
Abstract
Within the scope of SAMPL7 challenge for predicting physical properties, the Integral Equation Formalism of the Miertus-Scrocco-Tomasi (IEFPCM/MST) continuum solvation model has been used for the blind prediction of n-octanol/water partition coefficients and acidity constants of a set of 22 and 20 sulfonamide-containing compounds, respectively. The log P and pKa were computed using the B3LPYP/6-31G(d) parametrized version of the IEFPCM/MST model. The performance of our method for partition coefficients yielded a root-mean square error of 1.03 (log P units), placing this method among the most accurate theoretical approaches in the comparison with both globally (rank 8th) and physical (rank 2nd) methods. On the other hand, the deviation between predicted and experimental pKa values was 1.32 log units, obtaining the second best-ranked submission. Though this highlights the reliability of the IEFPCM/MST model for predicting the partitioning and the acid dissociation constant of drug-like compounds compound, the results are discussed to identify potential weaknesses and improve the performance of the method.
Similar content being viewed by others
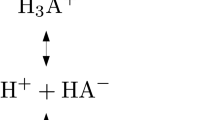
Introduction
Lipophilicity and (de)protonation are physicochemical properties that play a fundamental role to understand the biological activity of drugs [1,2,3,4]. From a pharmacokinetic point of view, these properties exert a marked influence on the ADME-Tox profile of drugs, affecting solubility in physiological fluids and permeability through biological barriers, as well as the excretion rate from the human body [5]. With regard to drug pharmacodynamics, lipophilicity affects recognition and binding of drugs to their macromolecular targets, since the global hydrophobic character is related to the changes in (de)solvation involved in ligand binding, whereas a complementarity between the 3D distribution of hydrophobic/hydrophilic regions in the drug and the binding pocket should reinforce the drug-target interaction [6,7,8]. On the other hand, the (de)protonation of a compound can clearly exert influence on the bioavailability of a molecule, affecting not only the biodistribution of the bioactive compound in the organism, but altering the interaction pattern that may be formed with specific residues in the binding pocket [9, 10].
The n-octanol/water partition coefficient (log P) is the physicochemical parameter generally adopted to quantify the lipophilicity of a compound, and can be experimentally determined from the partitioning between aqueous and n-octanol phases. From a computational point of view, log P can be estimated from the transfer free energy (\(\Delta \Delta {\text{G}}^{{{\text{w}} \to {\text{o}}}}\); Scheme 1) of the molecule between these two solvents, which in turn can be derived from the solvation free energy in n-octanol (\(\Delta{\text{G}}_{\text{solv}}^{\text{o}}\)) and water (\(\Delta{\text{G}}_{\text{hyd}}^{\text{w}}\)). The ionization equilibrium of a titratable compound is quantified by the negative logarithm of the acid dissociation constant (pKa), which reflects the population of acidic and basic species. This quantity can be related to the free energy change for the ionization of the compound in water (\(\Delta{\text{G}}_{\text{aq}}\); Scheme 1), which in turn can be calculated combining the free energy change for this process in the gas phase with the solvation free energies of protonated (HX) and deprotonated (X−) species of the compound and the solvation free energy of the proton [11, 12].

Thermodynamic cycles used to determine (left) the transfer free energy of a neutral (HX) compound between n-octanol and water, and (right) the pKa estimation of a titratable compound, where HX and X− stand for the acidic and basic species, respectively
The availability of computational tools able to provide accurate estimates of log P and pKa is valuable to provide useful guides in the search of novel hit compounds and the drug development process [13, 14]. This may deserve special interest in the screening of large libraries of compounds, as the experimental measurement of these properties would be demanding and often facing experimental challenges for specific classes of compounds. In this context, we present here the results obtained in the context of the SAMPL7 blind challenge [15]. Given the fundamental role of the solvation free energy in the computational prediction of both log P and pKa, our computational strategy exploits the B3LYP/6-31G(d) parametrized version [16, 17] of the quantum mechanical IEFPCM/MST solvation model [18], which relies on the Integral Equation Formalism of the Polarizable Continuum model [19, 20]. Here, we report the results obtained for predicting the log P and pKa for a group of sulfonamide-containing compounds. The results are discussed in light of the experimental data provided by the organizers of SAMPL7 [21] and the theoretical estimates reported by others groups, as well as with the IEFPCM/MST results obtained in previous editions of this contest [22, 23].
