
Abstract
Purpose
To describe the ocular clinical characteristics of a group of Mexican patients with lamellar ichthyosis (LI) arising from TGM1 pathogenic variants.
Methods
Ophthalmological exploration, pedigree analysis and genetic screening were performed in patients with an established clinical diagnosis of lamellar ichthyosis from families located in a small community in the Southeast of Mexico.
Results
Nine patients with LI in five families were identified. There were six affected females. All patients (9/9) demonstrated eye lid abnormalities with eight patients showing lid margin abnormalities. Madarosis was present in only three individuals and corneal scarring was documented in two. All nine individuals carried biallelic TGM1 variants, either homozygously or as compound heterozygous.
Conclusion
Ocular anomalies are common in individuals with TGM1-related LI. The occurrence of a variety of private or rare mutations hampers the identification of a genotype–phenotype correlation for ocular anomalies in this disorder.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The term “ichthyosis” derives from the Greek word “ichthys," meaning fish, and it has been used for over 200 years to describe a group of diseases characterized by generalized desquamation of the skin, generalized “scales,” dry skin, hyperkeratosis, and sometimes erythroderma [1, 2]. Ichthyoses can occur as an acquired or hereditary trait, with a congenital or late onset presentation and it can appear as an isolated entity or in association with other anomalies. While some forms of ichthyosis are clinically well defined and can be relatively easily to diagnose, precise diagnosis can be challenging to stablish due to their significant clinical variability. Hereditary ichthyosis is a Mendelian etiologically heterogeneous disease classified in two large groups: non-syndromic forms that manifest primarily in the skin, and syndromic forms, with extracutaneous associated anomalies [3]. The non-syndromic form comprises four entities: ordinary ichthyosis, autosomal recessive congenital ichthyosis (ARCI), keratopathic ichthyosis, and other less frequent forms of ichthyosis.
ARCI has an estimated prevalence of 1/200,000 individuals in the United States of America. Harlequin ichthyosis is the rarest form, and it presents in 1/1,000,000 births [4, 5]. Epidemiological studies in Norway and the Spanish community of Galicia have identified founder mutations responsible for the particularly high prevalence of ARCI in those populations, with 1/91,000 and 1/122,000, respectively [6, 7]. ARCI clinical manifestations include neonatal dehydration, ectropion, recurrent skin infections, hypohidrosis with severe intolerance to heat and eclabium. To date, there are nine known genes associated with ARCI although no clear genotype–phenotype relationship and different mutations in the same gene can present with different phenotypes [4].
Lamellar ichthyosis (LI), one of the least prevalent and most severe forms of congenital ichthyosis, is characterized by thick and gray or brown squama that covers all over body present at birth and persisting for life. The most commonly affected genes in LI are TGM1, ALOXE3, NIPAL4, CYP4F22 and ALOX12B. TGM1 mutations underlie 74–85% of LI cases [7,8,9].
Ocular manifestations of LI include exposure keratitis secondary to ectropion, unilateral megalocornea, enlarged corneal nerves, blepharitis, the absence of Meibomian glands, trichiasis, madarosis, and the absence of lacrimal puncta [10]. Al-Amry described ectropion of both the upper and lower eyelids and no conjunctival involvement as the most common ocular findings in a case series; they found ocular complications were not severe in most cases, according to previous reports [11]. Ocular surface changes include mild to moderate exposure keratopathy, secondary to lid abnormalities, trichiasis, and the absence of lacrimal punctum. Ectropion, however, does not improve spontaneously, and it tends to cause lagophthalmos with secondary corneal exposure, corneal ulceration, and in severe cases, perforation and phthisis bulbi [12]. Patients suffer from eye disease throughout life due to Meibomian gland dysfunction [13]. In this work, the ocular clinical characteristics in a series of Mexican patients with lamellar ichthyosis due to TGM1 mutations are described.
