
Abstract
Background
X-linked ichthyosis (XLI; OMIM# 308100) is a recessive keratinization disorder characterized by the presence of dark brown, polygonal, adherent scales on different parts of the body surface. It almost exclusively affects males and the estimated prevalence ranges from 1:2000–6000 in males worldwide. Extracutaneous manifestations are frequent including corneal opacities, cryptorchidism, neuropsychiatric symptoms or others. Up to 90% of XLI cases are caused by recurrent hemizygous microdeletion encompassing entire STS gene on chromosome Xp22.3, while only a minority of patients shows partial deletions or loss of function point mutations in STS. Larger deletions also involving contiguous genes are identified in syndromic patients.
Methods
Here, we report clinical and genetic findings of a large Pakistani family having 16 affected individuals including 2 females with XLI. Molecular karyotyping and direct DNA sequencing of coding region of the STS gene was performed.
Results
The clinical manifestations in affected individuals involved generalized dryness and scaling of the skin with polygonal, dark scales of the skin on scalp, trunk, limbs, and neck while sparing face, palms and soles. There were no associated extra-cutaneous features such as short stature, cryptorchidism, photophobia, corneal opacities, male baldness, and behavioral, cognitive, or neurological phenotypes including intellectual disability, autism or attention deficit hyperactivity disorder. Molecular karyotyping was normal and no copy number variation was found. Sanger sequencing identified a novel hemizygous nonsense mutation (c.287G > A; p.W96*), in exon 4 of STS gene in all affected male individuals. In addition, two XLI affected females in the family were found to be homozygous for the identified variant.
Conclusions
This study is useful for understanding the genetic basis of XLI in the patients studied, for extending the known mutational spectrum of STS, diagnosis of female carriers and for further application of mutation screening in the genetic counseling of this family.
Similar content being viewed by others

Background
Inherited ichthyoses are a heterogeneous group of skin disorders characterized by a defect of keratinization with the clinical appearance of scaling or hyperkeratosis or both. X-linked ichthyosis (XLI; OMIM #308100) is X-linked recessive skin disorder caused by a deficit in the steroid sulfatase enzyme (STS; EC 3.1.6.2). It has an estimated prevalence of 1:2000 to 1:6000 males worldwide; with no significant geographic or racial differences [1, 2]. Clinically, it is characterized by generalized dryness and scaling of skin in which the trunk, ears, neck, scalp and extremities are often involved; usually sparing palms and soles. Onset of symptoms is at birth or during the first months of life with the presence of mild-to-moderate dark, polygonal, adherent and regular scales of skin, more prominent on the extensor aspects of the limbs. XLI may occur solely as a skin disorder or it may be associated with several extracutaneous features such as corneal opacities, cryptorchidism, neuropsychiatric symptoms or others [3,4,5].
STS is a membrane-bound microsomal enzyme of 62 kDa capable of hydrolyzing 3-β-hydroxysteroid sulfates with ubiquitous expression in human tissues [6, 7]. In the skin, STS is expressed within the epidermis and play a role in steroid production and lipid metabolism. A deficiency of STS gives rise to excess accumulation of cholesterol sulfate in the stratum corneum that increases intercellular cohesion resulting in characteristic scale formation in XLI patients [8, 9]. The STS gene is located on distal short arm of X-chromosome (Xp22.3) region and spans more than 164 kb of genomic DNA. Almost ~ 90% of XLI patients harbor recurrent ∼2 Mb deletion of the entire STS gene and flanking sequences [1, 10,11,12,13,14]. The presence of variable nucleotide tandem repeat sequences on either side of the STS gene promotes mispairing at meiosis and unequal recombination, resulting in high frequency of these interstitial deletions observed in XLI patients [15].
Depending on the extent of the DNA deletion of Xp22.3, a patient may have XLI either isolated or also with other relevant phenotypical anomalies. Deletion of contiguous genes may result in syndromic conditions, e.g. Kallmann syndrome (OMIM #308700), X-linked recessive chondrodysplasia punctata (OMIM #302950), ocular albinism type I (OMIM #300500), and short stature (OMIM #300582) [1, 16, 17]. Partial deletions of the STS gene and point mutations are found only in a minority of patients [1, 11, 12, 18]. In the present study, we investigated a large Pakistani family with multiple affected members having clinical signs of XLI but without any detectable deletion in the STS gene. Instead a novel nonsense mutation (p.W96*) in STS gene was identified in the affected males in hemizygous state. Two females in the family exhibiting the diseased condition were also included and found to be homozygous for the mutation.
Methods
Patients
A large five generation consanguineous family from village near Rawalpindi, Pakistan, was investigated in the present study (Fig. 1A). The family has 16 affected individuals including 14 males and 2 females. The pedigree analysis showed recessive X-linked mode of inheritance. Collection of family and personal history was based on a specific questionnaire (Additional file 1). Patients were seen by a dermatologist and neurologist. EDTA-anticoagulated peripheral blood (5 ml) was drawn from 17 family members (Fig. 1A). Written informed consent was obtained from all subjects or their respective guardians prior to their participation and the study was approved by the Ethics Review Committee of the Faculty of Allied & Health Sciences, Imperial College of Business Studies, Lahore. Following molecular diagnosis confirmation, patients performed physical examinations, ophthalmologic and neuropsychiatric evaluations.