Methods
Test compounds
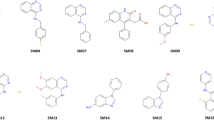
The dataset used in the SAMPL7 challenge contains 22 compounds (numbered SM25 to SM46; Fig. 1) provided by Carlo Ballatore and coworkers at UCSD (University of California, San Diego). Most of the compounds share chemical motifs, including the presence of a sulfonamide unit, a phenylethyl moiety (with the exception of compounds SM41- SM46), and a four-membered ring fused to the main chain, often containing oxygen and sulphur. Few compounds (SM41-SM46) include specific moieties, such as isoxazole (SM41-SM43) and triazole (SM44-SM46), in the main chain. Finally, besides the sulfonamide group, certain compounds contain sulfoxide (SM35-SM37) or sulfone (SM38-SM40) groups in their chemical structure. The smiles codes of the 22 compounds were obtained from the SAMPL7 website [15], and used to generate their 3D geometries with OpenBabel [24].

Dataset of 22 small molecules proposed in the SAMPL7 log P challenge
Log P computation
A preliminary sampling of the conformational preferences of the compounds was performed with Frog 2.14 [25]. Let us note that this program not only generates conformations at a reduced computational cost, but also exhibits a high performance in generating conformations close to the bioactive species, as noted in a rmsd 0.74 ± 0.44 Å for 85 drug-like compounds (Astex dataset), and a median rmsd below 1 Å for a subset of compounds containing up to 7 rotatable bonds [25]. On the basis of the structural complexity of the molecules, generation of conformations was limited to a maximum of 20 conformers, which were visually checked in order to eliminate redundant conformations. The geometry of the conformers in water and n-octanol was optimized at the B3LYP/6-31G(d) level of theory [26, 27] taking into account solvent effects on the geometrical parameters with the IEFPCM/MST model, which was implemented in a local version of Gaussian 16 [28]. The minimum energy nature of the optimized geometries in each solvent was verified upon inspection of the vibrational frequencies, and conformations displaying negative frequencies were discarded. Thermal corrections determined in water and n-octanol were subsequently added to estimate the relative free energy of conformations in the two solvents. Finally, single-point energy calculations in the gas phase were performed to estimate the solvation free energy of each conformation. Then, the log P was determined considering the Boltzmann-weighted population of the conformational families obtained in water and n-octanol.
pKa computation
The pKa of the deprotonation equilibria between acid and basic microstates was based on the thermodynamic cycle shown in Scheme 1. The ensemble of conformations determined in water for the set of compounds was used as starting geometries to build up the species involved in the deprotonation equilibria, according to the information provided by the SAMPL7 organizers for the different microstates [15]. The addition/removal of hydrogen atoms from the starting geometry of conformers was done manually using GaussView 6 (i.e., the graphical interface of Gaussian software) [29]. The geometries were optimized at the B3LYP/6-31G(d) level of theory taking into account hydration effects with the IEFPCM/MST model. The free energy difference between protonated and deprotonated species was estimated by combining the relative energies determined with single-point computations performed at the MP2/aug-cc-pVDZ level of theory [30] with solvation free energies and thermal corrections to the free energy calculated at the B3LYP/6-31G(d) in water. The pKa was determined using the experimental free energy of the proton in water (− 270.29 kcal/mol), which was determined by combining the gas phase free energy (− 6.28 kcal/mol), the free energy correction from 1 atm and 298 K to 1 M and 298 K state (1.89 kcal/mol), and the hydration free energy of the proton (− 265.9 kcal/mol) [31]. Finally, a Boltzmann weighting scheme was applied to account for the relative stabilities of the conformational species determined for the microstates involved in the deprotonation reaction, following the computational strategy adopted in previous studies [32, 33].
Raw data
The datasets generated during and/or analysed during the current study are available in the SAMPL7-IEF-PCM-MST GitHub repository [34].
