryotic proteins is primarily accomplished by protein phosphorylation of serine, threonine or tyrosine catalysed by “Hanks-type kinases”, i.e. serine/threonine and sometimes also serine/tyrosine protein kinases (STPKs). Bacterial STPKs were defined as “eukaryotic-type kinases”, but comprehensive phylostratigraphic analysis suggests that Hanks-type kinases present in eu-, prokaryotes and archaea all share a common evolutionary origin [34]. In general, the catalytic regions of bacterial, animal, and human STPKs and phosphorylases show strong homologies [35,36,37,38,39,40,41,42,43,44]. Penicillin-binding proteins (PBPs), being serine transferases [45], and STPKs are linked to an ancillary domain named penicillin-binding and serine/threonine kinase-associated (PASTA). An extracellular PBP domain characterizes PASTA proteins and an intracellular STPK domain that is similar to those of acyl serine transferases found in mammals [46]. PASTAs regulate bacterial metabolism, cell division, and cell wall homeostasis through the recognition of muropeptides and also sense and respond to hostile environmental stress factors such as an immune response or limited nutrient supply as well as antibiotic stress. Therefore, PASTA proteins play a central role in cell wall biosynthesis, virulence and β-lactam resistance [47,48,49,50]. PASTA domains are found in both, the C-terminus of PBPs as well as in bacterial STPKs [51,52,53,54,55]. While the STPK domain is well conserved, PASTA domains are quite divergent [56]. STPKs are involved in antibiotic-resistance as it influences the expression of low-affinity class B PBP5, PBP2X, and PBP2a being associated with cephalosporin-resistance in E. faecium [57,58,59], as well as penicillin-resistance in S. pneumoniae [60,61,62,63], and methicillin-resistance in S. aureus [64,65,66,67], respectively. Likewise, STPKs play a role during growth and β-lactam susceptibility of Corynebacterium spp. [68, 69], B. subtilis [70,71,72,73,74], M. tuberculosis [60, 75,76,77,78,79,80], and other species. Therefore it is not unexpected that several kinase inhibitors of diverse structural classes used in human medicine increased susceptibilities of various bacterial species to cell wall active antibacterial agents.
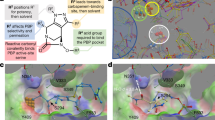
In general, PASTA kinase mutants are hypersusceptible to β-lactams and inhibition of kinases sensitizes in particular Gram-positive bacteria and also Gram-negative bacteria to β-lactams. Antibacterial activities of most of the agents were examined by monitoring time-kill curves so that minimal inhibitory concentrations (MICs) have rarely been reported. Screening of a natural compound library of low molecular weight kinase inhibitors revealed that most of the compounds inhibited in combination with a sub-MIC concentration of nafcillin growth of the methicillin-resistant S. aureus (MRSA) test strain. Staurosporine, an alkaloid isolated from Streptomyces staurosporeus exhibiting unspecific anti-cancer activity due to activation of caspase thus inducing apoptosis, was used as a comparator. The effect of staurosporine on β-lactam susceptibilities was however not due to caspase activation but attributable to a selective inhibition of PASTA kinases. Staurosporine caused in combination with a sub-MIC concentration of nafcillin a 68% growth inhibition, whereas several staurosporine derivatives inhibited the growth of the MRSA test stain to 100% [81, 82]. Staurosporine and the cyclin-dependent kinase inhibitor, AZD5438, sensitized L. monocytogenes to ampicillin, ceftriaxone, cephalexin, and lysostaphin 10- to 100-fold, whereas the activity of vancomycin was not enhanced by staurosporine [83]. Both, inhibition of the kinase activity and sensitization of L. monocytogenes to β-lactams were concentration dependent and parallel processes. An imidazopyridine aminofurazan (GSK690693) and a pyrazolopyridazine-derivative (GW779439X) sensitized L. monocytogenes [84] and MRSA [85, 86], respectively, to various β-lactams (Table 1) via inhibition of STPKs. Camostat, an inhibitor of transmembrane protease serine 2 (TMPRSS2), commercially available in Japan for treatment of chronic pancreatitis and postoperative reflux esophagitis, was found to inhibit viruses such as SARS-CoV 2 [87, 88] and Gram-positive bacteria. MICs of camostat and its derivatives gabexate and nafamostat ranged from ≥ 500 to 50 μM after an 18–20 h incubation in the absence of β-lactams. However, nafamostat exhibited a bactericidal activity during a 6 h incubation period reducing the inoculum by four log10 titres [89, 90].
Imidazopyridine aminofurazans (IPA) inhibited the kinase PknB of mycobacteria and potentiated activities of β-lactams against Mycobacterium spp. Incubation of M. smegmatis and M. chelonae with MIC50-concentrations of meropenem in combination with various IPAs resulted in an inhibition of the test strains by 2.0–0.5 and 8.0 mg/L meropenem, respectively as compared to MICs of 4.0 and 16 mg/L in the absence of IPAs. Likewise, incubation of M. abscessus to a fixed MIC50-concentration of ampicillin in combination with various IPAs resulted in an inhibition of the test strain by 12.5 mg/L as compared to a MIC of 125 mg/L in the absence of IPAs. The inhibitors potentiated activities of normally ineffective β-lactams against Nocardia spp. [91, 92]. Aminobenzimidazoles sensitized M. smegmatis and M. tuberculosis to 14 β-lactams reducing their MICs from ≥ 256 to 1 to 16 mg/L, except for cephalothin, cefadroxil, carbenicillin and piperacillin [93]. Other aminobenzimidazoles sensitized MRSA, methicillin-resistant S. epidermidis, and multidrug-resistant P. aeruginosa as well as A. baumanii not only to penicillin G, oxacillin, and methicillin but also to novobiocin, colistin, tobramycin, ciprofloxacin lowering MICs up to 512-fold [94,95,96]. Another aminobenzimidazole suppressed carbapenem-resistance in NDM-1 producing strains of K. pneumoniae [97]. Yet another aminobenzimidazole inhibited a histidine kinase two-component signalling system. It reconstituted colistin activity in multidrug-resistant A. baumannii and K. pneumoniae by down regulating the pmrCAB operon thus reverting phosphoethanolamine modification of lipid A which causes colistin resistance [98].
The tricyclic antihistamine loratadine inhibited biofilm formation and PASTA kinases Stk and Stk1 in S. epidermidis and S. aureus, respectively, resulting in increased activities of β-lactam antibiotics against MRSA and increased activities of both, β-lactams and vancomycin in vancomycin-resistant strains of S. aureus [3, 99]. A small-molecule quinazoline compound (Inh2-B1) inhibited specifically Stk1 in S. aureus. It reduced MBCs of ceftriaxone and cefotaxime for MRSA from ≥ 100 to ≤ 4 mg/L [100]. The finding that loratadine resensitized MRSA to vancomycin [99] is in contrast to previous reports [83,84,85] that STPK inhibitors revert activities of β-lactams only but not other antibiotic classes. This contradiction may be resolved by the finding that another STPK, i.e. Stp1, contributes to reduced vancomycin susceptibilities in S. aureus as this kinase regulates virulence and cell wall thickness [101, 102].
Several groups synthesized agents derived from protein kinase inhibitor pharmacophores with intrinsic antibacterial activities. An unsaturated crotonic acid derivative (IMB-YH-8) exerted an anti-mycobacterial activity in itself due to STPK inhibition [103]. This molecule inhibited selectively the kinases PknA and PknB of M. tuberculosis. PknB modulates the SigH regulatory pathways, which regulate a transcriptional network responding to various stresses. The MICs of IMB-YH-8 for M. tuberculosis ranged from 0.25 to 1.0 mg/L irrespective of whether the test strains were susceptible or mono- or multiply resistant to isoniazid, rifampin, streptomycin, ethambutol and ofloxacin [103]. The anthraquinone derivative mitoxantrone used in cancer therapy was found to inhibit PknB in M. tuberculosis, thereby preventing its growth [104]. Plant-derived trypsin inhibitors were active against Gram-positive as well as Gram-negative bacteria and exhibited anti-viral activities [105].
AMPKs are highly conserved in yeast, plants, and mammals. It is assumed that AMPKs evolved in the early eukaryote to control the output of carbohydrates produced by acquired bacterial endosymbionts that developed into mitochondria but were thought not to play a role in bacteria [106,107,108]. However, AMPKs were isolated from L. pneumophila [21], H. pylori [22,23,24], Mycobacterium spp., B. subtilis, L. monocytogenes, and rhizobial bacteria [109,110,111,112] and were involved in sensitization of resistant bacteria to antibiotics, too [10, 70, 83,84,85]. Bacterial AMPKs may be inhibited by metformin.
Antibiotic phosphorylating kinases confer resistance to aminoglycosides, macrolides, as well as phenicols and are organized in the antibiotic kinome consisting of the antibiotic resistome and the microbial kinome [113, 114]. It has been shown that the aminoglycoside modifying enzymes O-phosphotransferases are structurally and functionally ortholog to eukaryotic STPKs [115,116,117,118,119]. Pro- and eukaryotic kinases, which are most likely to be simultaneously inhibited by a common ligand, were identified based on the similarity in their ligand-binding profiles rather than via their sequence similarity. A survey of 150,000 kinase inhibitory values, comprising more than 3800 compounds tested against a panel 172 kinases revealed that pyrazolo-pyrimidines had nanomolar affinity against many STPKs involved in cancerogenesis [120] and two of them (pyrazolo-pyrimidines) as well as the flavone quercetin inhibited bacterial O-phosphotransferases and reverted aminoglycoside resistance [121,122,123]. Quercitin occupied the ATP binding site and formed several hydrogen bonds with the phosphotransferase APH(2″)-IVa. Furthermore, flavonoids such as quercitin chelate with metal cations thus inhibiting aminoglycoside-acetyltransferase activity and sensitizing aminoglycoside-resistant strains to these agents [124]. Apart from inhibition of aminoglycoside-phosphotransferases and aminoglycoside-acetyltransferases flavonoids interact unspecifically with bacterial membranes and efflux pumps, so that they exert antibacterial activities in themselves [27, 125] and exert additive or synergistic combination effects in particular with cell-wall active agents [125]. Several STPK substrates were phosphorylated by O-phosphotransferases and vice versa, STPK inhibitors such as several isoquinoline-sulfonamides or the casein kinase-1 inhibitor CKI-7 inhibited O-phosphotransferases. Unfortunately, these inhibitors were active in a cell-free system only but did not reverse aminoglycoside-resistance in living bacteria [126, 127]. The phosphoinositide 3-kinases inhibitor wortmanin inhibited APH(2″)-Ia because of structural homologies of lipid kinases and aminoglycoside modifying enzymes [128].
These data demonstrate that pro- and eukaryotic STPKs share common ancestors and exhibit significant structural and functional homology. Therefore, non-antibiotic inhibitors of eukaryotic STPKs inhibited specifically just one or two bacterial STPKs thus reverting antibiotic resistance in Gram-positive as well as Gram-negative bacteria. It is an open question why the inhibitors just interact with specific bacterial STPKs, e.g. with mycobacterial PknA and PknB out of 11 STPKs, two tyrosine-protein phosphatases and one serine/threonine protein phosphatase, and leave the others unaffected. However, this phenomenon may offer the chance to synthesize inhibitors of structurally and functionally related targets but being sufficiently different to allow for selectivity for pro- but not eukaryotic STPKs and in parallel inhibiting specifically just the relevant bacterial STPK.
Disabling bacterial pathogenesis by targeting host cell serine/tyrosine kinases
Enteropathogenic E. coli (EPEC) attach to epithelial cells and express several virulence factors such as the bacterial outer membrane protein intimin which mimics a ligand-receptor interaction. One essential factor causing adherence of EPEC to host intestinal epithelial cells is “translocated intimin receptor” (Tir) which spans the host plasma membrane and binds intimin on the bacterial surface to the epithelial cell resulting in tight adherence of the bacterium to the host cell and formation of lesions. These lesions are characterized by a loss of intestinal microvilli and the formation of actin-filled membranous pedestals that protrude beneath the adherent bacterium. Phosphorylation of Tir by multiple host cell tyrosine kinases, in particular members of the Abl family of tyrosine kinases, is crucial for the formation of actin pedestals and its inhibition or deletion results in a loss of virulence [129, 130]. Pyrido-pyrimidine compounds are developed to treat cancers caused by dysregulated Abl and were found to inhibit bacterial Abl family tyrosine kinases thus blocking pedestal formation and consequently virulence [130].
In general, Abl family tyrosine kinases play an important role in the pathogenesis of e.g. S. flexneri, H. pylori, S. enterica, P. aeruginosa, C. trachomatis, M. tuberculosis, Anaplasma phagocytophilum, and also viruses and parasites. A recent, comprehensive summary [131] has presented data demonstrating that Abl family tyrosine kinases phosphorylate microbial factors required for pathogen entry into-, release from-, and/or motility within host cells. Imatinib and other FDA-approved ATP-competitive inhibitors of Abl family tyrosine kinases affected host cell cytoskeletal dynamics required for cellular protrusions being essential for adhesion of bacteria to and release from host cells thus reducing the virulence of the pathogens.
Inhibition of bacterial serine/threonine phosphatases
As phosphorylation of STPKs is stable partner serine/threonine phosphatases are needed to reverse the regulation. Recently, a serine/threonine phosphatase has been identified in E. coli and has been characterized biochemically [132]. It shows significant homologies to human phosphatase 2C (PP2C) phosphatases [35, 133,134,135,136], so that bacterial homologs are referred to as eukaryote like serine/threonine phosphatases (eSTPs). It is well documented that inhibitors of human phophatases cyclosporin A, tacrolimus (FK506), and rapamycin exhibit anti-fungal and viral but no anti-bacterial activities. However, highly conserved FK506 binding proteins (FKBPs) exist in Legionella spp., Chlamydia spp., N. meningitidis, and P. aeruginosa [137]. FKBPs of these bacterial species function as outer membrane virulence factors. These virulence factors may possibly be inhibitable by cyclosporin A, tacrolimus, and rapamycin as these phosphatase inhibitors bind to FKBPs with high affinity. Tacrolimus and rapamycin inhibited intracellular survival of Legionella spp. theoretically via inhibition of virulence [138] thus indicating that inhibitors of human serine/threonine phosphatases may attenuate bacterial virulence. This finding was probably not followed up consistently as it is generally believed that cyclosporin A, tacrolimus, and rapamycin are not antibacterially active.
