
Abstract
Fluorescence-Activated Cell Sorting (FACS) is a powerful method with many applications in microalgal research, especially for screening and selection of cells with improved phenotypes. However, the technology requires review of gene expression changes responsible for enhanced phenotypes in sorted populations. Phaeodactylum tricornutum cells were sorted using FACS with excitation/emission parameters targeted to favouring the industrially-relevant carotenoid fucoxanthin. The resulting cultures showed significantly higher growth rate (1.10 ×), biomass (1.30 ×), chlorophyll a levels (1.22 ×) and fucoxanthin content (1.28 ×) relative to the wild-type strain. RNA-seq was used to elucidate the underlying molecular-level regulatory changes associated with these traits and represents the first study do so on FACS-sorted microalgal cultures. Transcriptome analysis corroborated evidence of increased chlorophyll a and fucoxanthin, showing enrichment for the genes/pathways for tetrapyrrole biosynthesis and for suites of genes directly related to photosynthesis. Only three genes were upregulated in the MEP (non-mevalonate) pathway to carotenoid biosynthesis pathway, suggesting either a strong influence of IDI, CRTISO5 and ZEP1 on fucoxanthin biosynthesis or a post-transcriptional or post-translational mechanism for the observed increase in fucoxanthin content.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Microalgal biotechnology is a developing area of research with enormous potential as a contributing technology for a renewable agricultural industry (Fabris et al. 2020; Sahu et al. 2020). Microalgae are single-celled eukaryotic organisms heavily responsible for Earth’s oxygen production, nutrient cycling, and primary production (Moroney 2001; Singh and Ahluwalia 2013; Prata et al. 2019). The potential of these organisms as contributors to sustainable industry lies in their high growth rates, ability to be grown under a wide range of conditions, high nutritional value, use of non-arable land, and as a source of a suite of valuable compounds (Spolaore et al. 2006; Brennan and Owende 2010; Dragone et al. 2010; Khan et al. 2018). Microalgae have proven effective in many areas including the sequestration of carbon, as fertiliser in agriculture and as feed in aquaculture (Singh and Ahluwalia 2013; Renuka et al. 2018; Shah et al. 2018). Valuable compounds like pigments are also being increasingly targeted using microalgae from a biotechnological perspective (Sun et al. 2023).
Microalgal pigments assist in the capture and transfer of light energy within the photosynthetic apparatus, but are currently used by humans as health compounds in the nutraceuticals and cosmetics industry, as well as for aquaculture feed (Nwoba et al. 2020; Pérez-Legaspi et al. 2020). The genera Chlorella, Spirulina, Haematoccocus and Dunaliella are the main biofactories for production of chlorophylls, phycocyanin, astaxanthin and β-carotene (Silva et al. 2020).
Fucoxanthin is the main carotenoid pigment in the diatom Phaeodactylum tricornutum, and a target for biotechnological projects for its health benefits and use in terrestrial farming and aquaculture (Xia et al. 2013; Zhang et al. 2015; Karpiński and Adamczak 2019; Bae et al. 2020). Fucoxanthin is currently produced in Israel and France and the highest yields have been observed in P. tricornutum, Odontella aurita, and Tisochrysis lutea (Pajot et al. 2022).
Phaeodactlum tricornutum is a model microalgal species with a sequenced genome and an advanced genetic toolkit. Fucoxanthin content in WT P. tricornutum can be orders of magnitude higher than in fucoxanthin-producing macroalgal species (up to 59.2 mg g−1 under specific culture conditions (McClure et al. 2018; Khaw et al. 2022; Pocha et al. 2022)). In addition, a variety of genetic manipulation techniques have been employed to improve baseline pigment content in P. tricornutum. Utilisation of physical and chemical mutagens have generated mutants with fucoxanthin content 1.7 × greater than WT, and genetic engineering efforts have produced transformant lines with 1.45 × WT fucoxanthin content by overexpressing the phytoene synthase gene (Kadono et al. 2015; Yi et al. 2015, 2018).
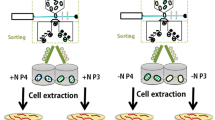
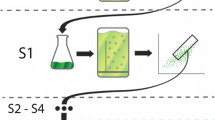
Approaches that produce large libraries of mutants require high-throughput screening methods to select for elite strains. Fluorescence Activated Cell Sorting (FACS) is a high-throughput technique and has been used to screen for pigments in microalgae (Pereira et al. 2018; Gao et al. 2020, 2021). This method has also been used previously to isolate a population with a 1.25 × increase in fucoxanthin using P. tricornutum (Fan et al. 2021). Screening of cell populations for pigment content by FACS eliminates the need for a time-consuming solvent extraction step and provides fluorescence, size, and cell volume data on a single-cell level (Gerashchenko 2020). In addition to high resolution measurements, FACS has the ability to sort cells based on combinations of these factors and with highly specific customised gating arrangements. The technology can therefore not only be used as a screening method for various compounds, but also can be used as a method of artificial selection to separate populations with desirable phenotypes without the need for mutagenesis or complex laboratory evolution methodology. While these approaches have been undertaken as outlined above, there is very little exploration of the phenotypic and genetic differences of FACS-sorted (F) populations. Consequently, further study is needed to elucidate the true potential of FACS for microalgal research. This work included a theoretical industry-based growth regime for optimising fucoxanthin content in order to elucidate differences between WT and F populations. Furthermore, RNAseq analysis was incorporated to investigate changes occurring in the transcriptomes of F cells versus WT P. tricornutum cells (Fig. 1). Cells sorted by the highest fluorescence using fucoxanthin gating were anticipated to exhibit elevated fucoxanthin content and differential gene expression in the carotenoid pathway, as well as related upstream pathways like the non-mevalonate (MEP) pathway. Additionally, other relevant pathways known to influence fucoxanthin biosynthesis, such as phytoene synthase (PSY), were expected to be involved. This would provide a comprehensive gene library potentially responsible for enhancing fucoxanthin production.