Results and discussion
Log P prediction
The predicted log P values are listed in Table 1. The root-mean square deviation (rmsd) between IEFPCM/MST results and experimental data is 1.03 log units, which places our results among the most accurate values in the comparison with both physical (rank 2nd) and global (comprising all submissions within empirical and physical categories; rank 8th) methods [21], taking into account the small differences observed between methods with rmsd ≤ 1 (see Supporting Information Fig. S1). The best ranked QM-based solvation models (see Supporting Information Fig. S2) were the Cosmotherm version of COSMO-RS [35] (ID COSMO RS, rmsd = 0.78), our method (ID TFE IEFPCM MST, rmsd = 1.03), the NHLBI TZVP model (ID TFE NHLBI TZVP QM, rmsd = 1.55), which combined B3LYP/Def2-TZVP computations in the gas phase with solvent effects determined using the SMD solvation model [36], the 3D integral equation theory with a cluster embedding approach [37] (ID EC RISM wet, rmsd = 1.84), and another model that combined B3LYP computations with dispersion corrections in the gas phase with the SMD model [36] (ID TFE b3lyp3d, rmsd = 2.19), reflecting a performance similar to the trends found in the SAMPL6 challenge [38].
The largest deviations (> 1.50 log P units) between predicted and experimental log P values are found for SM36 and SM42 (see Table 1). These deviations are in line with the analysis of the compounds that presented the highest mean absolute error between computed and experimental values (see Supporting Information Fig. S3), since SM42 and SM36 are in ranks 1 and 5, respectively. Upon exclusion of these compounds, the rmsd is reduced to 0.72 log P units, and the correlation between calculated and experimental values improves from 0.52 to 0.76 (see Fig. 2).

Comparison between experimental and IEFPCM/MST n-octanol/water log P for the SAMPL7 dataset. Red points represent the compounds with the largest errors in the original submission. Statistical analyses are shown for (top left) all compounds and (bottom right) after exclusion of SM36 and SM42
Compared to SM35 and SM41, SM36 and SM42 imply the replacement of a methyl group by a phenyl substituent, which would increase the hydrophobicity of the compound. This trend is reflected in the experimental log P values for pairs SM41-SM42, SM29-SM30, SM32-SM33, SM38-SM39 and SM44-SM45, where the methyl-phenyl replacement leads to an average increase of 1.02 log P units. In this context, the pair SM35-SM36 shows a distinctive trait, as the log P is decreased by − 0.12. In fact, more than 80% of submissions predicted the log P of SM36 and SM42 to be larger compared to the log P of SM35 and SM41, respectively (see Supporting Information Fig. S4).
Finally, we have compared the predictions performed for the SAMPL7 dataset with the results obtained in the SAMPL6 edition, which comprised a series of 11 fragment-like small molecules [38]. Upon exclusion of SM36, the comparison yields an overall rmsd of 0.66 log P units (see Fig. 3). Therefore, assuming that the reported accuracy for log P determination is ~ 1 log unit, present results lend support to the reliability of the IEF-PCM/MST model and encourage future efforts for achieving a better description of solvation effects.

Comparison between experimental and IEFPCM/MST n-octanol/water log P for the combined dataset including the 11 fragment-like small molecules in the SAMPL6 log P challenge (blue) and 22N-acylsulfonamides in the SAMPL7 log P challenge (lightblue). The red point represents the compound with the largest error in the final dataset. Statistical analyses are shown for (top left) all compounds and (bottom right) after exclusion of SM36
Without detracting from our values, among the set of methods presented in the current edition of log P SAMPL7 challenge, one may notice that methods based on Machine Learning (ML) have led to a better match with the experimental values provided by the organization. In our view, these type techniques present great advantages, since they allow a very quick estimation due to their low computational cost, making them suitable for large compound screening campaigns. However, the reliability of these methods may be affected by the chemical coverage of the data used in their training. In this context, QM-based methods seem better suited to provide a detailed analysis of the structural and energetic features of compounds, though this requires a significantly larger computational cost, which may be necessary in the analysis of compounds containing novel chemical scaffolds. Keeping in mind the vast diversity of the chemical space [40], it may be expected that integration of QM and ML techniques will be very powerful to enhance the quality and reliability of ML models in the prediction of physicochemical properties, enabling large-scale exploration of the chemical space [41, 42].