Calcium channel blockers
Voltage-dependent calcium channels (Cavs) couple membrane energetics with Ca2+-signalling thereby regulating essential physiological processes. Cavs are highly conserved and are phylogenetically related to bacterial voltage-gated sodium channels (BacNaVs). Previously it was thought that bacteria lack Cavs [139,140,141,142,143,144]. A bacterial sodium channel from Bacillus halodurans was characterized. Commercially available calcium channel blockers inhibited this NaChBac channel. The dose-response curves for dihydropyridines were comparable to those for mammalian ion channels [140]. Furthermore, first prokaryotic calcium channels were identified recently. The newly characterized two BacNav homologs, CavMr from Meiothermus ruber and NavPp from Plesiocystis pacifica, are selective for Ca2+, and selective for Na+ with Ca2+-dependent inhibition, respectively [145]. The amino acid sequences of mammalian Cavs and bacterial BacNavs are very similar and the quaternary structures and functional determinants of BacNavs are well defined [146,147,148,149,150] so that they are used as a model for gating and ion permeation.
It has been described that some calcium channel blockers exhibit quite low in vitro antibacterial activities in the absence of antibiotics with MICs ranging from 10 to 200 mg/L [151,152,153,154,155,156,157] (Table 2). Verapamil was tested against M. tuberculosis and M. abscessus only and was inactive [156, 157]. However, verapamil exhibited bactericidal activity against P. aeruginosa at a concentration of 0.98 μg/mL although its MIC value was as high as 12,768 μg/mL [158]. Also, lacidipine and nifedipine were bactericidally active at twice their MICs and reduced inocula of V. cholera and S. aureus [152] or Shigella spp. and S. typhimurium [153], respectively, by 5–8 log10 titres within 12 h. Likewise, verapamil, nifedipine, nisoldipine, and nitrendipine inhibited E. coli chemotaxis in the micromolar range. At concentrations around tenfold higher than that needed for inhibition of chemotaxis, each of these antagonists inhibited motility. While ≥ 1 mM of verapamil did not reduce viable counts of E. coli, 5 μM each of nifedipine, nisoldipine, and nimodipine reduced survival of the test strain to 50% within 30 min [159]. Verapamil, nitrendipine, and nifedipine reduced spore germination in B. megaterium at a concentration of 1 mM each [160]. Thus, calcium channel blockers affected bacterial physiology at relatively low concentrations and exhibited even a bactericidal activity during the 30 min and 12 h incubation period, respectively, without exhibiting a relevant—if any—effect on the discrete endpoint MIC. Concentrations of calcium antagonists exhibiting in vitro activities against Gram-positive and Gram-negative bacteria should be related to serum concentrations following single oral doses as specified in Table 2. This comparison indicates that even the lowest MICs exceed mean maximal serum concentrations significantly by more than one order of magnitude. However, verapamil, nitrendipine, nisoldipin, nimodipine, nifedipine, and lacidipine affected physiological functions and even viability of the test strains at low concentrations. Similarly, lactividine, amlodipine and verapamil showed protective effects in vivo. Concentrations achieved in experimental animals were significantly lower than their MICs as the agents were administered at human-equivalent and thus subinhibitory doses [152,153,154,155, 161, 162]. Lactividine protected animals from death in a mouse model of V. cholera infection; it reduced viable counts dose dependently and inhibited cholera toxin production in vivo [152, 154]. Amlodipine acted as a tissue protectant in experimentally induced S. aureus rhinosinusitis. Both, monotherapy with cefazolin or amlodipine reduced numbers of macrophages in epithelial cells by 57% and 39%, respectively [155]. Coadministration of verapamil with sub-inhibitory doses of bedaquiline achieved an equivalent anti-mycobacterial effect as the full dose of bedaquilin. This adjunctive effect of verapamil may permit the administration of lower doses of bedaquilin [156, 157].
Calcium antagonists improved in vitro activities of tetracycline and fluoroquinolones against S. aureus, Enterobacteriaceae and non-fermenters irrespective of whether the strains were tetracycline- or fluoroquinolone-resistant [158,159,160]. It also improved activities of ofloxacin, rifampicin, and bedaquilin, but not isoniazid or amikacin against M. tuberculosis as well as M. abscessus (Table 3) [161,162,163,164,165,166,167,168,169]. Verapamil also increased the intraphagocytic activities of isoniazid and rifampicin [168] as well as bedaquilin and moxifloxacin [168] against drug-susceptible and drug-resistant M. tuberculosis and also the intracellular activity of azithromycin against L. monocytogenes [170, 171] and that of daptomycin against S. aureus [172]. Resistance phenotypes but not resistance mechanisms were described for these test strains so that it may be probable but it is not proven that antibiotic resistances were caused by efflux mechanisms which were inhibited by calcium channel blockers. Likewise, amlodipine enhanced the activities of imipenem against multidrug-resistant A. baumanii by inhibiting expression of the resistance nodulation cell division efflux pump AdeABC [173].
Verapamil is assumed to be an inhibitor of P-glycoprotein and efflux pumps in M. tuberculosis and other bacterial species, so that its potentiating activity was attributed to an intraphagocytic and/or intrabacterial drug accumulation [157, 169,170,171,172,173,174,175,176,177] and also increased bioavailability due to interaction with CYP3A4 [157, 178]. Interestingly, the ABC transporter isolated from Lactococcis lactis, LmrA, exports intracellular amphiphilic compounds. When LmrA was expressed in human lung fibroblasts it substituted for P-glycoprotein and conferred multidrug resistance on these human cells. This effect was due to almost identical biochemical characteristics of LmrA and P-glycoprotein showing the same substrate affinities. The activities of both were equally well affected by verapamil [179,180,181,182]. Homologs of LmrA have been found in a variety of bacterial pathogens, suggesting that this resistance mechanism is ancient and plays a crucial role in procaryotes and that its functional homolog, the P-glycoprotein, mediates drug resistance in eukaryotes [183]. In agreement with this theory is the finding that E. coli strains lacking the AcrAB pump are hypersusceptible to calcium channel blockers suggesting that verapamil and other calcium channel blockers are substrates of this bacterial pump. Furthermore, subinhibitory concentrations of verapamil abolished in E. coli the proton motive force, decreased intracellular ATP concentrations, and reduced the growth rate without affecting the synthesis of DNA, RNA and proteins in general or RecA protein in particular. This finding excludes the possibility that cell division could have been inhibited due to an induction of the SOS response. However, verapamil may likely have perturbed the integrity of the bacterial membrane and may have disrupted the assembly of the FtsZ complex [184]. In addition, verapamil affected transport activity of the OpuA (Glycine-betaine transporter) protein from Lactococcus lactis reconstituted into membrane vesicles [183], due to its accumulation in the inner leaflet of the vesicle. These data not only support the theory that extrusion of drugs represents an ancient resistance mechanism with homologous P-glycoproteins structures in pro- and eukaryotes with which calcium channel blockers interact but also suggest that in addition calcium channel blockers affect membrane functions [185, 186].
The theory of an augmented activity of antibiotics due on inhibition of efflux pumps by calcium-channel blockers in both, pro- and eukaryotes, resulting in increased intracellular concentrations has been questioned. The alternative model is based on the physicochemical characteristics of verapamil with a pKa value of 9.68 and a logP value of 5.23. Therefore, verapamil is a lipophilic weak base with a protonated tertiary amine group at neutral pH. At a pH of almost 10 verapamil exists in its least ionized state, so that it has a high affinity to neutral lipid bilayers. At a neutral pH and in particular at a pathophysiologically relevant slightly acidic pH, however, verapamil is positively charged, so that it adheres with high affinity to and inserts into negatively charged lipid membranes thus disrupting membrane energetics and inducing membrane stress response [187, 188]. This interaction with membranes due to physicochemical mechanisms explains why verapamil reduced viable counts of M. tuberculosis by 8 log10 titres within 15 h although bacteria did not replicate and were deprived of nutrients. In addition, membrane disruption was not due to increased intramycobacterial drug accumulation [188]. These data suggest that augmentation of antibiotic activities was not caused by direct inhibition of efflux pumps by verapamil resulting in drug accumulation, but rather by dissipation of the proton motive force, which in turn affects most efflux pumps so that drug accumulation was an indirect consequence of perturbed membrane energetics resulting in altered efflux pump function. In agreement with this conclusion, it was shown that incorporation of verapamil [188, 189] and dihydropyridines [190,191,192,193,194] into the cell- and model membranes affected P-glycoprotein activity. In general, modulation of membrane functions due to physicochemical interactions of dugs with bacterial- and mammalian membranes, respectively, are prevalent amongst almost every antibiotic class provided the agents interact as catamphiphilic drugs with membranes [195, 196]. Physicochemical constants summarized in Table 2 indicate that lipophilicity and ionization vary considerably amongst the calcium channel blockers. Therefore, the P-glycoprotein inhibitory potential of the most lipophilic calcium antagonist verapamil is more pronounced than that of less lipohilic dihydropyridines (e.g. IC50-values of nitrendipine and verapamil, respectively, were 18.2 μM versus 2.8 μM for digoxin transport); analogous data were obtained for other dihydropyridines as compared to verapamil [197,198,199,200]. These data demonstrate that all the calcium-antagonists tested interacted primarily with membranes before drug binding to L-type calcium channel receptors thus secondarily affecting P-glycoprotein functions [187, 188, 193]. It has to be considered that ionization of agents depends on environmental pH values which vary according to pathophysiologically relevant conditions [196]. Furthermore, the pH gradient in microbes varies significantly with the environmental pH, so that also the membrane potential varies with external pH [197,198,199,200,201,202]. Thus, membrane disruption is mutually dependent from the ionization of the agent and the environmental pH, so that the real level of inhibition is a function of the specific condition at the focus of infection and may be variable and hardly predictable.
Calcium channel blockers exert multiple actions in pro- and eukaryotes. In particular, they augmented antibacterial activity and reversed antibiotic resistance in Gram-positive and Gram-negative bacteria. Thus, calcium channel blockers could be used as an adjunct to antibacterial therapy. Antibiotic-calcium channel blocker combinations may result in a beneficial additive or even synergistic antibacterial effect and/or resistance reversal provided dose regimens are optimally adjusted.
The discrepancies between conclusions to be drawn on the basis of the static endpoint parameter MIC demonstrating that calcium channel blockers are devoid of a relevant in vitro antibacterial activity and their pronounced in vitro bactericidal activity and interference with cellular functions as well as their beneficial in vivo efficacy in experimental animals give rise to the question if MIC testing is an appropriate method to analyse antibacterial effects of non-antibiotics. These doubts are reasonable as bacterial voltage-gated calcium flux is correlated with physiological processes like chemotaxis, synthesis of pathogenicity factors, sporulation and modulation of the transcriptome [41, 201, 202]. Therefore, endpoints for a phenotypic examination or functional analysis of antibacterial activities of non-antibiotic drugs should mirror both, the mode of action of and the physiological processes triggered by the non-antibiotic drug. Dynamic physiological processes cannot be described by the static endpoint MIC.
Inhibitors of angiotensin-converting enzyme and other zinc-containing enzymes
The angiotensin I converting enzyme (ACE, or kininase II) is a bivalent zinc-dependent dipeptidyl carboxypeptidase catalysing the conversion of angiotensin I into angiotensin II. Displacement of zinc from the active site inactivates ACE [203,204,205]. The thiol- and carboxyl-group, respectively, of ACE inhibitors such as captopril, elanapril and lisinopril, bind directly to the catalytic zinc. Removal of the thiol-group of captopril or its replacement with a carboxylic acid led to complete loss of activity [206,207,208]. Metallo-β-lactamases (MBLs), too, are characterized by conserved zinc ion binding sites in their active centres. Therefore, it was hypothesized that ACE inhibitors could probably bind to MBLs. Several studies have confirmed that ACE inhibitors form complexes with MBLs [209,210,211,212,213] due to binding of their thiol groups to both active site Zn2+ ions of MBLs of various bacterial species (summarized in [209]). Furthermore, captopril has structural similarity to the MBL degraded penicillin, so that captopril bound to MBLs most similar to that of hydrolysed β-lactams [209]. Such functional homologies suggest that the active sites of MBLs and ACE, respectively, and the mode of binding to their active centres may represent conserved structures. The IC50 values of the most active D-stereoisomer of captopril for various MBLs ranged from 0.07 μM for VIM-2, 1.7 μM for VIM-4, 7.2 μM for IMP-1, 20.1 μM for NDM-1 up to 262.8 μM for SPM-1 [209, 210]. The high affinity of D-captopril to VIM-2 was due to additional interactions observed between VIM-2 and the carboxyl-group of captopril as well as due to additional hydrogen bonds between VIM-2 and other MBLs with captopril [209,210,211,212,213,214]. The combination effect of D-captopril with meropenem was tested against a panel of E. coli, K. pneumoniae, S. marcescens, and P. aeruginosa strains producing VIM-2, VIM-4, IMP-4 and NDM-1 β-lactamases or NDM-1 β-lactamase in combination with CTX-M or TEM-type β-lactamases. Meropenem MICs for these strains ranged from 1 to 512 mg/L as compared to < 0.125 mg/L for the wild type strain. D-captopril reduced the MICs of meropenem for the resistant strains by two- to four dilution steps [209] so that 8 out of 11 strains with MICs greater than the resistant breakpoint of 8 mg/L became susceptible. Using a disc diffusion test it was demonstrated that l-captopril inhibited the growth of E. coli and S. enterica at ≥ 25 mg/mL [215]. Ramipril inhibited M. chelonae at 1.3 mg/L and M. abscessus at ≥ 5.2 mg/L [216]. Some investigational captopril derivatives as well as some approved thiol-containing drugs such as thiorphan, dimercaprol, and tiopronin inhibited activities of NDM-1, VIM-1, and IMP-7 at concentrations lower than those of captopril [217,218,219].