Conceptual diagram of workflow used in this study: from Fluorescence-Activated Cell Sorting (FACS) to culturing, culture measurement, pigment detection using High-Performance Liquid Chromatography (HPLC), and RNA sequencing and analysis
Materials and methods
Stock culturing
Axenic Phaeodactylum tricornutum (CCAP 1055/1) stock cultures were grown in Artificial Sea Water (ASW) medium according to Darley and Volcani (1969) under fluorescent light (150 µmol photons m−2 s−1) with a 24:0 light:dark cycle in shaking tissue culture flasks (140 rpm) kept at 21 °C (Climo-Shaker ISF1-XC, Kuhner, Switzerland).
Sorting with FACS
FACS was performed using a BD FACSMelody (BD Biosciences, USA) with the PerCP-Cy5.5 channel, as this laser arrangement had high correlation to fucoxanthin (Ex/Em 561/710 using CytoFlex LX, Beckman Coulter, USA) in our previous work (Macdonald Miller et al. 2021). To isolate cell populations, forward scatter area (FSC-A) was first plotted against forward scatter height (FSC-H) to separate singlets after which FSC-A was plotted against SSC-A to exclude debris. The fusiform cell morphology was the sole cell type observed throughout experimentation and therefore consideration of either oval or triradiate cells when performing FACS was not taken. Gated single cells were then plotted using PerCP-Cy5.5 fluorescence against FSC-A for further gating for fucoxanthin content. The gating strategy for selecting single cells using FACS is outlined: while manually generating a curve to follow the shape of the distribution and thereby select for high-fluorescing cells irrespective of cell volume (Supplementary Fig. 1B, it was found that despite doubling average fluorescence, the mean FSC was approximately equal between the top 1% and the total population. However, a flat line / rectangle (Supplementary Fig. 1A) selected for higher cell volume in the top 1% (~ 3.6 × mean FSC) but this allowed for much higher fluorescence selection (~ 3.2 × mean fluorescence) than the total population. It was later observed that FSC and SSC were similar or identical between WT and F (FACS) cultures, indicating that the rectangular gating strategy was appropriate. Therefore, a straight line gate was used for sorting single cells into 384-well microplates. A rectangular gate was created which included the entire population to be sorted, and then was set at a threshold of ~ 1% top fluorescing cells. Cells were sorted to a total of 100,000 into 5 mL of ASW at exponential phase and this was replicated 5 times.
Culture data
In order to simulate a theoretical product-favoured growth arrangement, sorted populations were grown to an inoculation density of 50,000 cells mL−1 and subcultured into fresh ASW under fluorescent light at 150 µmol photons m−2 s−1 with a 24:0 light cycle in shaking tissue culture flasks (140 rpm) kept at 21 °C. Daily 1:1 dilutions were performed for 3 days to provide optimal growth conditions for maximising biomass, after which the flasks were placed in 10 µmol photons m−2 s−1 without dilution for a further 5 days to maximise carotenoid content, as fucoxanthin has been observed to increase under conditions of low light as a light harvesting pigment (McClure et al. 2018; Seth et al. 2021). Cell counts and fluorescence were measured daily using flow cytometry and biomass was measured at the start and end of the experimental period. At the end of the experimental period, cultures were centrifuged at 4 °C, washed with chilled (4 °C) Milli-Q water, flash-frozen in liquid nitrogen, and stored at -80 °C for further analysis. All samples were centrifuged within 1 h of each other and always remained chilled throughout collection.
HPLC
Freeze-dried pellets were weighed and re-suspended at 2–3 mg dry biomass per mL of chilled ethanol before being sonicated with 1-s pulses using an ultrasonic homogeniser (Qsonica Q125) at 100% amplitude and filtered once extraction was confirmed. Extracts were filtered using 0.2 µm PTFE syringe filters and stored at -80 °C until analysis. High Performance Liquid Chromatography (HPLC) was conducted using an Agilent Technologies 1290 Infinity, equipped with a binary pump with integrated vacuum degasser, thermostated column compartment modules, Infinity 1290 auto-sampler and PDA detector. Column separation was performed using a 4.6 mm × 150 mm Zorbax Eclipse XDB-C8 reverse-phase column (Agilent Technologies) and guard column using a gradient of TBAA (tetrabutyl ammonium acetate):methanol mix (30:70) (solvent A) and methanol (Solvent B) as follows: 0–22 min, from 5 to 95% B; 22–29 min, 95% B; 29–31 min, 5% B; 31–40 min, column equilibration with 5% B. Column temperature was maintained at 55 °C. A complete pigment profile from 270 to 700 nm was recorded using PDA detector with 3.4 nm bandwidth. Sample concentrations were determined using a calibration curve of peak area to known concentrations of pure pigment standards.