pKa prediction
Only physical methods contributed to predicting the pKa values for the 22 sulfonamide-containing compounds included in the blind test. Table 2 reports the pKa values estimated from IEFPCM/MST computations and submitted to SAMPL7. Compared to the values available with the SAMPL7 repository [39], the difference between the originally submitted results and those estimated by the organizers from the microstates reported in our original submission is in general within 0.10 pKa units, except for SM37, where the difference increases up to 3.90 pKa units (detailed values are available in Supporting Information Table S1). The origin of this difference was due to a mistake in the relative free energy reported by us for the negatively charged microstate of compound SM37, as we had flipped the values for microstates SM37_micro004 and SM37_micro005 in the file submitted to the SAMPL7 website. This mistake led to a different macroscopic pKa value between the one calculated automatically by the organizers and the one reported in the original submission. For these reasons, we have kept the macroscopic pKa value of the original submission in Table 2.
The rmsd between predicted and experimental pKa values is 1.32 log units, which places our results among the best-ranked submissions (rank 2nd, Supporting Information Fig. S5). The largest deviations (> 1.50 in pKa units) involve four compounds: SM25, SM27, SM37 and SM42. Exclusion of these compounds reduces the rmsd to 0.98 pKa units, and the correlation between calculated and experimental values changes from 0.86 to 0.92 (see Fig. 4).

Comparison between experimental and IEFPCM/MST pKa for the SAMPL7 Dataset. Red points denote compounds with the largest errors in the original submission. Statistical analyses are shown for (top left) all compounds and (bottom right) after exclusion of SM25, SM27, SM37 and SM42
To explore the potential sources of these deviations, we compared the results obtained for SM25, SM27, SM37 and SM42 with the values reported by the contributors ranked 1st (ID EC_RISM) and 3rd (ID TVZP_QM) in the blind test (see Table 3). The results show that EC_RISM provides a range of values (5.42–10.17) that compares well with the experimental data (4.49–10.45), whereas our results are distributed in a slightly larger range (4.86 to 12.34). In contrast, the TVZP_QM values are in a narrower range (6.77–7.65). We then checked the workflow used to compute the macroscopic pKa and found a mistake in the definition of the Boltzmann weights for the conformations sampled for the main microstates of compound SM25 (Fig. 5), which caused a 3.94 units decrease in the pKa value (pKa = 3.30), remaining at 1.19 units from the experimental value.

Microstates involved in the error of SM25 pKa estimate
This analysis points out the need to perform an adequate sampling of the conformational states available for the different species involved in the deprotonation reaction [44, 45]. In particular, since our approach relied on the sampling performed for the neutral compounds (see above), the population of conformers obtained for ionized species may be inaccurate for some compounds, affecting the final estimate of the macroscopic pKa. Nevertheless, one must also keep in mind the intrinsic errors of the gas phase and solvation contributions to the aqueous free energy change for the deprotonation of the different microstates. At this point, the uncertainty of the IEFPCM/MST model in predicting the hydration free energy for simple neutral molecules amounts, on average, to 0.7 kcal/mol, but can be sensibly larger for charged compounds [46, 47]. This would then represent an additional difficulty for the proper estimation of the free energy change determined for microscopic deprotonation equilibria, challenging the ability of QM-based continuum solvation models to yield pKa estimates with an uncertainty below 1 pKa unit.
Overall, the results support the suitability of our QM-based approach for computing log P and pKa properties. SAMPL6 blind challenge mainly relied on rigid compounds [38], but SAMPL7 presented more complex compounds considering both chemical diversity and flexibility [21]. In the blind challenges mentioned above, the Frog tool has been used to explore the conformational space in our QM workflow mainly due to the good balance between computational cost and accuracy of the conformer ensemble [25]. Ongoing research in our group is seeking to explore protocols for characterizing the conformer generation based on multilevel strategies [45], since the proper sampling of the conformational space is a crucial issue that can directly impact the reliable prediction of physicochemical properties [48,49,50]. The other two critical components of our QM approach are the calculation of the internal energy of the generated conformers and the inclusion of solvation effects, which are relevant in determining the accuracy of the relative stabilities of conformers in condensed phases. For example, extrapolation of the MP2 energies to complete basis set or the inclusion of higher-level electron correlation corrections, like coupled cluster with single and double substitutions (CCSD), could improve the accuracy of our protocol by several tenths of kcal/mol when computing deprotonation free energies or relative conformer stabilities [33, 51]. The improvement of solvation effects is more complicated, as there is no systematic strategy to improve the accuracy of the results given the empirically parametrized nature of continuum models. Nevertheless, the performance obtained in the SAMPL6 and SAMPL7 challenges shows close agreement with the results obtained in previous studies [16, 22, 32, 52] for rigid compounds, thus lending confidence to the computational protocol used in this study.