Apart from the functional homology between captopril and β-lactams, ACE is an evolutionary highly conserved protein. DNA sequence analysis revealed that ACE-homologos could be identified in a variety of different eukaryotic phyla and in procaryotes [220,221,222,223,224,225,226,227]. Some of these ACE-like enzymes have been purified and biochemically characterized in vitro. The bacterial ACE-like enzymes from E. coli (EcDCP), Xanthamonas citri (XcACE), and dipeptidyl carboxypeptidases from Pseudomonas spp., P. maltophilia, Corynebacterium equi, B. subtilis and B. pumilus have retained their ability to hydrolyse angiotensin I, among other ACE substrates, and were inhibited by relevant ACE inhibitors. Sequence identities between the enzymes vary from 91 to 73% for XcACE and human N-ACE or ACE2, and 20–8% for EcDCP and the human enzymes, respectively [227,228,229]. Although the overall sequence similarity may be low in some cases, the active site is highly conserved, so that ACE-inhibitors inactivate the enzymes irrespective of their origin effectively. Regardless of the functional and genetic homologies between pro- and eukaryotic ACE homologs, the function of the prokaryotic ACE-like enzymes has still to be defined in most cases. A dipeptidyl-carboxypeptidase with defined activity has been isolated from S. gordonii. The enzyme was a functional homolog of human ACE and was eightfold more active than the recombinant human ACE. The bacterial enzyme was able to hydrolyse the alpha- and beta-chains of fibrinogen. However, 1 μM each of captopril, lisinopril and enalapril, did not inhibit the bacterial enzyme activity. This discrepancy may be due to the fact that human recombinant ACE is a zinc-dependent dipeptidyl-carboxy-peptidase while the bacterial enzyme also behaves as a dipeptidyl-carboxypeptidase, but without an absolute requirement for metal ions, as neither EDTA nor EGTA inhibited the activity of the bacterial enzyme. On the other hand, divalent metal ions caused a more than twofold increase in enzyme activity.
Carbonic anhydrases (classes α, β, and γ) are essential metalloenzymes which play crucial roles in the entire animate system. Their physiological roles, mechanisms of action and agents used clinically as carbonic anhydrase inhibitors have been described in comprehensive reviews [230,231,232]. The active site of these enzymes consists of Zn2+ with which metal complexing anions or sulfonamides like acetazolamide, methazolamide, ethoxzolamide, dichlorophenamide, dorzolamide and brinzolamide, used clinically in glaucoma patients to lower the intraocular pressure, interact [232, 233]. Carbonic anhydrases could be isolated from a large variety though not from all bacterial species [234] and were inhibited by the sulfonamides acetazolamide and methazolamide (summarized in [231]). Inhibition of carbonic anhydrases leads to growth impairment of bacteria, viruses, and yeasts, but species-specific sulfonamide derivatives have been synthesized (summarized in [233]). However, bacterial species being devoid of carbonic anhydrases were nevertheless inhibited by sulfonamides [235] probably due to their classical mode of action as a PABA antagonist. Clinical efficacy of acetazolamide 500 mg b.i.d. for 4 days was examined in a pilot study in eight volunteers with active H. pylori infection. The urea breath test reverted in none of the patients to negative [236] possibly due to an effect of acetazolamide on acid secretion, which may have prevented an effect on H. pylori. Thus, inhibition of bacterial carbonic anhydrases by sulfonamides may represent a third target for novel sulfonamides in addition to their antagonism with para-aminobenzoate and their function as alternate substrates for pteridine resulting in an inhibition of dihydropteroate synthase. Captopril and elanapril inhibited carbonic anhydrases, too, but not as effectively as sulfonamides [237].
Efflux pump inhibitors and non-steroidal anti-inflammatory drugs in particular
Efflux pumps are highly conserved structures in pro-and eukaryotes. Comprehensive reviews have summarized a plethora of data describing interactions of a broad variety of agents with efflux pumps [238,239,240,241,242,243,244]. In general, non-steroidal anti-inflammatory drugs (NSAIDS) and acetylsalicylic acid (aspirin®), its major metabolite salicylic acid, and acetaminophen (paracetamol®) in particular, interact not only with efflux pumps but exert pleiotropic antibacterial activities against bacteria many of which are equivocal [245,246,247,248,249]. Notably, the effect of NSAIDS on bacterial efflux pumps was contradictory as growth in the presence of (acetyl-) salicylic acid either caused intrinsic multiple drug resistance and increased virulence or reduced resistance and virulence, so that growth in the presence of salicylate can be both beneficial and detrimental. The reader is kindly referred to the comprehensive review articles for more detailed information [245,246,247,248,249].
Conclusions and open questions
Data summarized above support the hypothesis that prokaryotic proteins may represent targets for non-antibiotics because of a common evolutionary origin of the corresponding pro- and eukaryotic proteins. The phylogenetically related and structurally highly conserved targets provide the rational basis for antibacterial actions of non-antibiotics, and vice versa interactions of antibiotics with mammalian targets [30]. Advances in structural biology, genetics, bioinformatics etc. resulted in the availability of an enormous pool of information enabling the analysis of the ancestry of pro- and eukaryotic genes and proteins. Comparisons of gene- or protein-sequences provide information about structural and/or functional convergencies or divergencies. Although pro- or eukaryotic enzymes may differ structurally hence being non-homologous they may be functionally homologous catalysing the same biological reactions because of their highly conserved active centres [250] as exemplified e.g. by eukaryotic ACE and the homologous enzymes in bacteria [227,228,229]. Bacterial—as well as mammalian active centres of STPKs, voltage-gated ion channels, ACE and MBLs are ancient and highly conserved so that agents interfering with activities of mammalian proteins inhibited the bacterial counterparts as well. Representative examples constituting key physiological reactions are summarized in Table 4. Protease inhibitors inhibited bacterial STPKs to which PBPs are linked [81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112] so that these drugs synergized with β-lactams and attenuated bacterial virulence. Calcium antagonists blocked not only pro- and eukaryotic ion channels [150,151,152,153,154,155,156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172] but interacted with negatively charged cell membranes thus disrupting membrane energetics and inducing membrane stress response [186, 187], so that P-glycoprotein like bacterial pumps were affected. ACE-inhibitors and sulfonamides interacted with zinc-metalloenzymes such as eukaryotic ACE and prokaryotic MBLs [209,210,211,212,213,214,215,216,217,218,219] or eu- and prokaryotic carbonic anhydrases [230,231,232,233,234,235].
These examples demonstrate that non-antibiotics most frequently support and augment antibacterial activities of antibiotics but exerted an antibacterial activity on their own in rare cases only. As none of the non-antibiotics discussed above interacted antagonistically with various antibiotics—except NSAIDs with frequently unpredictable synergistic or antagonistic interactions [245,246,247,248,249]—they could be administered in combination to improve antibacterial therapy and to reverse antibiotic resistance. Furthermore, the data summarized above could possibly provide models for target interactions with utility for future drug design. Screening could be based on the strategy to identify proteins of convergent evolution as these could represent essential targets.
Open questions are if protease-inhibitor and β-lactam hybrids could be synthesized targeting in parallel STPK—as well as PBP-domains of PASTA proteins?
Could Zn2+ chelating ACE-inhibitors and Zn2+ chelating antibiotics [30] mutually affect each other positively or negatively? Could a combination of non-antibiotics with antibiotics both carrying mitochondrial liabilities increase or decrease their individual effects on mitochondrial functions? Calcium antagonists [251,252,253,254,255] and ACE-inhibitors [256,257,258,259,260] reduced respiration and ATP synthesis. Sorafenib directly impaired mitochondrial function at clinically relevant concentrations [261] whereas imatinib lacked direct mitochondrial effects but affected mitochondrial functions via altered kinase and other signalling pathways [261, 262]. Likewise, ciclosporin A, rapamycin, and tacrolimus did not directly induce mitochondrial dysfunction but decreased energy production [263, 264]. Thus, almost all of the non-antibiotics mentioned above affect mitochondrial functions being either part of their modes of action or potential toxicities. Many antibiotics cause mitochondrial dysfunction, too, thus explaining their anti-neoplastic activities [30], or possibly promoting tumorigenesis [265,266,267,268], obesity [269], and psychiatric disorders [270, 271]. Thus, antibiotics as well as non-antibiotics exert beneficial or detrimental anti-mitochondrial activities. The question if combinations of non-antibiotics with antibiotics may increase or decrease drug inherent anti-mitochondrial effects of the respective drug thus affecting their efficacy and/or toxicity has not yet been assessed. It might be reasonably assumed that such drug combinations have been prescribed frequently to certain populations like cancer- or geriatric patients in whom polypharmacy is frequent but evidently having been without peculiarities so far [272].
Other open questions such as drug/drug interactions, impact on the microbiomes, etc. are beyond the scope of this review article but will need to be kept in focus. In summary, drug development could benefit from new perspectives on protein evolution. The currently available data permit an immediate implementation of well-justified combination therapies and may indicate perspectives for the future.
References
Cederlund H, Mårdh PA. Antibacterial activities of non-antibiotic drugs. J Antimicrob Chemother. 1993;32:355–65. https://doi.org/10.1093/jac/32.3.355.
Vandevelde NM, Tulkens PM, Van Bambeke F. Modulating antibiotic activity towards respiratory bacterial pathogens by co-medications: a multi-target approach. Drug Discov Today. 2016;21:1114–29. https://doi.org/10.1016/j.drudis.2016.04.001.
Brown D. Antibiotic resistance breakers: can repurposed drugs fill the antibiotic discovery void? Nat Rev Drug Discov. 2015;14:821–32. https://doi.org/10.1038/nrd4675.
Miró-Canturri A, Ayerbe-Algaba R, Smani Y. Drug repurposing for the treatment of bacterial and fungal infections. Front Microbiol. 2019;10:41. https://doi.org/10.3389/fmicb.2019.00041.
Lagadinou M, Onisor MO, Rigas A, Musetescu DV, Gkentzi D, Assimakopoulos SF, Panos G, Marangos M. Antimicrobial properties of non-antibiotic drugs in the era of increased bacterial resistance. Antibiotics. 2020;9:107. https://doi.org/10.3390/antibiotics9030107.
Kristiansen JE, Hendricks O, Delvin T, Butterworth TS, Aagaard L, Christiansen JB, Flores VC, Keyzer H. Reversal of resistance in microorganisms by help of non-antibiotics. J Antimicrob Chemother. 2007;59:1271–9. https://doi.org/10.1093/jac/dkm071.
Kalayci J. Antimicrobial properties of various non-antimicrobial drugs against microorganisms. Bioanal Biomed. 2016;8:4. https://doi.org/10.4172/1948-593X.1000e142.
Tyski S. Non-antibiotics-drugs with additional antimicrobial activity. Acta Pol Pharm. 2003;60:401–4 (PMID: 15005425).
Brown MW. In vitro antibacterial activity of drugs against human intestinal anaerobic bacteria. J Pharm Sci. 1975;64:700–2. https://doi.org/10.1002/jps.2600640431.
Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE, Brochado AR, Fernandez KC, Dose H, Mori H, Patil KR, Bork P, Typas A. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature. 2018;555:623–8. https://doi.org/10.1038/nature25979.
Vila AV, Collij V, Sanna S, Sinha T, Imhann F, Bourgonje AR, Mujagicv Z, Jonkers DMAE, Masclee AAM, Fu J, Kurilshikov A, Wijmenga C, Zhernakova A, Weersma RK. Impact of commonly used drugs on the composition and metabolic function of the gut microbiota. Nat Commun. 2020;11:362. https://doi.org/10.1038/s41467-019-14177.
Bruno G, Zaccari P, Rocco G, Scalese G, Panetta C, Porowska B, Pontone S, Severi C. Proton pump inhibitors and dysbiosis: current knowledge and aspects to be clarified. World J Gastroenterol. 2019;25:2706–19. https://doi.org/10.3748/wjg.v25.i22.2706.
Sjöström JE, Fryklund J, Kühler T, Larsson H. In vitro antibacterial activity of omeprazole and its selectivity for Helicobacter spp. are dependent on incubation conditions. Antimicrob Agents Chemother. 1996;40:621–6. https://doi.org/10.1128/AAC.40.3.621.
Biasco G, Miglioli M, Barbara L, Corinaldesi R, Febo GD. Omeprazole, Helicobacter pylori, gastritis, and duodenal ulcer. Lancet. 1989. https://doi.org/10.1016/s0140-6736(89)92021-7.
Hunt RH. Hp and pH: implications for the eradication of Helicobacter pylori. Scand J Gastroenterol. 1993;28:12–6. https://doi.org/10.3109/00365529309098335.
Mainguet P, Delmee M, Debongnie JC. Omeprazole, Campylobacter pylori, and duodenal ulcer. Lancet. 1989. https://doi.org/10.1016/s0140-6736(89)90569-2.
An H, He L. Current understanding of metformin effect on the control of hyperglycemia in diabetes. J Endocrinol. 2016;228:R97-106. https://doi.org/10.1530/JOE-15-0447.
Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60:1577–85. https://doi.org/10.1007/s00125-017-4342-z.
Logie L, Harthill J, Patel K, Bacon S, Hamilton DL, Macrae K, McDougall G, Wang HH, Xue L, Jiang H, Sakamoto K, Prescott AR, Rena G. Cellular responses to the metal-binding properties of metformin. Diabetes. 2012;61:1423–33. https://doi.org/10.2337/db11-0961.
Repiščák P, Erhardt S, Rena G, Paterson MJ. Biomolecular mode of action of metformin in relation to its copper binding properties. Biochemistry. 2014;53:787–95. https://doi.org/10.1021/bi401444n.
Kajiwara C, Kusaka Y, Kimura S, Yamaguchi T, Nanjo Y, Ishii Y, Udono H, Standiford TJ, Tateda K. Metformin mediates protection against Legionella pneumonia through activation of AMPK and mitochondrial reactive oxygen species. J Immunol. 2018;200:623–31. https://doi.org/10.4049/jimmunol.1700474.
Courtois S, Bénéjat L, Izotte J, Mégraud F, Varon C, Lehours P, Bessède E. Metformin can inhibit Helicobacter pylori growth. Future Microbiol. 2018;13:1575–83. https://doi.org/10.2217/fmb-2018-0184.
Lee JH, Park J, Park MR, Na YH, Cho SJ. A comparative study of Helicobacter pylori growth on different agar-based media. Korean J Helicobacter Up Gastrointest Res. 2017;17:208–12. https://doi.org/10.7704/kjhugr.2017.17.4.208.
Tseng CH. Metformin and Helicobacter pylori infection in patients with type 2 diabetes. Diabetes Care. 2018;41:e42–3. https://doi.org/10.2337/dc17-2551.
Malik F, Mehdi SF, Ali H, Patel P, Basharat A, Kumar A, Ashok F, Stein J, Brima W, Malhotra P, Roth J. Is metformin poised for a second career as an antimicrobial? Diabetes Metab Res Rev. 2018;34:e2975. https://doi.org/10.1002/dmrr.2975.