RNA extraction
RNA extractions were performed in triplicate using RNAse-free reagents. Flash-frozen pellets were resuspended in 1.5 mL of Trizol reagent and pipetted until homogenous before incubating at room temperature for 5 min. Then, 300 µL chloroform (Sigma-Aldrich, #C2432) was added and mixed by inverting tubes for 15 s before a second incubation at room temperature for 15 min. Tubes were centrifuged at 4 °C and 12,000 RCF for 15 min. The aqueous phase was transferred to a clean 2 mL tube and an equal volume of molecular grade ethanol was added (Sigma-Aldrich, #E7023) before being inverted several times to mix. This was then transferred to a spin column in a 2 mL collection tube 650 µL at a time and centrifuged at 8,000 RCF for 30 s with discarding of flow-through, and this step was repeated until all of the liquid had passed through the column. Further purification was performed using a Qiagen RNeasy kit using the protocol recommended by manufacturer (Qiagen, USA). RNA concentration was measured using a NanoDrop spectrophotometer (Thermo Fisher Scientific, USA) and quality was evaluated via the presence of intact 18S and 28S ribosomal RNA bands using an Agilent 2100 BioAnalyzer (Agilent Technologies) prior to sequencing. Samples with RINS > 8.8 were included in sequencing.
Sequencing and assembly
Sequencing was undertaken at the Ramaciotti Centre for Genomics (University of New South Wales, Sydney, Australia). First, samples were converted to libraries using Illumina Stranded mRNA prep, ligation. Ribonucleic acid at 500 ng was used as input to the library prep with 10 PCR cycles. Libraries were pooled equimolar and the final pool was cleaned using 0.9 × AMPure XP beads. Sequencing was performed on an Illumina NextSeq 500 150 cycle High Output platform (Illumina, USA) in 2 × 75 bp strand-specific format. Fastq files were trimmed using TrimGalore! (Babraham Bioinformatics, v0.6.7) using default parameters. Quality control of fastq files was performed using FastQC v0.11.9 (Andrews 2010) pre- and post-trimming, and reports compiled by MultiQC v1.11 (Ewels et al. 2016). Fastq reads were then pseudo-aligned to the P. tricornutum ASM15095v2 transcriptome which was downloaded from ensemble genomes (http://protists.ensembl.org/Phaeodactylum_tricornutum/Info/Index) using kallisto v0.44.0 (Bray et al. 2016) with the strand specific –rf-stranded option, to give read counts for each transcript. Counts were then combined for multiple transcripts of the same gene to give gene-level counts. Genes were filtered for those with at least 10 counts in at least 3 samples. Gene-level differential expression was performed using Limma (Smyth 2005) and Voom (Law et al. 2014). All gene counts presented are Voom-transformed counts.
Pathway enrichment analysis
Degust (Powell 2015) was used to automatically generate an MDS (multi-dimensional scaling) plot to visualise differences between gene expression between WT and F cultures. Genes with a logFC (log twofold change) threshold of 1 and p-value < 0.05 were input to ShinyGO v0.77 (Ge et al. 2020) for enrichment of three GO aspects: Molecular Function (MF), Cellular Component (CC) and Biological Process (BP) with a minimum pathway size of 10.
Statistics and visualisation
One-way ANOVA with confidence level of 0.05 was performed followed by Tukey’s post-hoc using GraphPad Prism version 9.0.2 for Windows (GraphPad Software, USA) for culture characteristics and pigment content data. Degust v4.2 (Powell 2015) was used to analyse Voom (variance modeling at the observational level) counts. Data visualisation was performed using Prism, Degust, Clustergrammer (Fernandez et al. 2017) and Kaluza Flow Cytometry Analysis Software version 2.1 (Beckman Coulter, USA).
Results
Growth and pigment phenotypes of FACS-sorted cultures
Because cell volume did not significantly increase in the F cultures by the end of the experimental period (Supplementary Fig. 3), artificial selection for cell volume can be excluded as the reason for increased fucoxanthin and chlorophyll a content in FACS cultures. Despite selecting for both fluorescence and cell volume, FACS and WT cultures throughout the experiment were similar for both geometric mean FSC and SSC (Supplementary Fig. 3) and despite an initial F culture increase in FSC-A of 3.6 × WT, this number decreased to 1.04 × WT by day 8 of the experimental period.
FACS-sorted cultures had significantly higher cell counts than WT cultures at every time point, with the exception of day 0 (p < 0.0003 for days 1—3 and p < 0.01 for days 4 – 8 using t-tests). The highest mean cell count for F cultures was 4.7 × 106 mL−1 at day 2, whereas the maximum for WT was 3.1 × 106 mL−1 at day 3 (Fig. 2a). Cultures sorted with FACS also exhibited a significantly higher (p = 0.025) mean specific growth rate during the optimal growth phase during days 0–3, which was 1.16 ± 0.06 for F cultures and 1.05 ± 0.05 for WT cultures (Fig. 2b).