Methods
Ophthalmological evaluation was performed in subjects with established clinical diagnosis of lamellar ichthyosis. All participants gave written informed consent prior to inclusion in the study. Examination was performed by a single ophthalmologist and included best-corrected visual acuity determination, slit-lamp biomicroscopy, fundoscopy, and applanation tonometry. A geneticist investigated systemic anomalies. Family history and pedigrees were collected as well as oral mucosa cells for genomic DNA isolation (Gentra Puregene Buccal Cell—Qiagen, Hilden, Germany) and subsequent mutational screening of the TGM1 gene by PCR amplification direct Sanger sequencing using the program PrimerQuest® IDT, Coralville, U.S.A.). All procedures were performed in the Genetics Laboratory of the Research Unit at the Institute of Ophthalmology “Conde de Valenciana” in Mexico City. The study was performed under adherence of the ethical foundations of the Declaration of Helsinki and approved by the Ethics Commission of the Institute of Ophthalmology “Conde de Valenciana.”
Results
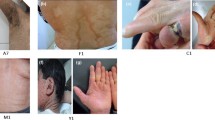
Nine patients from five LI families were identified. The subjects pertained to 5 families settled in a small community of the Veracruz state (south Mexico). All of the patients (9/9) presented with the classic LI phenotype of dark brown scales distributed throughout the body. LI was present in all of them at birth, and none presented atypical features of the disease. The genealogical tree analysis supported autosomal recessive transmission in all families. Six out of nine patients (67%) were females. The youngest patient was 1 year old, while the oldest one was 27 years old. All patients (9/9) demonstrated palpebral abnormalities (Figs. 1, 2, 3, 4, 5 and 6). Seven (77%) presented ectropion, while lagophthalmos was present in four patients (44%). Eyelid shortening was exhibited in four patients (44%). Euryblepharon was observed in two patients (22%). Unilateral spontaneous eversion was present in two patients (22%), one on the left inferior eyelid and the other on the upper right eyelid. Shortening of the anterior lamella was present in two patients (22%). Eight patients (88%) showed lid margin abnormalities, being the most common lid margin keratinization (5/9, 55%), followed by lacrimal punctum abnormalities as a group (4/9, 44%). Of these, two patients (22%) demonstrated lacrimal punctum keratinization in both eyes: One patient showed inferior lacrimal punctum obstruction in one eye and another patient exhibited lacrimal punctum stenosis in both eyes (Figs. 5 and 6). Three patients (33%) presented with eyelash abnormalities, two with inferior madarosis in both eyes, while the third presented inferior madarosis only in the right eye. One of the patients as mentioned above also displayed unilateral superior trichiasis. Two patients (22%) displayed Meibomian gland dysfunction and one patient exhibited entropion. Regarding conjunctival abnormalities, four patients (44%) revealed a papillary reaction in both eyes. The cornea was affected in two patients (22%), one presenting a central corneal opacity in the right eye and a paracentral nasal opacity in the left eye. In contrast, the other patient displayed a central opacity only in the left eye. Clinical data are summarized in Table 1.

Patient 2 demonstrates spontaneous upper lid eversion in both eyes with eyelid closure

Patient 6 exhibits lid margin keratinization, lower lid madarosis and ectropion

Patient 5 exhibits lid margin keratinization and ectropion

Patient 9 exhibits lid margin keratinization, lid madarosis and euryblepharon

Patient 4 demonstrates corneal opacity secundary to the lid pathology

Patient 4 demonstrates lacrimal punctum keratinization, corneal scarring, and exposure keratopathy
TGM1 genetic analysis identified that seven subjects (66%) were homozygous for the c.427C > T (p.Arg143Cys) variant in exon 3, one patient was homozygous for the c.760G > A (p.Asp254Asn) variant in exon 5, and one subject was compound heterozygous for the c.427C > T/c.760G > A variants. This was an unexpected finding as all patients originated from the same small community and homozygosity for a single variant was assumed a priori. A summary of the clinical characteristics and the pathogenic TGM1 variants in all nine patients is shown in Table 1.