Pollak M. The effects of metformin on gut microbiota and the immune system as research frontiers. Diabetologia. 2017;60:1662–7. https://doi.org/10.1007/s00125-017-4352-x.
Cushnie TT, Lamb AJ. Antimicrobial activity of flavonoids. Int J Antimicrob Agents. 2005;26:343–56. https://doi.org/10.1016/j.ijantimicag.2005.09.002.
Wu YW, Ouyang J, Xiao XH, Gao WY, Liu Y. Antimicrobial properties and toxicity of anthraquinones by microcalorimetric bioassay. Chinese J Chem. 2006;24:45–50. https://doi.org/10.1002/cjoc.200690020.
Kemegne GA, Mkounga P, Ngang JJE, Kamdem SLS, Nkengfack AE. Antimicrobial structure activity relationship of five anthraquinones of emodine type isolated from Vismia laurentii. BMC Microbiol. 2017;17:41. https://doi.org/10.1186/s12866-017-0954-1.
Dalhoff A. Selective toxicity of antibacterial agents—still a valid concept or do we miss chances and ignore risks? Submitted to this journal. 2020
Tarrant MK, Cole PA. The chemical biology of protein phosphorylation. Ann Rev Biochem. 2009;78:797–825. https://doi.org/10.1146/annurev.biochem.78.070907.103047.
Weiser DC, Shenolikar S. Use of protein phosphatase inhibitors. Curr Protoc Mol Biol. 2003;62:18–10. https://doi.org/10.1002/0471142727.mb1810s62.
Hunter T. Signaling–2000 and beyond. Cell. 2000;100:113–27. https://doi.org/10.1016/S0092-8674(00)81688-8.
Stancik IA, Šestak MS, Ji B, Axelson-Fisk M, Franjevic D, Jers C, Domazet-Loso T, Mijakovic I. Serine/threonine protein kinases from bacteria, archaea and eukarya share a common evolutionary origin deeply rooted in the tree of life. J Mol Biol. 2018;430:27–32. https://doi.org/10.1016/j.jmb.2017.11.004.
Pereira SF, Goss L, Dworkin J. Eukaryote-like serine/threonine kinases and phosphatases in bacteria. Microbiol Mol Biol Rev. 2011;75:192–212. https://doi.org/10.1128/MMBR.00042-10.
Bakal CJ, Davies JE. No longer an exclusive club: eukaryotic signaling domains in bacteria. Trends Cell Biol. 2000;10:32–8. https://doi.org/10.1016/S0962-8924(99)01681-5.
Kennelly PJ. Protein kinases and protein phosphatases in prokaryotes: a genomic perspective. FEMS Microbiol Lett. 2002;206:1–8. https://doi.org/10.1111/j.1574-6968.2002.tb10978.x.
Kennelly PJ. Protein Ser/Thr/Tyr phosphorylation in the archaea. J Biol Chem. 2014;289:9480–7. https://doi.org/10.1074/jbc.R113.529412.
Palm D, Goerl R, Burger KJ. Evolution of catalytic and regulatory sites in phosphorylases. Nature. 1985;313:500–2. https://doi.org/10.1038/313500a0.
Schinzel R, Nidetzky B. Bacterial α-glucan phosphorylases. FEMS Microbiol Lett. 1999;171:73–9. https://doi.org/10.1111/j.1574-6968.1999.tb13414.x.
Chao JD, Wong D, Av-Gay Y. Microbial protein-tyrosine kinases. J Biol Chem. 2014;289:9463–72. https://doi.org/10.1074/jbc.R113.520015.
Grangeasse C, Nessler S, Mijakovic I. Bacterial tyrosine kinases: evolution, biological function and structural insights. Philos Trans R Soc Lond B Biol Sci. 2012;367:2640–55. https://doi.org/10.1098/rstb.2011.0424.
Macek B, Gnad F, Soufi B, Kumar C, Olsen JV, Mijakovic I, Mann M. Phosphoproteome analysis of E. coli reveals evolutionary conservation of bacterial Ser/Thr/Tyr phosphorylation. Mol Cell Proteom. 2008;7:299–307. https://doi.org/10.1074/mcp.M700311-MCP200.
Boekhorst J, van Breukelen B, Heck AJ, Snel B. Comparative phosphoproteomics reveals evolutionary and functional conservation of phosphorylation across eukaryotes. Genome Biol. 2008;9:R144. https://doi.org/10.1186/gb-2008-9-10-r144.
Mkaouar H, Akermi N, Kriaa A, Abraham A, Jablaoui A, Soussou S, Mokdad-Gargouri R, Maguin E, Rhimi M. Serine protease inhibitors and human wellbeing interplay: new insights for old friends. PeerJ. 2019;7:e7224. https://doi.org/10.7717/peerj.7224.
Goffin C, Ghuysen JM. Multimodular penicillin-binding proteins: an enigmatic family of orthologs and paralogs. Microbiol Mol Biol Rev. 1998;62:1079–93 (PMID: 9841666).
Yeats C, Finn RD, Bateman A. The PASTA domain: a β-lactam-binding domain. Trends Biochem Sci. 2002;27:438–40. https://doi.org/10.1016/S0968-0004(02)02164-3.
Calvanese L, Falcigno L, Squeglia F, D’Auria G, Berisio R. PASTA in penicillin binding proteins and serine/threonine kinases: a recipe of structural, dynamic and binding properties. Curr Med Chem. 2017a;24:4038–56. https://doi.org/10.2174/0929867324666170216112746.
Calvanese L, Falcigno L, Squeglia F, D’Auria G, Berisio R. Structural and dynamic features of PASTA domains with different functional roles. J Biomol Struct Dyn. 2017b;35:2293–300. https://doi.org/10.1080/07391102.2016.1217274.
Calvanese L, Falcigno L, Squeglia F, Berisio R, D’Auria G. PASTA sequence composition is a predictive tool for protein class identification. Amino Acids. 2018;50:1441–50. https://doi.org/10.1007/s00726-018-2621-8.
Ogawara H. Distribution of PASTA domains in penicillin-binding proteins and serine/threonine kinases of Actinobacteria. J Antibiot. 2016;69:660–85. https://doi.org/10.1038/ja.2015.138.
Irazoki O, Hernandez S, Cava F. Peptidoglycan muropeptides: release, perception and functions as signalling molecules. Front Microbiol. 2019;10:500. https://doi.org/10.3389/fmicb.2019.00500.
Pensinger DA, Schaenzer AJ, Sauer JD. Do shoot the messenger: PASTA kinases as virulence determinants and antibiotic targets. Trends Microbiol. 2018;26:56–69. https://doi.org/10.1016/j.tim.2017.06.010.
Shah IM, Laaberki MH, Popham DL, Dworkin J. A eukaryotic-like Ser/Thr kinase signals bacteria to exit dormancy in response to peptidoglycan fragments. Cell. 2008;135:486–96. https://doi.org/10.1016/j.cell.2008.08.039.
Manuse S, Fleurie A, Zucchini L, Lesterlin C, Grangeasse C. Role of eukaryotic-like serine/threonine kinases in bacterial cell division and morphogenesis. FEMS Microbiol Rev. 2015;40:41–56. https://doi.org/10.1093/femsre/fuv041.
Canova MJ, Molle V. Bacterial serine/threonine protein kinases in host-pathogen interactions. J Biol Chem. 2014;289:9473–9. https://doi.org/10.1074/jbc.R113.529917.
Jones G, Dyson P. Evolution of Transmembrane protein kinases implicated in coordinating remodeling of Gram-positive peptidoglycan: inside versus outside. J Bacteriol. 2006;188:7470–6. https://doi.org/10.1128/JB.00800-06.
Desbonnet C, Tait-Kamradt A, Garcia-Solache M, Dunman P, Coleman J, Arthur M, Rice LB. Involvement of the eukaryote-like kinase-phosphatase system and a protein that interacts with penicillin-binding protein 5 in emergence of cephalosporin resistance in cephalosporin-sensitive class A penicillin-binding protein mutants in Enterococcus faecium. MBio. 2016;7:e02188-e2215. https://doi.org/10.1128/mBio.02188-15.
Labbe BD, Kristich CJ. Growth-and stress-induced PASTA kinase phosphorylation in Enterococcus faecalis. J Bacteriol. 2017;199:e00363-e417. https://doi.org/10.1128/JB.00363-17.
Kristich CJ, Wells CL, Dunny GM. A eukaryotic-type Ser/Thr kinase in Enterococcus faecalis mediates antimicrobial resistance and intestinal persistence. Proc Nat Acad Sci. 2007;104:3508–13. https://doi.org/10.1073/pnas.0608742104.
Barthe P, Mukamolova GV, Roumestand C, Cohen-Gonsaud M. The structure of PknB extracellular PASTA domain from Mycobacterium tuberculosis suggests a ligand-dependent kinase activation. Structure. 2010;18:606–15. https://doi.org/10.1016/j.str.2010.02.013.
Schweizer I, Peters K, Stahlmann C, Hakenbeck R, Denapaite D. Penicillin-binding protein 2x of Streptococcus pneumoniae: the mutation Ala707Asp within the C-terminal PASTA2 domain leads to destabilization. Microb Drug Resist. 2014;20:250–7. https://doi.org/10.1089/mdr.2014.0082.
Peters K, Schweizer I, Beilharz K, Stahlmann C, Veening JW, Hakenbeck R, Denapaite D. Streptococcus pneumoniae PBP2x mid-cell localization requires the C-terminal PASTA domains and is essential for cell shape maintenance. Mol Microbiol. 2014;92:733–55. https://doi.org/10.1111/mmi.12588.
Maestro B, Novaková L, Hesek D, Lee M, Leyva E, Mobashery S, Sanz JM, Branny P. Recognition of peptidoglycan and β-lactam antibiotics by the extracellular domain of the Ser/Thr protein kinase StkP from Streptococcus pneumoniae. FEBS Lett. 2011;585:357–63. https://doi.org/10.1016/j.febslet.2010.12.016.
Ohlsen K, Donat S. The impact of serine/threonine phosphorylation in Staphylococcus aureus. Int J Med Microbiol. 2010;300:137–41. https://doi.org/10.1016/j.ijmm.2009.08.016.
Tamber S, Schwartzman J, Cheung AL. Role of PknB kinase in antibiotic resistance and virulence in community-acquired methicillin-resistant Staphylococcus aureus strain USA300. Infect Immun. 2010;78:3637–46. https://doi.org/10.1128/IAI.00296-10.
Paracuellos P, Ballandras A, Robert X, Kahn R, Hervé M, Mengin-Lecreulx D, Cozzone AJ, Duclos B, Gouet P. The extended conformation of the 2.9-Å crystal structure of the three-PASTA domain of a Ser/Thr kinase from the human pathogen Staphylococcus aureus. J Mol Biol. 2010;404:847–58. https://doi.org/10.1016/j.jmb.2010.10.012.
Mohamed SB, Adlan TA, Khalafalla NA, Abdalla NI, Ali ZS, Munir KAA, Hassan MM, Elnour MAB. Proteomics and docking study targeting penicillin-binding protein and penicillin-binding Protein2a of methicillin-resistant Staphylococcus aureus strain SO-1977 isolated from Sudan. Evol Bioinform. 2019;2019(15):1176934319864945. https://doi.org/10.1177/1176934319864945.
Fiuza M, Canova MJ, Zanella-Cléon I, Becchi M, Cozzone AJ, Mateos LM, Kremer L, Gil JA, Molle V. From the characterization of the four serine/threonine protein kinases (PknA/B/G/L) of Corynebacterium glutamicum toward the role of PknA and PknB in cell division. J Biol Chem. 2008;283:18099–112. https://doi.org/10.1074/jbc.M802615200.
Schultz C, Niebisch A, Schwaiger A, Viets U, Metzger S, Bramkamp M, Bott M. Genetic and biochemical analysis of the serine/threonine protein kinases PknA, PknB, PknG and PknL of Corynebacterium glutamicum: evidence for non-essentiality and for phosphorylation of OdhI and FtsZ by multiple kinases. Mol Microbiol. 2009;74:724–41. https://doi.org/10.1111/j.1365-2958.2009.06897.x.
Yan J, Zou W, Fang J, Huang X, Gao F, He Z, Zhang K, Zhao N. Eukaryote-like Ser/Thr proteinkinase PrkA modulates sporulation via regulating the transcriptional factor σK in Bacillus subtilis. Front Microbiol. 2015;6:382. https://doi.org/10.3389/fmicb.2015.00382.
Pompeo F, Foulquier E, Serrano B, Grangeasse C, Galinier A. Phosphorylation of the cell division protein GpsB regulates PrkC kinase activity through a negative feedback loop in Bacillus subtilis. Mol Microbiol. 2015;97:139–50. https://doi.org/10.1111/mmi.13015.
Pompeo F, Byrne D, Mengin-lecreulx D, Galinier A. Dual regulation of activity and intracellular localization of the PASTA kinase PrkC during Bacillus subtilis growth. Sci Rep. 2018;8:1660. https://doi.org/10.1038/s41598-018-20145-2.
Libby EA, Goss LA, Dworkin J. The eukaryotic-like Ser/Thr kinase PrkC regulates the essential WalRK two-component system in Bacillus subtilis. PLoS Genet. 2015;11:e1005275. https://doi.org/10.1371/journal.pgen.1005275.
Macek B, Mijakovic I, Olsen JV, Gnad F, Kumar C, Jensen PR, Mann M. The serine/threonine/tyrosine phosphoproteome of the model bacterium Bacillus subtilis. Mol Cell Proteom. 2007;6:697–707. https://doi.org/10.1074/mcp.M600464-MCP200.
Ortiz-Lombardia M, Pompeo F, Boitel B, Alzari PM. Crystalstructure of the catalytic domain of the PknB serine/threonine kinase from Mycobacterium tuberculosis. J Biol Chem. 2003;278:13094–100. https://doi.org/10.1074/jbc.M300660200.
Young TA, Delagoutte B, Endrizzi JA, Falick AM, Alber T. Structure of Mycobacterium tuberculosis PknB supports a universal activation mechanism for Ser/Thr protein kinases. Nat Struct Biol. 2003;10:168–74. https://doi.org/10.1038/nsb897.
Av-Gay Y, Everett M. The eukaryotic-like Ser/Thr protein kinases of Mycobacterium tuberculosis. Trends Microbiol. 2000;8:238–44. https://doi.org/10.1016/S0966-842X(00)01734-0.