Biomass, growth and pigment data for WT and F cultures of P. tricornutum, with (a) cell counts measured using flow cytometry, (b) specific growth rate (µ), (c) Dry weight chlorophyll a and fucoxanthin pigment content measured using HPLC, and (d) biomass (mg per L) All graphs depict mean with standard deviation, asterisks denote statistical significance, n = 5
After a period of low light from days 3 to 8, mean chlorophyll a and fucoxanthin content were both significantly higher in F cultures (p = 0.024 and 0.008, respectively) compared to WT (Fig. 2c). Chlorophyll a content was 30.7 ± 2.6 for F cultures and 25.2 ± 1.5 mg g−1 for WT, while fucoxanthin was 22.5 ± 1.9 mg g−1 for F cultures and 17.6 ± 1.3 mg g−1 for WT. These results show that not only did the F cultures grow faster under optimal conditions compared to WT cultures; they also had a higher chlorophyll a and fucoxanthin content after a period of low light.
Gene expression
We next performed RNA-seq on three samples for each F and control wild-type (WT), aligning to the reference P. tricornutum transcriptome to obtain gene expression levels. The MDS plot of normalised gene expression shows seperation F and WT samples in dimension 1 (explaining 70% of sample variance), suggesting a difference in RNA expression in these conditions (Fig. 3).

Multi-Dimensional Scaling (MDS) plot showing similarities between RNA samples (leading LogFC dimension 1 compared to leading LogFC dimension 2. WT = wild-type, F = FACS). Made using Degust
A total of 312 genes were upregulated and 63 downregulated over a threshold of 1 logFC (p-value < 0.05) in F cultures compared to WT cultures over the whole transcriptome, however, all exhibited a false discovery rate (FDR) value between 0.11 and 0.18. The highlighted 385 genes were used for gene enrichment analysis.
Gene enrichment analysis
Fifty-seven pathways were overrepresented in the gene set (FDR < 0.05) in F cultures compared to WT cultures as indicated by the ShinyGO platform; 18 related to biological processes, 19 to cellular components, and 20 related to molecular function (Fig. 4, Supplementary Tables 1–3).

Gene Ontology (GO) enrichment analysis results with a minimum pathway size of 10 and FDR cut-off = 0.05. Pink bars indicate pathways with 25 – 50% of genes in that pathway being represented by the gene set, blue indicates 50% of genes being represented. GO term IDs are in parentheses
Of the 18 pathways related to biological processes, 12 had at least 25% of genes present being overexpressed. One pathway, the protoporphyrinogen IX metabolic process pathway (GO: 0046501), exhibited 50% of genes present being overexpressed. Three out of 19 pathways related to cellular components had at least 25% of genes represented while 2 out of 20 related to molecular function had 25% of genes represented. All pathways with a minimum of 25% genes overexpressed corresponded to fold enrichment values between 10 – 15 (defined as the percentage of genes in a gene set belonging to a given pathway, divided by the corresponding percentage in the background). Meanwhile, the pathways that contained genes most overrepresented in the dataset were related to biological processes and were porphyrin-containing compound biosynthetic process (GO: 0006779) and protoporphyrinogen IX metabolic process pathway (GO: 0046501) at 17.3 and 19.6 fold enrichment, respectively. Gene Ontology (GO) ID terms directly associated with the tetrapyrrole biosynthetic pathway (protoporphyrinogen IX, porphyrins, pigments, tetrapyrroles, and heme pathways) were significantly enriched, as well as were other GO ID terms associated directly with pigments and photosynthesis (Fig. 4, Supplementary Tables 1–3).
Expression in pathways related to growth and pigmentation
We identified several differentially regulated genes -1 < logFC > 1 (increased and decreased abundance of transcripts) and p < 0.05) within pathways related to pigmentation and growth. Despite FDR values between 0.11 – 0.18, 10 of the 13 genes in the tetrapyrrole pathway were above 1 logFC in the F cultures compared to WT cultures (Fig. 5). One gene in the MEP pathway (Phatr3_J12533—isopentenyl-diphosphate delta-isomerase or IDI), which provides precursors for carotenoid biosynthesis as a fundamental step in isoprenoid biosynthesis, was found to be more abundant (1.07 logFC) (Berthelot et al. 2012; Jaramillo-Madrid et al. 2020). Two genes, Phatr3_J9210 and Phatr3_J45845, corresponding to the carotenoid biosynthesis genes CRTISO5 (carotenoid isomerase) and ZEP1 (zeaxanthin epoxidase), were also higher by 1.04 and 1.80 logFC, respectively.

Genes differentially expressed over a threshold of 1 logFC and p < 0.05 in pathways related to pigment biosynthesis and growth. Nineteen genes were differentially regulated between F and WT populations
Five genes involved in carbon metabolism (Phatr3_J55079, Phatr3_J45997, Phatr3_J29157, Phatr3_J49339 and Phatr3_J49098) corresponding to PK2, PK4 and PK6 (pyruvate kinase), PGK1 (phosphoglycerate kinase) and PYC2 (precursor of carboxylase pyruvate carboxylase) are all related to glycolysis and had increased abundance between 1.08 to 2.32 logFC. Another gene involved in carbon metabolism (Phatr3_J51970) had a -1.08 logFC. A gene related to branched-chain amino acid degradation (Phatr3_J11811) showed decreased abundance (logFC -1.06), while a gene related to amino acid biosynthesis (Phatr3_J26256) showed increased abundance (logFC).
Given the significant increase in fucoxanthin content and growth observed in F cultures (Fig. 2b, c), it was expected that genes involved in carotenoid precursor supply and biosynthesis would be significantly more abundant. However, relatively few genes in these pathways showed increased abundance (Fig. 6a). Instead, the tetrapyrrole to chlorophyll a pathway (Fig. 6b) was upregulated and overrepresented in the enrichment analysis as indicated by the ShinyGO platform. Rate-limiting steps include GSAT (glutamate-1-semialdehyde aminotransferase, Phatr3_J36347) and ALAD (ALA dehydratase, Phatr3_J41746), of which F cultures exhibited 1.49 and 0.55 logFC compared to WT, respectively.