Discussion
LI is the rarest and the most severe form of autosomal recessive ichthyoses. Individuals are born as collodion babies, with the membrane gradually exfoliating a few weeks after birth, only to be replaced by a scaling rash. The rash is generalized with skin flexures, palms, and soles affected [14]. Ocular anomalies are frequent in LI patients although a genotype–phenotype correlation has not been established yet.
As reported in the literature, ectropion of both the upper and lower eyelids is the most common finding in LI patients, with an estimated frequency between 45 and 80% [11]. Arnold was the first to report the association in 1834, and it occurs only in the lamellar type of ichthyosis [14]. The ectropion of LI is cicatricial in nature and appears to be a result of excessive dryness of the skin and subsequent contracture [15]. It is frequently bilateral and the lower lid is more severely affected. In the present group of patients, ectropion was observed in 7/9 patients and always in a bilateral fashion. Singh et al. reported a LI case with entropion, an unusual finding in the disease since lash ptosis usually occurs in conditions where the anterior lamella is loose [14]. Interestingly, one of our patients exhibited superior entropion of the right eye. The mechanism for this anomaly is ostensibly the same as that which causes ectropion, i.e., chronic inflammation in the anterior lamella of the eyelid skin [14]. A characteristic of ichthyosis is trans-epidermal dehydration and loss of elasticity and contraction of palpebral skin; thus, vertical shortening is seen in the anterior lamella with consequent ectropion arises [16, 17].
Conjunctival anomalies develop only in the autosomal recessive type of ichthyosis [18]. Buller et al. reported conjunctival changes, describing the conjunctiva of the lower lids as mildly swollen, having a smooth rather than glazed appearance, and presenting several longitudinal ridges without any follicular reaction [19] . Katowitz et al. reported keratinization and papillary formation of the conjunctiva [20] and Singh et al. reported no conjunctival involvement with LI [14]. In the present series, four patients (44%) exhibited a tarsal papillary reaction.
Ocular surface complications in LI are usually attributed to cornea exposure secondary to ectropion and lagophthalmos [14, 15]. In our series, two patients (22%) exhibited corneal opacities. One of them had bilateral opacities in eyes that were also affected by lagophthalmos and lid shortening and therefore can be attributed to exposure keratopathy. The other patient displayed a corneal opacity in only one eye that showed spontaneous inferior eyelid eversion. The absence of corneal opacities in the rest of the patients can be explained Bell´s phenomena. None of the patients demonstrated severe ocular complications, as has previously been reported by several authors [11]. Further clinical examination is still needed to elucidate the full spectrum of ocular surface alterations in LI. Of particular interest would be meibography and tear film analysis, epithelial mapping, and impression cytology of the corneal and conjunctival epithelium. Like the skin, meibomian glands are of ectodermal origin and are likely affected in LI [13]. Meibomian gland dysfunction can be presumed to be present in all patients. These patients benefit from the constant use of an appropriate ocular lubricant and lifestyle modifications. Lipid-containing lubricant eye drops to restore the balance of the tear film are largely recommended [13].
The management and treatment of LI patients must be multidisciplinary and according to the severity with topical and/or oral agents. It involves the use of hydrating and lubricating agents, keratolytics (e.g., salicylic acid, urea, and lactic acid), and modulators of keratinocyte differentiation (e.g., retinoic acid) [16].
In general management of lid malposition with exposure keratopathy requires surgical intervention involving the use of skin grafts, often full layer grafts [21]. If the exposure keratopathy is severe the use of soft bandage contact lenses, amniotic membrane grafts, and tarsorrhaphy should also be considered in order to prevent potentially sight threatening complications such as corneal perforation [11].
Our patients were all treated judiciously as none of them presented with severe ophthalmic disease. Artificial tears and lifestyle modifications including daily bathing with water or mild cleanser and the application of plain emollients directly after bathing, as well as frequently throughout the day, help to seal in moisture [16]. Theoretically bathing aids to hydrate and promote shedding of the stratum corneum, therefore reducing the thickness of scaling and improving overall skin. They were all prescribed moisturizing cream with 7.5% urea that is both keratolytic and moisturizer [22].