Dasgupta A, Datta P, Kundu M, Basu J. The serine/threonine kinase PknB of Mycobacterium tuberculosis phosphorylates PBPA, a penicillin-binding protein required for cell division. Microbiology. 2006;152:493–504. https://doi.org/10.1099/mic.0.28630-0.
Turapov O, Loraine J, Jenkins CH, Barthe P, McFeely D, Forti F, Ghisotti D, Hesek D, Lee M, Bottrill AR, Vollmer W, Mobashery S, Cohen-Gonsaud M, Mukamolova GV. The external PASTA domain of the essential serine/threonine protein kinase PknB regulates mycobacterial growth. Open Biol. 2015;5:150025. https://doi.org/10.1098/rsob.150025.
Lombana TN, Echols N, Good MC, Thomsen ND, Ng HL, Greenstein AE, Falick AM, King DS, Alber T. Allosteric activation mechanism of the Mycobacterium tuberculosis receptor Ser/Thr protein kinase. PknB Struct. 2010;18:1667–77. https://doi.org/10.1016/j.str.2010.09.019.
Rajagopal L, Howard K, Whidbey C. Kinase inhibitors capable of increasing the sensitivity of bacterial pathogens to β-lactam antibiotics. World Intellectual Property Organization. International Publication Number WO 2013/066469 A2; 2013.
Vornhagen J, Burnside K, Whidbey C, Berry J, Qin X, Rajagopal L. Kinase inhibitors that increase the sensitivity of methicillin resistant Staphylococcus aureus to β-lactam antibiotics. Pathogens. 2015;4:708–21. https://doi.org/10.3390/pathogens4040708.
Pensinger DA, Aliota MT, Schaenzer AJ, Boldon KM, Israr-ul HA, Vincent WJ, Knight B, Reniere ML, Striker R, Sauer JD. Selective pharmacologic inhibition of a PASTA kinase increases Listeria monocytogenes susceptibility to β-lactam antibiotics. Antimicrob Agents Chemother. 2014;58:4486–94. https://doi.org/10.1128/AAC.02396-14.
Schaenzer AJ, Wlodarchak N, Drewry DH, Zuercher WJ, Rose WE, Striker R, Sauer JD. A screen for kinase inhibitors identifies antimicrobial imidazopyridine aminofurazans as specific inhibitors of the Listeria monocytogenes PASTA kinase PrkA. J Biol Chem. 2017;292:17037–45. https://doi.org/10.1074/jbc.M117.808600.
Schaenzer AJ, Wlodarchak N, Drewry DH, Zuercher WJ, Rose WE, Ferrer CA, Sauer JD, Striker R. GW779439X and its pyrazolopyridazine derivatives inhibit the serine/threonine kinase Stk1 and act as antibiotic adjuvants against β-lactam-resistant Staphylococcus aureus. ACS Infect Dis. 2018;4:1508–18.
Striker RT, Sauer JD, Wlodarchak N. Use of kinase inhibitors to increase the susceptibility of Gram-positive bacteria to beta lactam antibiotics. United States Patent Application. Pub. No.: US 2015/0307495 A1; 2015.
Kawase M, Shirato K, van der Hoek L, Taguchi F, Matsuyama S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J Virol. 2012;86:6537–645. https://doi.org/10.1128/JVI.00094-12.
Hoffman M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Müller MA, Drosten C, Pöhlmann S. SARS-Cov-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–80. https://doi.org/10.1016/j.cell.2020.02.052.
Layton GT, Clements JM, Chandler SR. Serine protease inhibitors for use in the treatment of bacterial infections. World Intellectual Property Organization. International Publication Number WO 2008/142397 A1; 2008.
Layton GT, Clements JM, Chandler SR. Compositions containing serine protease inhibitors. UK Patent Application GB 2450780 A; 2009.
Wlodarchak N, Teachout N, Beczkiewicz J, Procknow R, Schaenzer AJ, Satyshur K, Pavelka M, Zuercher B, Drewry D, Sauer JD, Striker R. In silico screen and structural analysis identifies bacterial kinase inhibitors which act with β-lactams to inhibit mycobacterial growth. Mol Pharm. 2018;15:5410–26. https://doi.org/10.1021/acs.molpharmaceut.8b00905.
Wlodarchak N, Teachout N, Procknow R, Beczkiewicz J, Schaenzer A, Satyshur K, Pavelka M, Zuercher B, Drewry D, Sauer JD, Striker R. Repurposed kinase inhibitors and β-lactams as a novel therapy for antibiotic resistant bacteria. bioRxiv. 2017. https://doi.org/10.1101/199422.
Nguyen TV, Blackledge MS, Lindsey EA, Minrovic BM, Ackart DF, Jeon AB, Obregon-Henao A, Basasaba RJ, Melander C. The discovery of 2-aminobenzimidazoles that sensitize Mycobacterium smegmatis and M. tuberculosis to β-lactam antibiotics in a pattern distinct from β-lactamase inhibitors. Angew Chem Int Ed. 2017;56:3940–4. https://doi.org/10.1002/anie.201612006.
Harris TL, Worthington RJ, Melander C. Potent small-molecule suppression of oxacillin resistance in methicillin-resistant Staphylococcus aureus. Angew Chem Int Ed. 2012;51:11254–7. https://doi.org/10.1002/anie.201206911.
Rogers SA, Huigens RW III, Cavanagh J, Melander C. Synergistic effects between conventional antibiotics and 2-aminoimidazole-derived antibiofilm agents. Antimicrob Agents Chemother. 2010;54:2112–8. https://doi.org/10.1128/AAC.01418-09.
Brackett CM, Melander RJ, An IH, Krishnamurthy A, Thompson RJ, Cavanagh J, Melander C. Small-molecule suppression of β-lactam resistance in multidrug-resistant gram-negative pathogens. J Med Chem. 2014;57:7450–8. https://doi.org/10.1021/jm501050e.
Worthington RJ, Bunders CA, Reed CS, Melander C. Small molecule suppression of carbapenem resistance in NDM-1 Producing Klebsiella pneumonia. ACS Med Chem Lett. 2012;3:357–61. https://doi.org/10.1021/ml200290p.
Harris TL, Worthington RJ, Hittle LE, Zurawski DV, Ernst RK, Melander C. Small molecule downregulation of PmrAB reverses lipid A modification and breaks colistin resistance. ACS Chem Biol. 2014;9:122–7. https://doi.org/10.1021/cb400490k.
Cutrona N, Gillard KR, Ulrich RJ, Seemann M, Miller HB, Blackledge MS. From antihistamine to anti-infective: loratadine inhibits regulatory PASTA kinases in Staphylococci to reduce biofilm formation and potentiate β-lactam antibiotics and vancomycin in resistant strains of S. aureus. ACS Infect Dis. 2019;5:1397–410. https://doi.org/10.1021/acsinfecdis.9b00096.
Kant S, Asthana S, Missiakas D, Pancholi V. A novel STK1-targeted small-molecule as an “antibiotic resistance breaker” against multidrug-resistant Staphylococcus aureus. Sci Rep. 2017;7:5067. https://doi.org/10.1038/s41598-017-05314-z.
Cheung A, Duclos B. Stp1 and Stk1: the Yin and Yang of vancomycin sensitivity and virulence in vancomycin-intermediate Staphylococcus aureus strains. J Infect Dis. 2012;205:1625–7. https://doi.org/10.1093/infdis/jis255.
Cameron DR, Ward DV, Kostoulias X, Howden BP, Moellering RC Jr, Eliopoulos GM, Peleg AY. The serine/threonine phosphatase Stp1 con-tributes to reduced susceptibility to vancomycin and virulence in S. aureus. J Infect Dis. 2012;205:1677–87. https://doi.org/10.1093/infdis/jis252.
Xu J, Wang JX, Zhou JM, Xu CL, Huang B, Xing Y, Wang B, Luo R, Wang YC, You XF, Lu Y, Lu LY. A novel protein kinase inhibitor IMB-YH-8 with anti-tuberculosis activity. Sci Rep. 2017;7:5093. https://doi.org/10.1038/s41598-017-04108-7.
Wehenkel A, Fernandez P, Bellinzoni M, Catherinot V, Barilone N, Labesse G, Jackson M, Alzari PM. The structure of PknB in complex with mitoxantrone, an ATP-competitive inhibitor, suggests a mode of protein kinase regulation in mycobacteria. FEBS Lett. 2006;580:3018–22. https://doi.org/10.1016/j.febslet.2006.04.046.
Shamsi TN, Fatima S. Protease inhibitors as ad-hoc antibiotics. Open Pharm Sci J. 2016;3:131–7. https://doi.org/10.2174/1874844901603010131.
Carling D, Aguan K, Woods A, Verhoeven AJ, Beri RK, Brennan CH, Sidebottom C, Davison MD, Scott J. Mammalian AMP-activated protein kinase is homologous to yeast and plant protein kinases involved in the regulation of carbon metabolism. J Biol Chem. 1994;269:11442–8 (PMID: 7908907).
Lin SC, Hardie DG. AMPK: sensing glucose as well as cellular energy status. Cell Metab. 2018;27:299–313. https://doi.org/10.1016/j.cmet.2017.10.009.
Chantranupong L, Wolfson RL, Sabatini DM. Nutrient-sensing mechanisms across evolution. Cell. 2015;161:67–83. https://doi.org/10.1016/j.cell.2015.02.041.
Fischer C, Geourjon C, Bourson C, Deutscher J. Cloning and characterization of the Bacillus subtilis prkA gene encoding a novel serine protein kinase. Gene. 1996;168:55–60. https://doi.org/10.1016/0378-1119(95)00758-X.
Eichenberger P, Jensen ST, Conlon EM, Van Ooij C, Silvaggi J, Gonzalez-Pastor JE, Fujita M, Ben-Yehuda S, Stragier P, Liu JS, Losick R. The σE regulon and the identification of additional sporulation genes in Bacillus subtilis. J Mol Biol. 2003;327:945–72. https://doi.org/10.1016/S0022-2836(03)00205-5.
Lima A, Duran R, Schujman GE, Marchissio MJ, Portela MM, Obal G, Pritsch O, de Mendoza D, Cervanansky C. Serine/threonine protein kinase PrkA of the human pathogen Listeria monocytogenes: biochemical characterization and identification of interacting partners through proteomic approaches. J Prot. 2011;74:1720–34. https://doi.org/10.1016/j.jprot.2011.03.005.
Lipa P, Janczarek M. Phosphorylation systems in symbiotic nitrogen-fixing bacteria and their role in bacte-rial adaptation to various environmental stresses. Peer J. 2020;8:e8466. https://doi.org/10.7717/peerj.8466.
D’Costa VM, McGrann KM, Hughes DW, Wright GD. Sampling the antibiotic resistome. Science. 2006;311:374–7. https://doi.org/10.1126/science.1120800.
Kannan N, Taylor SS, Zhai Y, Venter JC, Manning G. Structural and functional diversity of the microbial kinome. PLoS Biol. 2007;5:e17. https://doi.org/10.1371/journal.pbio.0050017.
Hon WC, McKay GA, Thompson PR, Sweet RM, Yang DS, Wright GD, Berghuis AM. Structure of an enzyme required for aminoglycoside antibiotic resistance reveals homology to eukaryotic protein kinases. Cell. 1997;89:887–95. https://doi.org/10.1016/S0092-8674(00)80274-3.
Daigle DM, McKay GA, Thompson PR, Wright GD. Aminoglycoside antibiotic phosphotransferases are also serine protein kinases. Chem Biol. 1999;6:11–8. https://doi.org/10.1016/S1074-5521(99)80016-7.
Elizarov SM, Sergienko OV, Sizova IA, Danilenko VN. Dependence of aminoglycoside 3′-phosphotransferase VIII activity on serine/threonine protein kinases in Streptomyces rimosus. Mol Biol. 2005;39:226–33. https://doi.org/10.1007/s11008-005-0033-9.
Sizova IA, Hegemann P, Furmann M, Danilenko VN. Streptomyces rimosus aminoglycoside 3″-phosphotransferase VIII: comparisons with aminoglycoside 3″-phosphotransferases of aminoglycoside-producing strains and with eukaryotic protein kinases. Mol Biol. 2002;36:18–25. https://doi.org/10.1023/A:1014282003679.
Shakya T, Wright GD. Nucleotide selectivity of antibiotic kinases. Antimicrob Agents Chemother. 2010;54:1909–13. https://doi.org/10.1128/AAC.01570-09.
Metz JT, Johnson EF, Soni NB, Merta PJ, Kifle L, Hajduk PJ. Navigating the kinome. Nat Chem Biol. 2011;7:200–2. https://doi.org/10.1038/nchembio.530.
Shakya T. The antibiotic kinome. Uncovering the antibiotic kinome with small molecules (Doctoral dissertation). McMaster University, Hamilton, ON, USA. 2011. https://hdl.handle.net/11375/9890
Shakya T, Stogios PJ, Waglechner N, Evdokimova E, Eijm L, Blanchard JE, McArthur AG, Savchenko A, Wright GD. A small molecule discrimination map of the antibiotic resistance kinome. Chem Biol. 2011;18:1591–601. https://doi.org/10.1016/j.chembiol.2011.10.018.
Stigios PJ, Spanogiannopoulos P, Evdokimova E, Egorova O, Shakaya T, Todorovics N, Caprettas A, Wright GD, Savchenko A. Structure-guided optimization of protein kinase inhibitors reverses aminoglycoside antibiotic resistance. Biochem J. 2013;454:191–200. https://doi.org/10.1042/BJ20130317.
Li Y, Green KD, Johnson BR, Garneau-Tsodikova S. Inhibition of aminoglycoside acetyltransferase resistanceenzymes by metal salts. Antimicrob Agents Chemother. 2015;59:4148–56. https://doi.org/10.1128/AAC.00885-15.
Górniak I, Bartoszewski R, Króliczewski J. Comprehensive review of antimicrobial activities of plant flavonoids. Phytochem Rev. 2019;18:241–72. https://doi.org/10.1007/s11101-018-9591-z.
Daigle DM, McKay GA, Wright GD. Inhibition of aminoglycoside antibiotic resistance enzymes by protein kinase inhibitors. J Biol Chem. 1997;272:24755–8. https://doi.org/10.1074/jbc.272.40.24755.
Fong DH, Xiong B, Hwang J, Berghuis AM. Crystal structures of two aminoglycoside kinases bound with a eukaryotic protein kinase inhibitor. PLoS ONE. 2011;6:e19589. https://doi.org/10.1371/journal.pone.0019589.