Pigment pathways with coinciding gene expression values comparing F cultures with WT cultures of Phaeodactylum tricornutum.. (a) Carotenoid / fucoxanthin pathway from upstream MEP pathway and (b) tetrapyrrole pathway to chlorophyll a. Red-coloured terms indicate genes over threshold of 1 logFC, while grey terms indicate genes within threshold of 1 logFC. Asterisks denote genes with expression p-value < 0.05. Reactions involving more than one gene are shown as averages unless p-values are either side of 0.05; e.g. MgCh_H and MgCh_D are averaged to 1.98 and listed simply as MgCh as both had p < 0.05
Discussion
The combined results suggest that FACS-sorting can be a powerful tool for enhancing production of pigments as well as other target compounds in microalgal cultures. Recent work has opted for sorting with the relationship to cell volume incorporated into the sorting method (Fan et al. 2021; Gao et al. 2021). However, it is probable that differences in cell size during the initial sorting phase of the experiment are due to regular fluctuations over the Phaeodactylum life cycle, reducing the favouring of larger cells in sorted cultures downstream. Work on gating strategies for FACS-sorting in microalgal cultures is required to clarify best practices. Additionally, while such an analysis was beyond the reach of this work, further research should aim to incorporate assessment of phenotype stability post-sorting to understand the longevity of improvements in growth and pigment content in sorted populations.
Results from the gene enrichment analysis showed that all pathways with a minimum of 25% of genes represented (also corresponding to > tenfold enrichment) primarily exist to serve functions of photosynthesis. The pathway with highest significant gene content (50% with 19.6 fold enrichment) was the protoporphyrinogen IX metabolic process pathway (GO: 0046501), an upstream system responsible for chlorophyll biosynthesis. This is reinforced by the increased chlorophyll a content and logFC of > 1 for 10 out of 12 genes in the tetrapyrrole pathway to chlorophyll a for FACS cultures. Surprisingly, only 2 genes in the fucoxanthin pathway were significantly enriched with > 1 logFC (CRTISO5 and ZEP1). This gives mixed interpretations as CRTISO was previously seen to be downregulated under low light conditions, which was favorable for fucoxanthin biosynthesis, while ZEP had the opposite effect of being upregulated in correlation to increased fucoxanthin under low light (Kadono et al. 2015; Manfellotto et al. 2020; Kwon et al. 2021). More recent research has found that CRTISO5 is directly responsible for fucoxanthin biosynthesis, and that knockdown of this gene removed P. tricornutum ability to synthesize fucoxanthin altogether (Cao et al. 2023). Increased abundance of this gene in F cultures is therefore the likely cause of increased fucoxanthin content displayed in these cultures compared to WT.
It was reasoned that F cultures could display very large differences at the single gene level of expression but also that differences in gene expression could be smaller, yet distributed throughout multiple regions contributing to higher fucoxanthin. While there was a higher number of transcripts with increased abundance in the MEP to fucoxanthin pathway, the logFC increase was also generally higher. The 1.3 × WT increase of fucoxanthin in F cultures could therefore be a result of post-transcriptional or post-translational effects as seen elsewhere in P. tricornutum and other microalgae, but upregulation of one or more of CRTISO5, IDI and ZEP1 along the MEP to fucoxanthin pathway are likely contributors (Veluchamy et al. 2015; Huang et al. 2019; Bacova et al. 2020; Cao et al. 2023).
Tetrapyrroles are synthesized in plastids and regulate nuclear gene expression (Terry and Smith 2013; Larkin 2016). It could be that tetrapyrrole signaling is responsible for altered gene expression across the nucleus. Enzymes along the tetrapyrrole pathway, MgPMT (Mg Protoporphyrin IX Methyltransferase), MgCh (Magnesium Chelatase) and ALA (5-Aminolevulinic acid), were upregulated in a tobacco line with enhanced expression of the gene MTF (Mg-Protoporphyrin IX methyltransferase) under low light (Alawady and Grimm 2005). The expression level of MTF in F cultures was 2.18 logFC higher than in WT cultures. While the tetrapyrrole pathway was highly enriched in F cultures, this gene is a key step in regulating downstream products, along with ALA synthesis (GSAT) (Czarnecki and Grimm 2012; Zhang et al. 2021; Sinha et al. 2022; Wang et al. 2022). Both of these genes showed higher transcript abundance and may be the primary reason for increased chlorophyll a content in F cultures. It is likely that FACS cells will simply produce more pigments under all conditions, because cells exhibited higher chlorophyll a and fucoxanthin after experiencing low light conditions (days 4–8) despite being sorted under relatively high light (no adaptation before sorting).
The increased growth in F cultures is likely due in part to amplified photosynthetic activity from increased content of light harvesting pigments, as well as contributions from pyruvate kinase and phosphoglycerate kinase as vital contributors to carbon metabolism (Bosco et al. 2012; Ma et al. 2014). Genes associated with BCAA degradation have also been previously observed to be upregulated under conditions of TAG accumulation (Ge et al. 2014; Conte et al. 2018). Increased AA synthesis and BCAA degradation are likely increasing carbon metabolism alongside PK and PGK for increased growth of F populations (Pan et al. 2020).