After checking and considering the different drawbacks of our workflow, we consider that further improvements should be focused on two computational aspects that may affect the prediction of physicochemical properties. The first deals with obtaining a proper sampling of the conformational space available for drug-like compounds in water and n-octanol (or by extension other organic solvents), as it is reasonable to expect that distinct conformational ensembles will be adopted depending on the chemical features present in flexible compounds. In this context the exhaustiveness in sampling the whole conformational space can be calibrated through the analysis of the conformations sampled with other techniques, such as Molecular Dynamics simulations. The second is related to the capability of continuum solvation models to provide an accurate description of specific (i.e., hydrogen bonding) and nonspecific (i.e., bulk solvent electrostatic screening) interactions with solvent molecules, which is challenging for charged molecules. In this sense, the usage of cluster-continuum solvation models may lead to meaningful improvement with respect to pure continuum solvation models for modeling diverse chemical process in solution [53].
Conclusions
The results obtained in the SAMPL7 physical properties challenge has revealed the reliability of the IEFPCM/MST method to provide accurate estimates of both log P and pKa, which are relevant properties for understanding the pharmacokinetics of bioactive compounds. Nevertheless, the analysis of the results also points out that a major source of error comes from an improper weight of the conformational preferences of some compounds, particularly regarding the population distribution of ionized forms. In contrast, the prediction of the log P value resulted to have a marked deviation in one out of 22 compounds, though this marked deviation was also shared by a significant number of methods. Future modifications and improvements will be centered in finding an efficient approach for gaining better definition of the conformational space of flexible compounds in n-octanol and in water as well as to estimate the hydration free energies of charged species.
References
Testa B, Carrupt PA, Guillard P, Tsai RS (2008) Bioavailability prediction at early drug discovery stages: in vitro assays and simple physico-chemical rules. In: Pliska V, Testa B, van de Waterbeemd H (eds) Lipophilicity in drug action and toxicology. VCH, Weinheim, pp 49–71
Van de Waterbeemd H, Testa B (eds) (2009) Drug bioavailability: estimation of solubility, permeability, absorption and bioavailability. Wiley-VCH, Weinheim
Caron G, Ermondi G, Scherrer RA (2006) Lipophilicity, polarity and hydrophobicity. In: Taylor JB, Triggle DJ (eds) Comprehensive medicinal chemistry II. Elsevier Science, Oxford, pp 425–452
Muñoz-Muriedas J (2012) Bioavailability prediction at early drug discovery stages: in vitro assays and simple physico-chemical rules. In: Luque FJ, Barril X (eds) Physico-chemical and computational approaches to drug discovery. Royal Society of Chemistry, Cambridge, pp 104–127
Zhu L, Lu L, Wang S, Wu J, Shi J, Yan T, Xie C, Li Q, Hu M, Liu Z (2017) Oral absorption basics: pathways and physicochemical and biological factors affecting absorption. In: Qiu Y, Zhang GGZ, Mantri RV, Chen Y, Yu L (eds) Developing solid oral dosage forms: pharmaceutical theory and practice. Science Direct, Amsterdam, pp 297–329
Spyrakis F, Ahmed MH, Bayden AS, Cozzini P, Mozzarelli A, Kellogg GE (2017) The roles of water in the protein matrix: a largely untapped resource for drug discovery. J Med Chem 60:6781–6827
Cheng AC, Coleman RG, Smyth KT, Cao Q, Soulard P, Caffrey DR, Salzberg AC, Huabg ES (2007) Structure-based maximal affinity model predicts small-molecule druggability. Nat Biotechnol 25:71–75