Liu Y, Jiang N, Wu J, Dai W, Rosenblum JS. Polo-like kinases inhibited by wortmannin labeling site and downstream effects. J Biol Chem. 2007;282:2505–11. https://doi.org/10.1074/jbc.M609603200.
Phillips N, Hayward R, Koronakis V. Phosphorylation of the enteropathogenic E. coli receptor by the Src-family kinase c-Fyn triggers actin pedestal formation. Nat Cell Biol. 2004;6:618–25. https://doi.org/10.1038/ncb1148.
Swimm A, Bommarius B, Li Y, Cheng D, Reeves P, Sherman M, Veach D, Bornman W, Kalman D. Enteropathogenic Escherichia coli use redundant tyrosine kinases to form actin pedestals. Mol Biol Cell. 2004;15:3520–9. https://doi.org/10.1091/mbc.e04-02-0093.
Khatri A, Wang J, Pendergast AM. Multifunctional Abl kinases in health and disease. J Cell Sci. 2016;129:9–16. https://doi.org/10.1242/jcs.175521.
Rajagopalan K, Nagle E, Dworkin J. Identification and biochemical characterization ofa novel protein phosphatase 2C-like Ser/Thrphosphatase in Escherichia coli. J Bacteriol. 2018;200:e00225-e318. https://doi.org/10.1128/JB.00225-18.
Shi L. Manganese-dependent protein O-phosphatases in prokaryotesand their biological functions. Front Biosci. 2004;9:1382–97. https://doi.org/10.2741/1318.17.
Shi L, Potts M, Kennelly PJ. The serine, threonine, and/or tyrosine-specific protein kinases and protein phosphatases of prokaryotic organisms: a family portrait. FEMS Microbiol Rev. 1998;22:229–53. https://doi.org/10.1111/j.1574-6976.1998.tb00369.x.
Shi Y. Serine/threonine phosphatases: mechanism through struc-ture. Cell. 2009;139:468–84. https://doi.org/10.1016/j.cell.2009.10.006.
Ünal CM, Steinert M. Microbial peptidyl-prolyl cis/trans isomerases (PPIases): virulence factors and potential alternative drug targets. Microbiol Mol Biol Rev. 2014;78:544–71. https://doi.org/10.1128/MMBR.00015-14.
High KP. The antimicrobial activities of cyclosporine, FK506, and rapamycin. Transplantation. 1994;57:1689–700 (PMID: 7517076).
Shaya D, Findeisen F, Abderemane-Ali F, Arrigoni C, Wong S, Nurva SR, Loussouarn G, Minor DL Jr. Structure of a prokaryotic sodium channel pore reveals essential gating elements and an outer ion binding site common to eukaryotic channels. J Mol Biol. 2014;426:467–83. https://doi.org/10.1016/j.jmb.2013.10.010.
Ren D, Navarro B, Xu H, Yue L, Shi Q, Clapham DE. A prokaryotic voltage-gated sodium channel. Science. 2001;294:2372–5. https://doi.org/10.1126/science.1065635.
Koishi R, Xu H, Ren D, Navarro B, Spiller BW, Shi Q, Clapham DE. A superfamily of voltage-gated sodium channels in bacteria. J Biol Chem. 2004;279:9532–8. https://doi.org/10.1074/jbc.M313100200.
Irie K, Kitagawa K, Nagura H, Imai T, Shimomura T, Fujiyoshi Y. Comparative study of the gating motif and C-type inactivation in prokaryotic voltagegated sodium channels. J Biol Chem. 2010;285:3685–94. https://doi.org/10.1074/jbc.M109.057455.
Charalambous K, Wallace BA. NaChBac: the long lost sodium channel ancestor. Biochemistry. 2011;50:6742–52. https://doi.org/10.1021/bi200942y.
Tyson JR, Snutch TP. Molecular nature of voltage-gated calcium channels: structure and species comparison. WIREs Membr Transp Signal. 2013;2:181–206. https://doi.org/10.1002/wmts.91.
Shimomura T, Yonekawa Y, Nagura H, Tateyama M, Fujiyoshi Y, Irie K. A native prokaryotic voltage-dependent calcium channel with a novel selectivity filter sequence. eLife. 2020. https://doi.org/10.7554/eLife.52828.
Catterall WA, Swanson TM. Structural basis for pharmacology of voltage-gated sodium and calcium channels. Mol Pharmacol. 2015;88:141–50. https://doi.org/10.1124/mol.114.097659.
Yue L, Navarro B, Ren D, Ramos A, Clapham DE. The cation selectivityfilter of the bacterial sodium channel, NaChBac. J Gen Physiol. 2002;120:845–53. https://doi.org/10.1085/jgp.20028699.
Pozdnyakov I, Matantseva O, Skarlato S. Diversity and evolution of four-domain voltage-gated cation channels of eukaryotes and their ancestral functional determinants. Sci Rep. 2018;8:1–10. https://doi.org/10.1038/s41598-018-21897-7.
Tang L, Gamal El-Din T, Payandeh J, Martinez GQ, Heard TM, Scheuer T, Zeng N, Catterall WA. Structural basis for Ca2+ selectivity of a voltage-gated calcium channel. Nature. 2014;505:56–61. https://doi.org/10.1038/nature12775.
Tang L, Gamal El-Din TM, Swanson TM, Pryde DC, Scheuer T, Zheng N, Catteral WA. Structural basis for inhibition of a voltage-gated Ca2+ channel by Ca2+ antagonist drugs. Nature. 2016;537:117–21. https://doi.org/10.1038/nature19102.
Kumar KA, Ganguly K, Mazumdar K, Dutta NK, Dastidar SG, Chakrabarty AN. Amlodipine: a cardiovascular drug with powerful antimicrobial property. Acta Microbiol Pol. 2003;52:285–92 (PMID: 14743981).
Dasgupta A, Jeyaseeli L, Dutta NK, Mazumdar K, Karak P, Dastidar SG, Motohashi N, Shirataki Y. Studies on the antimicrobial potential of the cardiovascular drug lacidipine. In Vivo. 2007;21:847–50 (PMID: 18019422).
Pal T, Dutta NK, Mazumdar K, Dasgupta A, Jeyaseeli L, Dastidar SG. 2 Assessment of antibacterial activity of the cardiovascular drug nifedipine. Orient Pharm Exp Med. 2006;6:126–33. https://doi.org/10.3742/opem.2006.6.2.126.
Dasgupta A, Dastidar SG. Antibacterial and antitoxic effects of the cardiovascular drug lacidipine in an animal model. Indian J Med Res. 2012;135:913–6 (PMID: 22825612).
Tatar A, Korkmaz M, Yayla M, Polat E, Uslu H, Halici Z, Parlak SN. The potential role of amlodipine on experimentally induced bacterial rhinosinusitis. Braz J Otorhinolaryngol. 2017;83:619–26. https://doi.org/10.1016/j.bjorl.2016.08.006.
Gupta S, Tyagi S, Bishai WR. Verapamil increases the bactericidal activity of bedaquiline against Mycobacterium tuberculosis in a mouse model. Antimicrob Agents Chemother. 2015;59:673–6. https://doi.org/10.1128/AAC.04019-14.
Gupta S, Tyagi S, Almeida DV, Maiga MC, Ammerman NC, Bishai WR. Acceleration of tuberculosis treatment by adjunctive therapy with verapamil as an efflux inhibitor. Am J Respir Crit Care Med. 2013;188:600–7. https://doi.org/10.1164/rccm.201304-0650OC.
Treerat P, Widmer F, Middleton PG, Iredell J, George AM. In vitro interactions of tobramycin with various non antibiotics against Pseudomonas aeruginosa and Burkholderia cenocepacia. FEMS Microbiol Lett. 2008;285:40–50. https://doi.org/10.1111/j.1574-6968.2008.01219.x.
Tisa LS, Sekelsky JJ, Adler J. Effects of organic antagonists of Ca21, Na1, and K1 on chemotaxis and motility of Escherichia coli. J Bacteriol. 2000;182:4856–61. https://doi.org/10.1128/jb.182.17.4856-4861.2000.
Mitchell C, Skomurski JF, Vary JC. Effect of ion channel blockers on germination of Bacillus megaterium spores. FEMS Microbiol Lett. 1986;34:211–4. https://doi.org/10.1111/j.1574-6968.1986.tb01406.x.
Viljoen A, Raynaud C, Johansen MD, Roquet-Banères F, Herrmann JL, Daher W, Kremer L. Verapamil improves the activity of bedaquiline against Mycobacterium abscessus in vitro and in macrophages. Antimicrob Agents Chemother. 2019;63:e00705-e719. https://doi.org/10.1128/AAC.00705-19.
Coelho T, Machado D, Couto I, Maschmann R, Ramos D, von Groll A, Rossetti ML, Silva PA, Viveiros M. Enhancement of antibiotic activity by efflux inhibitors against multidrug resistant Mycobacterium tuberculosis clinical isolates from Brazil. Front Microbiol 2015; 6: 330. https://hdl.handle.net/10362/36925
Bazzaz FBS, Iranshahi M, Naderinasab M, Hajian S, Sabeti Z, Masumi E. Evaluation of the effects of galbanic acid from Ferula szowitsiana and conferol from F. badrakema, as modulators of multi-drug resistance in clinical isolates of Escherichia coli and Staphylococcus aureus. Res Pharm Sci. 2010;5:21–8 (PMID: 21589765).
Bazzaz BSF, Memariani Z, Khashiarmanesh Z, Iranshahi M, Naderinasab M. Effect of galbanic acid, a sesquiterpene coumarin from Ferula szowitsiana, as an inhibitor of efflux mechanism in resistant clinical isolates of Staphylococcus aureus. Braz J Microbiol. 2010;41:574–80. https://doi.org/10.1590/S1517-83822010000300006.
Martin A, Bouyakoub Y, Soumillion K, Mantu EN, Colmant A, Rodriguez-Villalobos H. Targeting bedaquiline mycobacterial efflux pump to potentially enhance therapy in Mycobacterium abscessus. Int J Mycobacteriol. 2020;9:71–5. https://doi.org/10.4103/ijmy.ijmy_181_19.
Ghajavand H, Kargarpour Kamakoli M, Khanipour S, Pourazar Dizaji S, Masoumi M, Rahimi Jamnani F, Fateh A, Siadat SD, Vaziri F. High prevalence of bedaquiline resistance in treatment-naive tuberculosis patients and verapamil effectiveness. Antimicrob Agents Chemother. 2019;63:e02530-e2618. https://doi.org/10.1128/AAC.02530-18.
Zhang Z, Yan J, Xu K, Ji Z, Li L. Tetrandrine reverses drug resistance in isoniazid and ethambutol dual drug-resistant Mycobacterium tuberculosis clinical isolates. BMC Infect Dis. 2015;15:153. https://doi.org/10.1186/s12879-015-0905-0.
Machado D, Pires D, Perdigão J, Couto I, Portugal I, Martins M, Amaral L, Anes E, Viveiros M. Ion channel blockers as antimicrobial agents, efflux inhibitors, and enhancers of macrophage killing activity against drug resistant Mycobacterium tuberculosis. PLoS ONE. 2016;11:e0149326. https://doi.org/10.1371/journal.pone.0149326.
Adams KN, Szumowski JD, Ramakrishnan L. Verapamil, and its metabolite norverapamil, inhibit macrophage-induced, bacterial efflux pump-mediated tolerance to multiple anti-tubercular drugs. J Infect Dis. 2014;210:456–66. https://doi.org/10.1093/infdis/jiu095.
Seral C, Michot JM, Chanteux H, Mingeot-Leclercq MP, Tulkens PM, Van Bambeke F. Influence of P-glycoprotein inhibitors on the accumulation of macrolides in J774 murine macrophages. Antimicrob Agents Chemother. 2003;47:1047–51. https://doi.org/10.1128/AAC.47.3.1047-1051.2003.
Seral C, Carryn S, Tulkens PM, Van Bambeke F. Influence of P-glycoprotein and MRP efflux pump inhibitors on the intracellular activity of azithromycin and ciprofloxacin in macrophages infected by Listeria monocytogenes or Staphylococcus aureus. J Antimicrob Chemother. 2003;51:1167–73. https://doi.org/10.1093/jac/dkg223.
Lemaire S, Van Bambeke F, Mingeot-Leclercq MP, Tulkens PM. Modulation of the cellular accumulation and intracellular activity of daptomycin towards phagocytized Staphylococcus aureus by the P-glycoprotein (MDR1) efflux transporter in human THP-1 macrophages and madindar by canine kidney cells. Antimicrob Agents Chemother. 2007;51:2748–57. https://doi.org/10.1128/AAC.00090-07.
Chao HuC, Li Y, Zhao Z, Wie S, Zhao Z, Chen H, Wu P. In vitro synergistic effect of amlodipine and imipenem on the expression of AdeABC efflux pump in multidrug-resistant Acinetobacter baumanii. PLoS ONE. 2018;13:e0198061. https://doi.org/10.1371/journal.pone.0198061.
Gupta S, Cohen KA, Winglee K, Maiga M, Diarra B, Bishai WR. Efflux inhibition with verapamil potentiates bedaquiline in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2014;58:574–6. https://doi.org/10.1128/AAC.01462-13.
Pule CM, Sampson SL, Warren RM, Black PA, van Helden PD, Victor TC, Louw GE. Efflux pump inhibitors: targeting mycobacterial efflux systems to enhance TB therapy. J Antimicrob Chemother. 2016;71:17–26. https://doi.org/10.1093/jac/dkv316.
Adams KN, Takaki K, Connolly LE, Edelstein PH, Cosma CL, Ramakrishnan L. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell. 2011;145:39–53. https://doi.org/10.1016/j.cell.2011.02.022.
Rodrigues L, Villellas C, Bailo R, Viveiros M, Ainsa JA. Role of the Mmr efflux pump in drug resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2013;7:751–7. https://doi.org/10.1128/AAC.01482-12.
Xu J, Tasneen R, Peloquin CA, Almeida DV, Li S-Y, Barnes-Boyle K, Lu Y, Nuermberger E. Verapamil increases the bioavailability and efficacy of bedaquiline but not clofazimine in a murine model of tuberculosis. Antimicrob Agents Chemother. 2018;62:e01692-e1717. https://doi.org/10.1128/AAC.01692-17.
van Veen HW, Venema K, Bolhuis H, Oussenko I, Kok J, Poolman B, Driessen AJM, Konings WN. Multidrug resistance mediated by a bacterial homolog of the human multidrug transporter MDR1. Proc Natl Acad Sci USA. 1996;1006(93):10668–72. https://doi.org/10.1073/pnas.93.20.10668.