Conclusion
Finally, biallelic TGM1 mutations were demonstrated in all nine patients, as expected for an autosomal recessive trait as LI. However, an unanticipated finding was the occurrence of 3 distinct TGM1 genotypes: 7 homozygous for c.427C > T(p.Arg143Cys), 1 homozygous for c.760G > A (p.Asp254Asn), and 1 compound heterozygous c.427C > T/c.760G > A. Both the c.427C > T and the c.760G > A variants have been previously published in LI patients [23, 24]. The identification of more than one pathogenic allele in “closed” populations has been described in several recessive diseases and has been attributed to the occurrence of sporadic waves of immigration with introduction of additional founders [25]. However, additional studies are required to confirm this possibility in the LI population described here.
References
Takeichi T, Akiyama M (2016) Inherited ichthyosis: non-syndromic forms. J Dermatol 43(3):242–251. https://doi.org/10.1111/1346-8138.13243
Frost P, Weinstein GD, Van Scott EJ (1966) The ichthyosiform dermatoses. II. Autoradiographic studies of epidermal proliferation. J Invest Dermatol 47(6):561–567. https://doi.org/10.1038/jid.1966.185
Oji V, Tadini G, Akiyama M, Blanchet Bardon C, Bodemer C, Bourrat E et al (2010) Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J Am Acad Dermatol 63(4):607–641. https://doi.org/10.1016/j.jaad.2009.11.020
Oji V, Traupe H (2009) Ichthyosis: clinical manifestations and practical treatment options. Am J Clin Dermatol 10(6):351–364. https://doi.org/10.2165/11311070-000000000-00000
Richard G (1993) Autosomal Recessive Congenital Ichthyosis. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, et al. (eds). GeneReviews® [Internet]. University of Washington, Seattle [cited 19 Feb 2023]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1420/
Pigg M, Gedde-Dahl T, Cox D, Hausser I, Anton-Lamprecht I, Dahl N (1998) Strong founder effect for a transglutaminase 1 gene mutation in lamellar ichthyosis and congenital ichthyosiform erythroderma from Norway. Eur J Hum Genet EJHG 6(6):589–596. https://doi.org/10.1038/sj.ejhg.5200224
Rodríguez-Pazos L, Ginarte M, Fachal L, Toribio J, Carracedo A, Vega A (2011) Analysis of TGM1, ALOX12B, ALOXE3, NIPAL4 and CYP4F22 in autosomal recessive congenital ichthyosis from Galicia (NW Spain): evidence of founder effects. Br J Dermatol 165(4):906–911. https://doi.org/10.1111/j.1365-2133.2011.10454.x
Pigg MH, Bygum A, Gånemo A, Virtanen M, Brandrup F, Zimmer AD et al (2016) Spectrum of autosomal recessive congenital ichthyosis in Scandinavia: clinical characteristics and novel and recurrent mutations in 132 patients. Acta Derm Venereol 96(7):932–937. https://doi.org/10.2340/00015555-2418
Israeli S, Goldberg I, Fuchs-Telem D, Bergman R, Indelman M, Bitterman-Deutsch O et al (2013) Non-syndromic autosomal recessive congenital ichthyosis in the Israeli population. Clin Exp Dermatol 38(8):911–916. https://doi.org/10.1111/ced.12148
Chakraborti C, Tripathi P, Bandopadhyay G, Mazumder DB (2011) Congenital bilateral ectropion in lamellar ichthyosis. Oman J Ophthalmol 4(1):35–36. https://doi.org/10.4103/0974-620X.77662
Al-Amry MA (2016) Ocular manifestation of Ichthyosis. Saudi J Ophthalmol Off J Saudi Ophthalmol Soc 30(1):39–43. https://doi.org/10.1016/j.sjopt.2015.12.004
Turgut B, Aydemir O, Kaya M, Türkçüoğlu P, Demir T, Celiker U (2009) Spontaneous corneal perforation in a patient with lamellar ichthyosis and dry eye. Clin Ophthalmol Auckl NZ 3:611–613. https://doi.org/10.2147/opth.s8407