Ginex T, Vazquez J, Gibert E, Herrero E, Luque FJ (2019) Lipophilicity in drug design. An overview of lipophilicity descriptors in 3D-QSAR studies. Fut Med Chem 11:1177–1193
Manallack DT (2007) The pKa distribution of drugs: application to drug discovery. Perspect Med Chem 1:25–38
Leeson PD, Springthorpe B (2007) The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov 6:881–890
Orozco M, Luque FJ (2000) Theoretical methods for the description of the solvent effect in biomolecular systems. Chem Rev 100:4187–4226
Jorgensen WL (2004) The many roles of computation in drug discovery. Science 303:1813–1818
Kujawski J, Popielarska H, Myka A, Drabińska B, Bernard M (2012) The log P parameter as a molecular descriptor in the computer-aided drug design–an overview. Comput Methods Sci Technol 18:81–88
Alongi KS, Shields GC (2010) Theoretical calculations of acid dissociation constants. a review article. Annu Rep Comput Chem 6:113–138
Soteras I, Curutchet C, Bidon-Chanal A, Orozco M, Luque FJ (2005) Extension of the MST model to the IEF formalism: HF and B3LYP parametrizations. J Mol Struct Theochem 727:29–40
Soteras I, Forti F, Orozco M, Luque FJ (2009) Performance of the IEF-MST solvation continuum model in a blind test prediction of hydration free energies. J Phys Chem B 113:9330–9334
Luque FJ, Curutchet C, Muñoz-Muriedas J, Bidon-Chanal A, Morreale A, Gelpí JL, Orozco M (2003) Continuum solvation models: Dissecting the free energy of solvation. Phys Chem Chem Phys 5:3827–3836
Cancès E, Mennucci B, Tomasi JA (1997) New integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics. J Chem Phys 107:3032
Mennucci B, Cancès E, Tomasi J (1997) Evaluation of solvent effects in isotropic and anisotropic dielectrics and in ionic solutions with a unified integral equation method: theoretical bases, computational implementation, and numerical applications. J Phys Chem B 101:10506–10517
Danielle TD, Tielker N, Zhang Y, Mao J, Gunner MR, Francisco K, Ballatore C, Kast SM, Mobley DL (2021) Evaluation of log P, pKa, and log D predictions from the SAMPL7 blind challenge. J Comput Aided Mol Des. https://doi.org/10.26434/chemrxiv.14461962.v1
Soteras I, Orozco M, Luque FJ (2010) Performance of the IEF-MST solvation continuum model in the SAMPL2 blind test prediction of hydration and tautomerization free energies. J Comput Aided Mol Des 24:281–291
Zamora WJ, Pinheiro S, German K, Ràfols C, Curutchet C, Luque FJ (2020) Prediction of the n-Octanol/water partition coefficients in the SAMPL6 blind challenge from MST continuum solvation calculations. J Comput Aided Mol Des 34:443–451
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open Babel. J Cheminform 3:1–14
Miteva MA, Guyon F, Tufféry P (2010) Frog2: efficient 3D conformation ensemble generator for small compounds. Nucleic Acids Res 38:622–627
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H et al (2016) Gaussian 16, revision B.01. Gaussian Inc, Wallingford CT
Dennington R, Keith TA, Millam JM (2016) GaussView 6.1. Semichem Inc., Shawnee Mission
Kendall RA, Dunning TH, Harrison RJ (1992) Electron affinities of the first-row atoms revisited. systematic basis sets and wave functions. J Chem Phys 96:6796–6806
Pliego JR, Miguel ELM (2013) Absolute single-ion solvation free energy scale in methanol determined by the lithium cluster-continuum approach. J Phys Chem B 117:5129–5135
Viayna A, Antermite SG, De Candia M, Altomare CD, Luque FJ (2020) Interplay between ionization and tautomerism in bioactive β-enamino ester-containing cyclic compounds: study of annulated 1,2,3,6-tetrahydroazocine derivatives. J Phys Chem B 124:28–37
Corbella M, Toa ZSD, Scholes GD, Luque FJ, Curutchet C (2018) Determination of the protonation preferences of bilin pigments in cryptophyte antenna complexes. Phys Chem Chem Phys 20:21404–21416