Conclusion
Sorting out the top 1% fluorescing P. tricornutum cells resulted in cultures with faster growth and higher pigment content. Analysis of RNA-seq data indicated that the underlying mechanisms for this improved functionality were upregulated pathways associated with photosynthesis, specifically upregulation of the tetrapyrrole pathway to chlorophyll a. The high utility of FACS as a method for artificial selection is clear, while the best gating strategies as well as accuracy for favouring specific pigments using FACS are yet to be determined and are likely species and condition specific. While this study provides a first comprehensive library of genes associated with a F population of cells showing increased production of fucoxanthin, future studies combining sequencing and FACS sorting of cells displaying different pigment fluorescence signatures under various conditions is clearly needed to elucidate the genetic mechanisms driving differences in microalgae pigment phenotypes. In conclusion, this study reveals the inherent presence of highly productive subpopulations within P. tricornutum culture without modification using mutagenic agents. By employing Fluorescence-Activated Cell Sorting (FACS), these high-performing algae can be identified and selectively sub-cultured. The F cultures exhibited significant improvements in growth rate, biomass, and fucoxanthin content compared to the wild-type strain, demonstrating FACS as a powerful method that can be used to select desirable phenotypes that are distinguishable from the original (both phenotypically and from a gene expression standpoint) with as little as one selection event.
Data availability
Data available upon reasonable request.
References
Alawady AE, Grimm B (2005) Tobacco Mg protoporphyrin IX methyltransferase is involved in inverse activation of Mg porphyrin and protoheme synthesis. Plant J 41:282–290
Andrews S (2010) FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Bacova R, Kolackova M, Klejdus B, Adam V, Huska D (2020) Epigenetic mechanisms leading to genetic flexibility during abiotic stress responses in microalgae: A review. Algal Res 50:101999
Bae M, Kim M-B, Park Y-K, Lee J-Y (2020) Health benefits of fucoxanthin in the prevention of chronic diseases. Biochim Biophys Acta -Mol Cell Biol Lipids 1865:158618
Berthelot K, Estevez Y, Deffieux A, Peruch F (2012) Isopentenyl diphosphate isomerase: a checkpoint to isoprenoid biosynthesis. Biochimie 94:1621–1634
Bosco MB, Aleanzi MC, Iglesias AÁ (2012) Plastidic phosphoglycerate kinase from Phaeodactylum tricornutum: on the critical role of cysteine residues for the enzyme function. Protist 163:188–203
Bray NL, Pimentel H, Melsted P, Pachter L (2016) Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34:525–527
Brennan L, Owende P (2010) Biofuels from microalgae—a review of technologies for production, processing, and extractions of biofuels and co-products. Renew Sust Energy Rev 14:557–577
Cao T, Bai Y, Buschbeck P, Tan Q, Cantrell MB, Chen Y, Jiang Y, Liu R-Z, Ries NK, Shi X (2023) An unexpected hydratase synthesizes the green light-absorbing pigment fucoxanthin. Plant Cell 35:3053–3072
Conte M, Lupette J, Seddiki K, Meï C, Dolch L-J, Gros V, Barette C, Rébeillé F, Jouhet J, Maréchal E (2018) Screening for biologically annotated drugs that trigger triacylglycerol accumulation in the diatom Phaeodactylum. Plant Physiol 177:532–552
Czarnecki O, Grimm B (2012) Post-translational control of tetrapyrrole biosynthesis in plants, algae, and cyanobacteria. J Exp Bot 63:1675–1687
Darley WM, Volcani B (1969) Role of silicon in diatom metabolism: a silicon requirement for deoxyribonucleic acid synthesis in the diatom Cylindrotheca fusiformis Reimann and Lewin. Exp Cell Res 58:334–342
Dragone G, Fernandes BD, Vicente AA, Teixeira JA (2010) Third generation biofuels from microalgae, current research, technology and education. Appl Microbiol Biotechnol 2:1355–1366
Ewels P, Magnusson M, Lundin S, Käller M (2016) MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32:3047–3048
Fabris M, Abbriano RM, Pernice M, Sutherland DL, Commault AS, Hall CC, Labeeuw L, McCauley JI, Kuzhiuparambil U, Ray P (2020) Emerging technologies in algal biotechnology: toward the establishment of a sustainable, algae-based bioeconomy. Front Plant Sci 11:279
Fan Y, Ding X-T, Wang L-J, Jiang E-Y, Van PN, Li F-L (2021) Rapid sorting of fucoxanthin-producing Phaeodactylum tricornutum mutants by flow cytometry. Mar Drugs 19:228
Fernandez NF, Gundersen GW, Rahman A, Grimes ML, Rikova K, Hornbeck P, Ma’ayan A (2017) Clustergrammer, a web-based heatmap visualization and analysis tool for high-dimensional biological data. Sci Data 4:1-12