Palamar M, Karaca I, Onay H, Ertam I, Yagci A (2018) Dry eye and Meibomian gland dysfunction with meibography in patients with lamellar ichthyosis. Contact Lens Anterior Eye J Br Contact Lens Assoc 41(2):154–156. https://doi.org/10.1016/j.clae.2017.06.001
Singh AJ, Atkinson PL (2005) Ocular manifestations of congenital lamellar ichthyosis. Eur J Ophthalmol 15(1):118–122. https://doi.org/10.1177/112067210501500118
Shindle RD, Leone CR Jr (1973) Cicatricial ectropion associated with lamellar ichthyosis. Arch Ophthalmol 89(1):62–64. https://doi.org/10.1001/archopht.1973.01000040064015
Limmer AL, Nwannunu CE, Patel RR, Mui UN, Tyring SK (2020) Management of Ichthyosis: a brief review. Skin Ther Lett 25(1):5–7 (PMID: 32023022)
Ozgur OR, Akcay L, Tutas N, Ozkurt Y (2011) Cicatricial upper and lower eyelid ectropion in an ichthyosis patient surgical correction. J Dermatol Case Rep 5(2):27–29. https://doi.org/10.3315/jdcr.2011.1068
Sever RJ, Frost P, Weinstein G (1968) Eye changes in ichthyosis. JAMA 206(10):2283–2286 (PMID: 5303230)
Buller F (1887) A rare form of ophthalmia granulosa associated with icthyosis. Trans Am Ophthalmol Soc 4:582–587 (PMID: 16691821)
Katowitz JA, Yolles EA, Yanoff M (1974) Ichthyosis congenita. Arch Ophthalmol 91(3):208–210
Karadağ R, Sevimli N, Karadağ AS, Wollina U (2020) Successful correction of ichthyosis-related ectropion by autografts. Dermatol Ther 33(6):e13851. https://doi.org/10.1111/dth.13851
Dorf ILH, Lunen MS, Koppelhus U (2021) Effect of topical treatment with 7.5% urea in Ichthyosis Vulgaris: a randomized, controlled, double blinded, split body study evaluating the effect of urea cream compared to the vehicle (moisturizing) cream. Skin Health Dis 1(4):e65. https://doi.org/10.1002/ski2.65
Laiho E, Ignatius J, Mikkola H, Yee VC, Teller DC, Niemi KM et al (1997) Transglutaminase 1 mutations in autosomal recessive congenital ichthyosis: private and recurrent mutations in an isolated population. Am J Hum Genet 61(3):529–538. https://doi.org/10.1086/515498
Cao D, Chu WK, Ng TK, Yip YWY, Young AL, Pang CP et al (2018) Cellular proliferation and migration of human pterygium cells: mitomycin versus small-molecule inhibitors. Cornea 37(6):760–766. https://doi.org/10.1097/ICO.0000000000001569
Piluso G, Politano L, Aurino S, Fanin M, Ricci E, Ventriglia VM et al (2005) Extensive scanning of the calpain-3 gene broadens the spectrum of LGMD2A phenotypes. J Med Genet 42(9):686–693. https://doi.org/10.1136/jmg.2004.028738
Acknowledgements
An abstract of this project was presented at the World Cornea Congress VIII (Cornea Society) in Chicago, U.S.A., during September 28–29, 2022. None of the authors have any conflict of interest to disclose.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design, writing, material preparation, data collection, and analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interests
The authors have no relevant financial or non-financial interests to disclose.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Consent to publication
The authors affirm that human research participants provided informed consent for publication of the images Figs. 1 , 2, 3, 4, 5, and 6.
Ethical approval
This study was performed in line with the principles of the Declaration of Helsinki.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Macriz-Romero, N., Vera-Duarte, G.R., Guerrero-Becerril, J. et al. Ophthalmic findings in patients with autosomal recessive lamellar ichthyosis due to TGM1 mutations in an isolated population. Int Ophthalmol 43, 3659–3665 (2023). https://doi.org/10.1007/s10792-023-02774-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10792-023-02774-3