Bolhuis H, van Veen HW, Molenaar D, Poolman B, Driessen AJM, Konings WN. Multidrug resistance in Lactococcus lactis: evidence for ATP-dependent drug extrusion from the inner leaflet of the cytoplasmic membrane. EMBO J. 1996;1996(15):4239–45. https://doi.org/10.1002/j.1460-2075.1996.tb00798.x.
van Veen HW, Callaghan R, Soceneantu L, Sardini A, Konings WN, Higgins CF. A bacterial antibiotic-resistance gene that complements the human multidrug-resistance P-glycoprotein gene. Nature. 1998;391:291–5. https://doi.org/10.1038/34669.
van Veen HW, Margolles A, Müller M, Higgins CF, Konings WN. The homodimeric ATP-binding cassette transporter LmrA mediates multidrug transport by an alternating two-site (two-cylinder engine) mechanism. EMBO J. 2000;19:2503–14. https://doi.org/10.1093/emboj/19.11.2503.
Poelarends GJ, Mazurkiewicz P, Putman M, Cool RH, van Veen HW, Konings WN. An ABC-type multidrug transporter of Lactococcus lactis possesses an exceptionally broad substrate specificity. Drug Resist Updates. 2000;3:330–4. https://doi.org/10.1054/drup.2000.0173.
Casaregola S, Chen M, Bouquin N, Norris V, Jacq A, Goldberg M, Margarson S, Tempete M, McKenna S, Sweetman H, Bernard S, McGurk G, Seror S, Holland IB. Analysis of a myosin-like protein and the role of calcium in the E. coli cell cycle. Res Microbiol. 1991;142:201–7. https://doi.org/10.1016/0923-2508(91)90031-5.
Andersen CL, Holland IB, Jacq A. Verapamil, a Ca2+ channel inhibitor acts as a local anesthetic and induces the sigma E dependent extra-cytoplasmic stress response in E. coli. Biochim Biophys Acta Biomembr. 2006;1758:1587–95. https://doi.org/10.1016/j.bbamem.2006.05.022.
Poolman B, Spitzer JJ, Wood JM. Bacterial osmosensing: roles of membrane structure and electrostatics in lipid–protein and protein–protein interactions. Biochim Biophys Acta. 2004;1666:88–104. https://doi.org/10.1016/j.bbamem.2004.06.013.
Pohl EE, Krylov AV, Block M, Pohl P. Changes of the membrane potential profile induced by verapamil and propranolol. Biochim Biophys Acta. 1998;1373:170–8. https://doi.org/10.1016/S0005-2736(98)00098-4.
Chen C, Gardete S, Jansen RS, Shetty A, Dick T, Rhee KY, Dartois V. Verapamil targets membrane energetics in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2018;62:e02107-e2117. https://doi.org/10.1128/AAC.02107-17.
Suwalsky M, Munoz M, Mennickent S, Sotomayor CP, Bolognin S, Zatta P. Structural effects of verapamil on cell membranes and molecular models. J Chil Chem Soc. 2010;55:1–4. https://doi.org/10.4067/S0717-97072010000100002.
Meier M, Blatter XL, Seelig A, Seelig J. Interaction of verapamil with lipid membranes and P-glycoprotein: connecting thermodynamics and membrane structure with functional activity. Biophys J. 2006;91:2943–55. https://doi.org/10.1529/biophysj.106.089581.
Mason RP, Moisey DM, Shajenko L. Cholesterol alters the binding of Ca2+ channel blockers to the membrane lipid bilayer. Mol Pharmacol. 1992;41:315–21 (PMID: 1531693).
Mason RP. Differential effect of cholesterol on membrane interaction of charged versus uncharged 1,4-dihydropyridine calcium channel antagonists: a biophysical analysis. Cardiovasc Drug Ther. 1995;9:45–54. https://doi.org/10.1007/BF00878572.
Herbette L, Vant-Erve YMH, Rhodes D. Interaction of 1,4 dihydropyridine calcium channel antagonists with biological membranes: lipid bilayer partitioning could occur before drug binding to receptors. J Mol Cell Cardiol. 1989;21:187–201. https://doi.org/10.1016/0022-2828(89)90861-4.
Watts JA. Effects of dihydropyridine calcium antagonists upon microvascular function following ischemia and oxidative stress. In: Busse WD, Garthoff B, Seuter F, editors. Dihydropyridines. Progress in pharmacology and therapy. Berlin: Springer; 1993. p. 46–59 (ISBN-13: 978-3-540-57308-1).
Pajeva IK, Wiese M, Cordes HP, Seydel JK. Membrane interactions of some catamphiphilic drugs and relation to their multidrug-resistance-reversing ability. J Cancer Res Clin Oncol. 1996;122:27–40. https://doi.org/10.1007/bf01203070.
Dalhoff A. Membrane interactions of antibacterial agents. Trend Clin Microbiol. 2018; 1: 04–48. https://www.gratisoa.org/journals/index.php/TCMY/article/view/1244/1173. Accessed 29 May 2020
Jouan E, Le Vée M, Mayati A, Denizot C, Parmentier Y, Fardel O. Evaluation of P-glycoprotein inhibitory potential using a rhodamine 123 accumulation assay. Pharmaceutics. 2016;8:12. https://doi.org/10.3390/pharmaceutics8020012.
Shi J, Zhang Y, Yeleswaram S. The relevance of assessment of intestinal P-gp inhibition using digoxin as an in vivo probe substrate. Nat Rev Drug Discov. 2011;10:75. https://doi.org/10.1038/nrd3028-c1.
Katoh M, Nakajima M, Yamazaki H, Yokoi T. Inhibitory potencies of 1, 4-dihydropyridine calcium antagonists to P-glycoprotein-mediated transport: comparison with the effects on CYP3A4. Pharm Res. 2000;17:1189–97. https://doi.org/10.1023/A:1007568811691.
Jaffrezou JP, Herbert JM, Levade T, Gau MN, Chatelain P, Laurent G. Reversal of multidrug resistance by calcium channel blocker SR33557 without photoaffinity labeling of P-glycoprotein. J Biol Chem. 1991;266:19858–64 (PMID: 1918089).
Martinac B, Saimi Y, Kung C. Ion channels in microbes. Physiol Rev. 2008;88:1449–90. https://doi.org/10.1152/physrev.00005.2008.
Brunia GN, Weekley A, Doddb BJTD, Kralja JM. Voltage-gated calcium flux mediates Escherichia coli mechanosensation. Proc Nat Acad Sci. 2017;114:9445–50. https://doi.org/10.1073/pnas.1703084114.
Ehlers MR, Riordan JF. Angiotensin-converting enzyme: zinc- and inhibitor-binding stoichiometries of the somatic and testis isozymes. Biochemistry. 1991;30:7118–26. https://doi.org/10.1021/bi00243a012.
de Souza MC, Diniz LF, Franco CHF, Diniz R. Synthesis and crystalline structure of zinc complexes with antihypertensive drug lisinopril. J Chem. 2018. https://doi.org/10.1155/2018/8910242 (Article ID 8910242).
de Souza MC, Franco CHJ, Pinheiro CB, Ciniz R. Conformational polymorphism of a zinc complex with enalapril antihypertensive drug. Polyhedron. 2014;81:290–7. https://doi.org/10.1016/j.poly.2014.06.025.
Natesh R, Schwager SL, Evans HR, Sturrock ED, Acharya KR. Structural details on the binding of antihypertensive drugs captopril and enalaprilat to human testicular angiotensin I-converting enzyme. Biochemistry. 2004;43:8718–24. https://doi.org/10.1021/bi049480n.
Natesh R, Schwager SL, Sturrock ED, Acharya KR. Crystal structure of the human angiotensin-converting enzyme–lisinopril complex. Nature. 2003;421:551–4. https://doi.org/10.1038/nature01370.
Hughes MA, Smith GL, Williams DR. The binding of metal ions by captopril (SQ 14225). Part I. Complexation of zinc(II), cadmium(II) and lead(II). Inorg Chim Acta. 1985;107:247–52. https://doi.org/10.1016/S0020-1693(00)82296-3.
Christie GL, Hughes MA, Rees SB, Williams DR. The binding of metal ions by captopril (SQ 14225). Part II. Complexation of copper(II). Inorg Chim Acta. 1988;151:215–25. https://doi.org/10.1016/S0020-1693(00)83470-2.
Brem J, van Berkel SS, Zollman D, Lee SY, Gileadi O, McHugh PJ, Walsh TR, McDonough MA, Schofield CJ. Structural basis of metallo-β-lactamase inhibition by captopril stereoisomers. Antimicrob Agents Chemother. 2016;60:142–50. https://doi.org/10.1128/AAC.01335-15.
Li GB, Abboud MI, Brem J, Someya H, Lohans CT, Yang SY, Spencer J, Wareham DW, MacDonough MA, Schofield CJ. NMR-filtered virtual screening leads to non-metal chelating metallo-β-lactamase inhibitors. Chem Sci. 2017;8:928–37. https://doi.org/10.1039/C6SC04524C.
Guo Y, Wang J, Niu G, Shui W, Sun Y, Zhou H, Zhang Y, Yang C, Lou Z, Rao Z. A structural view of the antibiotic degradation enzyme NDM-1 from a superbug. Protein Cell. 2011;2:384–94. https://doi.org/10.1007/s13238-011-1055-9.
García-Sáez I, Hopkins J, Papamicael C, Franceschini N, Amicosante G, Rossolini GM, Galleni M, Frere JM, Dideberg O. The 1.5-Å structure of Chryseobacterium meningosepticum zinc β-lactamase in complex with the inhibitor, d-captopril. J Biol Chem. 2003;278:23868–73. https://doi.org/10.1074/jbc.m301062200.
Somboro AM, Osei Sekyere J, Amoako DG, Essack SY, Bester LA. Diversity and proliferation of metallo-β-lactamases: a clarion call for clinically effective metallo-β-lactamase inhibitors. Appl Environ Microbiol. 2018;84:e00698-e718. https://doi.org/10.1128/AEM.00698-18.
Hinchliffe P, González MM, Mojica MF, González JM, Castillo V, Saiz C, Kosmopoulou M, Tooke CL, Li L, Mahler G, Bonomo RA, Vila AJ, Spencer J. Cross-class metallo-β-lactamase inhibition by bisthiazolidines reveals multiple binding modes. Proc Nat Acad Sci. 2016;113:E3745–54. https://doi.org/10.1073/pnas.1601368113.
Uda NR, Creus M. Selectivity of inhibition of N-succinyl-l, l-diaminopimelic acid desuccinylase in bacteria: the product of DapE-gene is not the target of l-captopril antimicrobial activity. Bioinorg Chem Appl. 2011. https://doi.org/10.1155/2011/306465.
Chopra S, Matsuyama K, Hutson C, Madrid P. Identification of antimicrobial activity among FDA-approved drugs for combating Mycobacterium abscessus and Mycobacterium chelonae. J Antimicrob Chemother. 2011;66:1533–6. https://doi.org/10.1093/jac/dkr154.
Klingler FM, Wichelhaus TA, Frank D, Cuesta-Bernal J, El-Delik J, Müller HF, Sjuts H, Goettig S, Koenigs A, Poest HM, Pogoryelov D, Proschak E. Approved drugs containing thiols as inhibitors of metallo-β-lactamases: strategy to combat multidrug-resistant bacteria. J Med Chem. 2015;58:3626–30. https://doi.org/10.1021/jm501844d.
Li N, Xu Y, Xia Q, Bai C, Wang T, Wang L, He D, Xie N, Li L, Wang J, Zhou HG, Xu F, Yang C, Zhang Q, Yin Z, Guo Y, Cehn Y. Simplified captopril analogues as NDM-1 inhibitors. Bioorg Med Chem Lett. 2014;24:386–9. https://doi.org/10.1016/j.bmcl.2013.10.068.
Yusof Y, Tan DT, Arjomandi OK, Schenk G, McGeary RP. Captopril analogues as metallo-β-lactamase inhibitors. Bioorg Med Chem Lett. 2016;26:1589–93. https://doi.org/10.1016/j.bmcl.2016.02.007.
Rivière G. Angiotensin-converting enzyme: a protein conserved during evolution. J Soc Biol. 2009;203:281–93. https://doi.org/10.1051/jbio/2009032.
Rivière G, Michaud A, Corradi HR, Sturrock ED, Acharya KR, Cogez V, Bohin JP, Vieau D, Corvol P. Characterization of the first angiotensin-converting like enzyme in bacteria: ancestor ACE is already active. Gene. 2007;399:81–90. https://doi.org/10.1016/j.gene.2007.05.010.
Bernstein KE, Ong FS, Blackwell WL, Shah KH, Giani JF, Gonzalez-Villalobos RA, Shen XZ, Fuchs S, Touyz RM. A modern understanding of the traditional and nontraditional biological functions of angiotensin-converting enzyme. Pharmacol Rev. 2013;65:1–46. https://doi.org/10.1124/pr.112.006809.
Macours N, Poels J, Hens K, Francis C, Huybrechts R. Structure, evolutionary conservation, and functions of angiotensin- and endothelin-converting enzymes. Int Rev Cytol. 2004;239:47–97. https://doi.org/10.1016/S0074-7696(04)39002-9.
Stevens J, Fanburg BL, Lanzillo JJ. Determination of peptidyl dipeptidase activity in 24 bacterial species. Can J Microbiol. 1990;36:56–9. https://doi.org/10.1139/m90-010.
Ogasawara W, Abe N, Hagio T, Okada H, Morikawa Y. Purification and characterization of a dipeptidyl carboxypeptidase from Pseudomonas sp. WO24. Biosci Biotechnol Biochem. 1997;61:858–63. https://doi.org/10.1271/bbb.61.858.
Nagamori Y, Kusaka K, Fujishima N, Okada S. Enzymatic properties of dipeptidyl carboxypeptidase from Bacillus pumilus. Agric Biol Chem. 1991;55:1695–9. https://doi.org/10.1080/00021369.1991.10870850.
Kramer GJ. Pharmacological and chemical probe development of the K-26 family of natural product angiotensin-I converting enzyme inhibitors. Dissertation 2014, Vanderbilt University, Nashville, Tennessee. https://etd.library.vanderbilt.edu/available/etd-10012014-134811/unrestricted/Kramer_Dissertation.pdf. Accessed 29 May 2020.