Gao F, Sá M, Cabanelas ITD, Wijffels RH, Barbosa MJ (2021) Improved fucoxanthin and docosahexaenoic acid productivities of a sorted self-settling Tisochrysis lutea phenotype at pilot scale. Bioresour Technol 325:124725
Gao F, Teles I, Ferrer-Ledo N, Wijffels RH, Barbosa MJ (2020) Production and high throughput quantification of fucoxanthin and lipids in Tisochrysis lutea using single-cell fluorescence. Bioresour Technol 318:124104
Ge F, Huang W, Chen Z, Zhang C, Xiong Q, Bowler C, Yang J, Xu J, Hu H (2014) Methylcrotonyl-CoA carboxylase regulates triacylglycerol accumulation in the model diatom Phaeodactylum tricornutum. Plant Cell 26:1681–1697
Ge SX, Jung D, Yao R (2020) ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36:2628–2629
Gerashchenko BI (2020) Fluorescence-activated cell sorting (FACS)-based characterization of microalgae. Mar Ecol Curr Future Dev 2:148–160
Huang R, Ding J, Gao K, Cruz de Carvalho MH, Tirichine L, Bowler C, Lin X (2019) A potential role for epigenetic processes in the acclimation response to elevated pCO2 in the model diatom Phaeodactylum tricornutum. Front Microbiol 9:3342
Jaramillo-Madrid AC, Ashworth J, Fabris M, Ralph PJ (2020) The unique sterol biosynthesis pathway of three model diatoms consists of a conserved core and diversified endpoints. Algal Res 48:101902
Kadono T, Kira N, Suzuki K, Iwata O, Ohama T, Okada S, Nishimura T, Akakabe M, Tsuda M, Adachi M (2015) Effect of an introduced phytoene synthase gene expression on carotenoid biosynthesis in the marine diatom Phaeodactylum tricornutum. Mar Drugs 13:5334–5357
Karpiński TM, Adamczak A (2019) Fucoxanthin—An antibacterial carotenoid. Antioxidants 8:239
Khan MI, Shin JH, Kim JD (2018) The promising future of microalgae: current status, challenges, and optimization of a sustainable and renewable industry for biofuels, feed, and other products. Microb Cell Fact 17:36
Khaw YS, Yusoff FM, Tan HT, Noor Mazli NAI, Nazarudin MF, Shaharuddin NA, Omar AR, Takahashi K (2022) Fucoxanthin production of microalgae under different culture factors: A systematic review. Mar Drugs 20:592
Kwon DY, Vuong TT, Choi J, Lee TS, Um J-I, Koo SY, Hwang KT, Kim SM (2021) Fucoxanthin biosynthesis has a positive correlation with the specific growth rate in the culture of microalga Phaeodactylum tricornutum. J Appl Phycol 33:1473–1485
Larkin RM (2016) Tetrapyrrole signaling in plants. Front Plant Sci 7:1586
Law CW, Chen Y, Shi W, Smyth GK (2014) voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15:R49
Ma Y-H, Wang X, Niu Y-F, Yang Z-K, Zhang M-H, Wang Z-M, Yang W-D, Liu J-S, Li H-Y (2014) Antisense knockdown of pyruvate dehydrogenase kinase promotes the neutral lipid accumulation in the diatom Phaeodactylum tricornutum. Microb Cell Fact 13:100
Macdonald Miller S, Abbriano RM, Segecova A, Herdean A, Ralph PJ, Pernice M (2021) Comparative study highlights the potential of spectral deconvolution for fucoxanthin screening in live Phaeodactylum tricornutum cultures. Mar Drugs 20:19
Manfellotto F, Stella GR, Falciatore A, Brunet C, Ferrante MI (2020) Engineering the unicellular alga Phaeodactylum tricornutum for enhancing carotenoid production. Antioxidants 9:757
McClure DD, Luiz A, Gerber B, Barton GW, Kavanagh JM (2018) An investigation into the effect of culture conditions on fucoxanthin production using the marine microalgae Phaeodactylum tricornutum. Algal Res 29:41–48
Moroney JV (2001) Algal photosynthesis. Encyclopedia of Life Sciences (eLS). https://doi.org/10.1038/npg.els.0000322
Nwoba EG, Ogbonna CN, Ishika T, Vadiveloo A (2020) Microalgal pigments: a source of natural food colors. In: Alam MA, Xu J-L, Wang Z (eds) Microalgae biotechnology for food, health and high value products. Springer, Cham, pp 81–123
Pajot A, Hao Huynh G, Picot L, Marchal L, Nicolau E (2022) Fucoxanthin from algae to human, an extraordinary bioresource: Insights and advances in up and downstream processes. Mar Drugs 20:222
Pan Y, Hu F, Yu C, Li C, Huang T, Hu H (2020) Amino acid catabolism during nitrogen limitation in Phaeodactylum tricornutum. Front Plant Sci 11:589026
Pereira H, Schulze PS, Schüler LM, Santos T, Barreira L, Varela J (2018) Fluorescence activated cell-sorting principles and applications in microalgal biotechnology. Algal Res 30:113–120
Pérez-Legaspi IA, Valadez-Rocha V, Ortega-Clemente LA, Jiménez-García MI (2020) Microalgal pigment induction and transfer in aquaculture. Rev Aquac 12:1323–1343
Pocha CKR, Chia WY, Chew KW, Munawaroh HSH, Show PL (2022) Current advances in recovery and biorefinery of fucoxanthin from Phaeodactylum tricornutum. Algal Res 65:102735
Powell D (2015) Degust: interactive RNA-seq analysis. https://degust.erc.monash.edu/