Klamt A (2018) The COSMO and COSMO-RS solvation models. Wiley Interdiscip Rev Comput Mol Sci 1:1–11
Marenich AV, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113:6378–6396
Kloss T, Heil J, Kast SM (2008) Quantum chemistry in solution by combining 3D integral equation theory with a cluster embedding approach. J Phys Chem B 112:4337–4343
Işık M, Bergazin TD, Fox T, Rizzi A, Chodera JD, Mobley DL (2020) Assessing the accuracy of octanol–water partition coefficient predictions in the SAMPL6 part II Log P challenge. J Comput Aided Mol Des 34:335–370
Reymond J-L, Awale M (2012) Exploring chemical space for drug discovery using the chemical universe database. ACS Chem Neurosci 3:649–657
Schütt KT, Gastegger M, Tkatchenko A, Müller K-R, Maurer RJ (2019) Unifying mechaine learning and quantum chemistry with a deep neural network for molecular wavefunctions. Nat Commun 10:5024
Tkatchenko A (2020) Machine learning for chemical discovery. Nat Commun 11:4125
Francisco KR, Varricchio C, Paniak TJ, Kozlowski MC, Brancale A, Ballatore C (2021) Structure property relationships of N-acylsulfonamides and related bioisosteres. Eur J Med Chem 218:113399
Kolár M, Fanfrlík J, Lepsík M, Forti F, Luque FJ, Hobza P (2013) Assessing the accuracy and performance of implicit solvent models for dug molecules: conformational ensemble approaches. J Phys Chem B 16:5950–5962
Juárez-Jiménez J, Barril X, Orozco M, Pouplana R, Luque FJ (2015) Assessing the suitability of the multilevel strategy for the conformational analysis of small ligands. J Phys Chem B 119:1164–1172
Cramer CJ, Truhlar DG (2008) A universal approach to solvation modeling. Acc Chem Res 41:760–768
Klamt A, Mennucci B, Tomasi J, Barone V, Curutchet C, Orozco M, Luque FJ (2009) On the performance of continuum solvation methods. A comment on universal approaches to solvation modeling. Acc Chem Res 42:489–492
Foloppe N, Chen I-J (2009) Conformational sampling and energetics of drug-like molecules. Curr Med Chem 16:3381–3413
Hawkins PCD (2017) Conformation generation: the state of the art. J Chem Inf Model 57:1747–1756
Poongavanam V, Danelius E, Peintner S, Alcaraz L, Caron G, Cummings MD, Wlodek S, Erdelyi M, Hawkins PCD, Ermondi G, Kihlberg J (2018) Conformational sampling of macrocyclic drugs in different environments: can we find the relevant conformations? ACS Omega 3:11742–11757
Pérez-Areales FJ, Betari N, Viayna A, Pont C, Espargaró A, Bartolini M, De Simone A, Alvarenga JFR, Pérez B, Sabaté R, Lamuela-Raventós RM, Andrisano V, Luque FJ, Muñoz-Torrero D (2017) Design, synthesis and multitarget biological profiling of second-generation anti-alzheimer rhein-huprine hybrids. Fut Med Chem 9:965–981
Zamora WJ, Curutchet C, Campanera JM, Luque FJ (2017) Prediction of pH-dependent hydrophobic profiles of small molecules from miertus-scrocco-tomasi continuum solvation calculations. J Phys Chem B 121:9868–9880
Pliego JR, Riveros JM (2019) Hybrid discrete-continuum solvation methods. Wires Comput Mol Sci 10:e1440
Acknowledgements
We acknowledge the computational facilities provided by the Consorci de Serveis Universitaris de Catalunya (CSUC) and the Barcelona Supercomputing Center (BCV-2020-1-0010). We appreciate the National Institutes of Health for its support of the SAMPL project via R01GM124270 to David L. Mobley (UC Irvine).
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original paper has been corrected: funding note is updated.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Viayna, A., Pinheiro, S., Curutchet, C. et al. Prediction of n-octanol/water partition coefficients and acidity constants (pKa) in the SAMPL7 blind challenge with the IEFPCM-MST model. J Comput Aided Mol Des 35, 803–811 (2021). https://doi.org/10.1007/s10822-021-00394-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-021-00394-6