Harty DW, Hunter N. Carboxypeptidase activity common to viridans group streptococci cleaves angiotensin I to angiotensin II: an activity homologous to angiotensin-converting enzyme (ACE). Microbiology. 2011;157:2143–51. https://doi.org/10.1099/mic.0.048710-0.
Harty DW, Farahani RM, Simonian MR, Hunter L, Hunter N. Streptococcus gordonii FSS2 challisin affects fibrin clot formation by digestion of the αC region and cleavage of the N-terminal region of the Bβ chains of fibrinogen. Thromb Haemost. 2012;108:236–46. https://doi.org/10.1160/TH12-01-0032.
Boone CD, Habibzadegan A, Gill S, McKenna R. Carbonic anhydrases and their biotechnological applications. Biomolecules. 2013;3:553–62. https://doi.org/10.3390/biom3030553.
Supuran CT, Capasso CT. An overview of the bacterial carbonic anhydrases. Metabolites. 2017;7:56. https://doi.org/10.3390/metabo7040056.
Supuran CT. Bacterial carbonic anhydrases as drug targets: toward novel antibiotics? Front Pharmacol. 2011;2:34. https://doi.org/10.3389/fphar.2011.00034.
Capasso C, Supuran CT. Inhibition of bacterial carbonic anhydrases as a novel approach to escape drug resistance. Curr Top Med Chem. 2017;17:1237–48. https://doi.org/10.2174/1568026617666170104101058.
Capasso C, Supuran CT. Chapter 3: Developing novel bacterial targets: carbonic anhydrases as antibacterial drug targets. In: Phoenix DA, Harris F, Dennison SR, editors. Novel antimicrobial agents and strategies. Weinheim: Wiley-VCH Verlag GmbH & CoKGaA; 2015. p. 31–46 (Print ISBN: 978-3-527-33638-8).
Nafi BM, Miles RJ, Butler LO, Carter ND, Kelly C, Jeffery S. Expression of carbonic anhydrase in Neisseriae and other heterotrophic bacteria. J Med Microbiol. 1990;32:1–7. https://doi.org/10.1099/00222615-32-1-1.
Shahidzadeh R, Opekun A, Shiotani A, Graham DY. Effect of the carbonic anhydrase inhibitor, acetazolamide, on Helicobacter pylori infection in vivo: a pilot study. Helicobacter. 2005;10:136–8. https://doi.org/10.1111/j.1523-5378.2005.00306.x.
Esmaeili S, Ashrafi-Kooshk MR, Adibi H, Khodarahmi R. Captopril/enalapril inhibit promiscuous esterase activity of carbonic anhydrase at micromolar concentrations: an in vitro study. Chem Biol Interact. 2017;265:24–35. https://doi.org/10.1016/j.cbi.2017.01.014.
Sharma A, Gupta VK, Pathania R. Efflux pump inhibitors for bacterial pathogens: from bench to bedside. Indian J Med Res. 2019;2019(149):129–45. https://doi.org/10.4103/ijmr.IJMR_2079_17.
Du D, Wang-Kan X, Neuberger A, van Veen HW, Pos KM, Piddock LJV, Luisi BF. Multidrug efflux pumps: structure, function and regulation. Nat Rev Microbiol. 2018;16:523–39. https://doi.org/10.1038/s41579-018-0048-6.
Fernández L, Hancock RE. Adaptive and mutational resistance: role of porins and efflux pumps in drug resistance. Clin Microbiol Rev. 2012;25:661–81. https://doi.org/10.1128/CMR.00043-12.
Van Bambeke F, Michot JM, Tulkens PM. Antibiotic efflux pumps in eukaryotic cells: occurrence and impact on antibiotic cellular pharmacokinetics, pharmacodynamics and toxicodynamics. J Antimicrob Chemother. 2003;51:1067–77. https://doi.org/10.1093/jac/dkg225.
Vargiu AV, Pos KM, Poole K, Nikaido H. Bad bugs in the XXIst century: resistance mediated by multi-drug efflux pumps in Gram-negative bacteria. Front Microbiol. 2016;7:833. https://doi.org/10.3389/fmicb.2016.00833.
Lamut A, Masic LP, Kikeli D, Tomasic T. Efflux pump inhibitors of clinically relevant multidrug resistant bacteria. Med Res Rev. 2019;39:2460–504. https://doi.org/10.1002/med.21591.
Laudy AE, Mrowka A, Krajewska J, Tyski S. The influence of efflux pump inhibitors on the activity of non-antibiotic NSAIDS against Gram-negative rods. PLoS ONE. 2016;11:e0147131. https://doi.org/10.1371/journal.pone.0147131.
Zimmermann P, Curtis N. Antimicrobial effects of antipyretics. Antimicrob Agents Chemother. 2017;61:e02268-e2316. https://doi.org/10.1128/AAC.02268-16.
Silva AADL, Silva PM. Non-antibiotic compounds: the activity of the NSAID diclofenac on bacteria—a review. Int J Curr Microbiol App Sci. 2018;7:340–51. https://doi.org/10.20546/ijcmas.2018.712.042.
Price CT, Lee IR, Gustafson JE. The effects of salicylate on bacteria. Int J Biochem Cell Biol. 2000;32:1029–43. https://doi.org/10.1016/S1357-2725(00)00042-X.
Blanco P, Hernando-Amado S, Reales-Calderon JA, Corona F, Lira F, Alcalde-Rico M, Bernardini A, Sanchez MB, Martinez JL. Bacterial multidrug efflux pumps: much more than antibiotic resistance determinants. Microorganisms. 2016;4:14. https://doi.org/10.3390/microorganisms4010014.
Galperin MY, Koonin EV. Divergence and convergence in enzyme evolution. J Biol Chem. 2012;287:21–8. https://doi.org/10.1074/jbc.R111.241976.
Baydoun AR, Markham A, Morgan RM, Sweetman AJ. Bay K 8644, modifier of calcium transport and energy metabolism in rat heart mitochondria: a new intracellular site of action. Br J Pharmacol. 1990;101:15. https://doi.org/10.1111/j.1476-5381.1990.tb12081.x.
Leblondel G, Allan P. Ca2+ uptake and energy supply of sheep heart mitochondria in presence of some calcium antagonists. Res Commun Chem Pathol Pharmacol. 1984;44:499–502 (PMID: 6611569).
Uzieliene I, Bernotiene E, Rakauskiene G, Denkovskij J, Bagdonas E, Mackiewicz Z, Porvaneckas N, Kvederas G, Mobasheri A. The antihypertensive drug nifedipine modulates the metabolism of chondrocytes and human bone marrow-derived mesenchymal stem cells. Front Endocrinol. 2019;10:756. https://doi.org/10.3389/fendo.2019.00756.
Park HH, Han MH, Choi H, Lee YJ, Kim JM, Cheong JH, Ryu JI, Lee KY, Koh SH. Mitochondria damaged by oxygen glucose deprivation can be restored through activation of the PI3K/Akt pathway and inhibition of calcium influx by amlodipine camsylate. Sci Rep. 2019;9:1–11. https://doi.org/10.1038/s41598-019-52083-y.
Tabata Y, Imaizumi Y, Sugawara M, Andoh-Noda T, Banno S, Chai M, Sone T, Yamazaki K, Ito M, Tsukahara K, Saya H, Hattori N, Kohyama J, Saya H. T-type calcium channels determine the vulnerability of dopaminergic neurons to mitochondrial stress in familial Parkinson disease. Stem Cell Rep. 2018;11:1171–84. https://doi.org/10.1016/j.stemcr.2018.09.006.
Ederer KA, Jin K, Bouslog S, Wang L, Gorman GS, Rowe GC, Abadir P, Raftery D, Moellering D, Promislow D, Jumbo-Luconi P, DeLuca M. Age-and genotype-specific effects of the angiotensin-converting enzyme inhibitor lisinopril on mitochondrial and metabolic parameters in Drosophila melanogaster. Int J Mol Sci. 2018;19:3351. https://doi.org/10.3390/ijms19113351.
De Cavanagh EMV, Inserra F, Ferder M, Ferder L. From mitochondria to disease: role of the renin-angiotensin system. Am J Nephrol. 2007;27:545–53. https://doi.org/10.1159/000107757.
Kojic Z, Gopcevic K, Marinkovic D, Tasic G. Effect of captopril on serum lipid levels and cardiac mitochondrial oxygen consumption in experimentally-induced hypercholesterolemia in rabbits. Physiol Res. 2011;60(Suppl 1):S177–84 (PMID: 21777025).
Kojic ZZ. The cardioprotective effects of ramipril during the course of experimental hypercholesterolemia in rabbits. J Clin Transl Sci. 2020;4:17–8. https://doi.org/10.1017/cts.2020.96.
Kancirová I, Jašová M, Waczulíková I, Ravingerová T, Ziegelhöffer A, Ferko M. Effect of antihypertensive agents-captopril and nifedipine-on the functional properties of rat heart mitochondria. Iranian J Basic Med Sci. 2016;19:615–23. https://doi.org/10.22038/ijbms.2016.7129.
Will Y, Dykens JA, Nadanaciva S, Hirakawa B, Jamieson J, Marroquin LD, Hynes J, Patyna S, Jessen BA. Effect of the multitargeted tyrosine kinase inhibitors imatinib, dasatinib, sunitinib, and sorafenib on mitochondrial function in isolated rat heart mitochondria and H9c2 cells. Toxicol Sci. 2008;106:153–61. https://doi.org/10.1093/toxsci/kfn157.
Emadi E, Abdoli N, Ghanbarinejad V, Mohammadi HR, Mobarakeh KM, Azarpira N, Mahboudi Z, Niknahad H, Heidari R. The potential role of mitochondrial impairment in the pathogenesis of imatinib-induced renal injury. Heliyon. 2019;5:e01996. https://doi.org/10.1016/j.heliyon.2019.e01996.
Nash A, Samoylova M, Leuthner T, Zhu M, Lin L, Meyer JN, Brennan TV. Effects of immunosuppressive medications on fitochondrial function. J Surg Res. 2020;249:50–7. https://doi.org/10.1016/j.jss.2019.12.010.
Yu JH, Lim SW, Luo K, Cui S, Quan Y, Shin YJ, Lee KE, Kim HL, Ko EJ, Chung BH, Kim JH, Chung SJ, Yang CW. Coenzyme Q10 alleviates tacrolimus-induced mitochondrial dysfunction in kidney. FASEB J. 2019;33:12288–98. https://doi.org/10.1096/fj.201900386RR.
Pacheu-Grau D, Gómez-Durán A, López-Pérez MJ, Montoya J, Ruiz-Pesini E. Mitochondrial pharmacogenomics: barcode for antibiotic therapy. Drug Discov Today. 2010;15:33–9. https://doi.org/10.1016/j.drudis.2009.10.008.
Zhang J, Haines C, Watson AJ, Hart AR, Platt MJ, Pardoll DM, Cosgrove SE, Gebo KA, Sears CL. Oral antibiotic use and risk of colorectal cancer in the United Kingdom, 1989–2012: a matched case–control study. Gut. 2019;68:1971–8. https://doi.org/10.1136/gutjnl-2019-318593.
Elliott RL, Jiang XP, Baucom CC. Antibiotic overusage causes mitochondrial dysfunction which may promote tumorigenesis. J Cancer Treat Res. 2017;5:62–5. https://doi.org/10.11648/j.jctr.20170504.11.
Elliott RL, Jiang XP, Baucom C, Lomnicka Z. Antibiotics friend and foe: from wonder drug to causing mitochondrial dysfunction, disrupting human microbiome and promoting tumorigenesis. Int J Clin Med 2018;9: 182–186. https://www.scirp.org/journal/ijcm
Andrade MJ, Jayaprakash C, Bhat S, Evangelatos N, Brand A, Satyamoorthy K. Antibiotics-induced obesity: a mitochondrial perspective. Public Health Genom. 2017;20:257–73. https://doi.org/10.1159/000485095.
Stefano G, Sanuel J, Kream RM. Antibiotics may trigger mitochondrial dysfunction inducing psychiatric disorders. Med Sci Monit. 2017;23:101–6. https://doi.org/10.12659/MSM.899478.
Obregon D, Parker-Athill EC, Tan J, Murphy T. Psychotropic effects of antimicrobials and immune modulation by psychotropics: implications for neuroimmune disorders. Neuropsychiatry. 2012;2:331–43. https://doi.org/10.2217/npy.12.41.
Will Y, Shields JE, Wallace KB. Drug-induced mitochondrial toxicity in the geriatric population: challenges and future directions. Biology. 2019;8:32. https://doi.org/10.3390/biology8020032.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that there is no conflict of interest.
Additional information
A companion paper entitled “Selective toxicity of antibacterial agents—still a valid concept or do we miss chances and ignore risks? “has described mechanisms of non-antibacterial effects of antibiotics in eukaryotes.
Addendum search strategy
Addendum search strategy
Publications addressing four topics were screened: first, phosphorylation and dephosphorylation with the keywords “Hanks-type kinases”, serine/threonine-, serine/tyrosine protein kinases (STPKs), serine/threonine phosphatases, penicillin-binding and serine/threonine kinase-associated (PASTA) protein, and the corresponding inhibitors. Second, initiation and propagation of electrical signalling by voltage-gated ion channels with the key words calcium antagonists, dihydropyridines, benzodiazepines, phenylalkylamines and the corresponding drugs, in particular nitrendipine and its derivatives, amlodipine, and verapamil. Third, angiotensin I converting enzyme (ACE, or kininase II) with the keywords ACE-inhibitors, in particular captopril. Fourth, evolutionary originin-, ancestry of prokaryotic, eukaryotic, archaeal proteins or active centres, homolog, ortholog, paralog. Additional keywords were antibiotics in general and sulfonamides, β-lactams, aminoglycosides, macrolides, chloramphenicol, oxazolidinones, tetracyclines, fluoroquinolones and the corresponding single agents of these drug-classes. Search strategy and selection criteria were based on the combination of keywords. Articles summarized in recent reviews were excluded from this synopsis and the reviews are quoted instead.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dalhoff, A. Are antibacterial effects of non-antibiotic drugs random or purposeful because of a common evolutionary origin of bacterial and mammalian targets?. Infection 49, 569–589 (2021). https://doi.org/10.1007/s15010-020-01547-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s15010-020-01547-9