Prata JC, da Costa JP, Lopes I, Duarte AC, Rocha-Santos T (2019) Effects of microplastics on microalgae populations: a critical review. Sci Total Environ 665:400–405
Renuka N, Guldhe A, Prasanna R, Singh P, Bux F (2018) Microalgae as multi-functional options in modern agriculture: current trends, prospects and challenges. Biotech Adv 36:1255–1273
Sahu SK, Mantri VA, Zheng P, Yao N (2020) Algae biotechnology: current status, potential and impediments. In: Kim S-K (ed) Encyclopedia of marine biotechnology. John Wiley & Sons, Chichester, pp 1–31
Seth K, Kumar A, Rastogi RP, Meena M, Vinayak V (2021) Bioprospecting of fucoxanthin from diatoms—Challenges and perspectives. Algal Res 60:102475
Shah MR, Lutzu GA, Alam A, Sarker P, Chowdhury K, Parsaeimehr A, Liang Y, Daroch M (2018) Microalgae in aquafeeds for a sustainable aquaculture industry. J Appl Phycol 30:197–213
Silva SC, Ferreira IC, Dias MM, Barreiro MF (2020) Microalgae-derived pigments: A 10-year bibliometric review and industry and market trend analysis. Molecules 25:3406
Singh UB, Ahluwalia A (2013) Microalgae: a promising tool for carbon sequestration. Mitig Adapt Strat Glob Change 18:73–95
Sinha N, Eirich J, Finkemeier I, Grimm B (2022) Glutamate 1-semialdehyde aminotransferase is connected to GluTR by GluTR-binding protein and contributes to the rate-limiting step of 5-aminolevulinic acid synthesis. Plant Cell 34:4623–4640
Smyth GK (2005) Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irzarry R, Huber W (eds) Bioinformatics and computational biology solutions using R and Bioconductor. Springer, NY, pp 397–420
Spolaore P, Joannis-Cassan C, Duran E, Isambert A (2006) Commercial applications of microalgae. J Biosci Bioeng 101:87–96
Sun H, Wang Y, He Y, Liu B, Mou H, Chen F, Yang S (2023) Microalgae-derived pigments for the food industry. Mar Drugs 21:82
Terry MJ, Smith AG (2013) A model for tetrapyrrole synthesis as the primary mechanism for plastid-to-nucleus signaling during chloroplast biogenesis. Front Plant Sci 4:14
Veluchamy A, Rastogi A, Lin X, Lombard B, Murik O, Thomas Y, Dingli F, Rivarola M, Ott S, Liu X (2015) An integrative analysis of post-translational histone modifications in the marine diatom Phaeodactylum tricornutum. Genome Biol 16:1–18
Wang P, Ji S, Grimm B (2022) Post-translational regulation of metabolic checkpoints in plant tetrapyrrole biosynthesis. J Exp Bot 73:4624–4636
Xia S, Wang K, Wan L, Li A, Hu Q, Zhang C (2013) Production, characterization, and antioxidant activity of fucoxanthin from the marine diatom Odontella aurita. Mar Drugs 11:2667–2681
Yi Z, Su Y, Xu M, Bergmann A, Ingthorsson S, Rolfsson O, Salehi-Ashtiani K, Brynjolfsson S, Fu W (2018) Chemical mutagenesis and fluorescence-based high-throughput screening for enhanced accumulation of carotenoids in a model marine diatom Phaeodactylum tricornutum. Mar Drugs 16:272
Yi Z, Xu M, Magnusdottir M, Zhang Y, Brynjolfsson S, Fu W (2015) Photo-oxidative stress-driven mutagenesis and adaptive evolution on the marine diatom Phaeodactylum tricornutum for enhanced carotenoid accumulation. Mar Drugs 13:6138–6151
Zhang H, Tang Y, Zhang Y, Zhang S, Qu J, Wang X, Kong R, Han C, Liu Z (2015) Fucoxanthin: a promising medicinal and nutritional ingredient. Evid-Based Compl Altern Med 2015:723515
Zhang K, Li J, Zhou Z, Huang R, Lin S (2021) Roles of alkaline phosphatase PhoA in algal metabolic regulation under phosphorus-replete conditions. J Phycol 57:703–707
Acknowledgements
The authors would like to humbly thank Dr. Unnikrishnan Kuzhiumparambil and Dr. Taya Lapshina for their indispensable expertise on HPLC.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions This work was supported by a UTS Doctoral Scholarship to S.M.
Author information
Authors and Affiliations
Contributions
Conceptualization, Sean Macdonald Miller and Mathieu Pernice; Data curation, Sean Macdonald Miller; Formal analysis, Sean Macdonald Miller, Brandon Signal and Vishal Gupta; Investigation, Sean Macdonald Miller; Methodology, Sean Macdonald Miller, Brandon Signal, Vishal Gupta and Mathieu Pernice; Project administration, Peter Ralph, Mathieu Pernice and Andrei Herdean; Software, Sean Macdonald Miller, Brandon Signal and Vishal Gupta; Supervision, Raffaela Abbriano, Andrei Herdean, Peter Ralph and Mathieu Pernice; Visualization, Sean Macdonald Miller; Writing – original draft, Sean Macdonald Miller; Writing – review & editing, all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Macdonald Miller, S., Herdean, A., Gupta, V. et al. Differential gene expression in a subpopulation of Phaeodactylum tricornutum with enhanced growth and carotenoid production after FACS-mediated selection. J Appl Phycol 35, 2777–2787 (2023). https://doi.org/10.1007/s10811-023-03102-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-023-03102-z