Identification of a novel nonsense mutation in STS gene (A) Pedigree of the family with X-linked ichthyosis. The proband (IV:23) is noted with an arrow. ■, affected male; ●, affected female; ◻, healthy male; ○, healthy female. (B) Sequencing chromatogram showing the wild type normal control hemizygous affected male and heterozygous female carrier, with mutation position marked by an arrow. (C) Schematic representation of exon-intron structure of STS gene; exons are designated as boxes E (1–10). DNA and protein sequence of the exon 4 where the mutation c.287G > A (p.W96*) resides is shown expanded. Nucleotide and amino acid numbering correspond to NM_000351 for the cDNA and NP_000342.2 for the protein. Nucleotides were numbered using A of the ATG translation initiation codon as + 1 nucleotide of the coding sequence. (D) A ribbon diagram showing the secondary and tertiary structures of the wild type STS enzyme. Sheets are drawn in yellow, helices in red and loop regions in green. (a) showing β-sheets, α-helices and coils. The structure (b) denotes the potential truncated polypeptide (STS enzyme) due to p.W96* mutation (Swiss-Model). (E) Schematic illustration of the domain graph of the encoded STS protein (Uniprot identifier: P08842), and the genetic variants. A full-length wild type STS protein is shown, with its N-terminus and C-terminus. Novel variant identified in our study is boxed in red alongside previously reported point mutations and a splice- site variant underlying XLI phenotype in The Human Gene Mutation Database (HGMD®; http://www.hgmd.cf.ac.uk)
Molecular karyotyping/comparative genomic hybridization array (aCGH)
Genomic DNA was isolated from whole blood using Gentra Puregene Blood Kit (Qiagen, Germantown, MD), according to the manufacturer’s protocol. We used Cytoscan HD (Affymetrix, Santa Clara, USA) for molecular karyotyping which contains 2.6 million markers, of which 750,000 are genotyping single nucleotide polymorphisms and 1.9 million are non-polymorphic probes for genome coverage. The Cytoscan HD can detect copy number variation (CNV) and data analysis was performed using Chromosome Analysis Suite version Cyto2.0.0.195. Calling of pathogenic CNVs was in accordance with the ACMG (American College of Medical Genetics and Genomics) guidelines.
Mutation detection
For mutational screening the ten coding exons and splice junction sites of STS gene were PCR amplified as described before [19]. Amplification by PCR was performed in a total volume of 25 μl, containing 20 ng DNA, 0.5 μM primers (Metabion, Germany), dNTPs at a final concentration of 0.2 mM, and HotStar Taq DNA Polymerase (Qiagen, Germantown, MD). PCR amplicons were sequenced using the Big Dye Terminator sequencing kit (Applied Biosystems [ABI], Foster City, CA). Samples were run on an ABI PRISM 3730 XL automated sequencer (ABI) and sequence analysis was performed using the SeqMan 6.1 module of the Lasergene (DNA Star Inc. WI, USA) software package and then compared to the reference GenBank sequence (accession number STS; NM_000351). To exclude the chances of neutral polymorphism a panel of 200 unrelated, unaffected, ethnically matched control individuals were screened. STS three-dimensional protein prediction models were viewed in a Swiss-Pdb Viewer, version 4.0.
Results
Clinical data
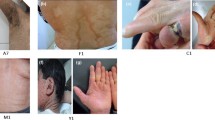
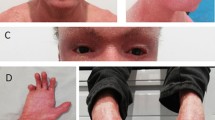
In all the patients from the family, the most prominent features included generalized dryness and dark brown scales prominent on the skin (Fig. 2); in which trunk, ears, scalp and limbs were affected. In almost all patients the abdomen was more severely affected than the back and other parts of the body. Scales were confluent and lighter on the scalp. Face, palms and soles were not involved. The hair and nails were normal. Preauricular small scales were also observed in young individuals. Their skin symptoms started by birth and during their youth and adolescence they were severely affected by this condition which consists of mild-to-moderate polygonal, dark brown, adherent and regular scales. The patients reported seasonal variation in disease severity. Their skin condition often exacerbated in cold weather but dramatically improves during the summer season, with the exception of very warm weather when insufficient sweating makes temperature regulation problematic with reduced heat tolerance. Physical examinations of the affected male members showed no micro-penises, cryptorchidism, small testes; ophthalmologic examination showed no corneal opacities or other any other abnormality. The growth, development and intellect of the affected members were normal. IQ of affected individuals was average in the range of 93 to 112.

Clinical findings of XLI patients. Photographs of skin showing thick, large, polygonal, dark-brown scales involving the trunk front and back, neck, arm and ear
Molecular data
Initial genetic workup for two affected male patients (IV:4 and IV:23, Fig. 1A) involved molecular karyotyping, which was found to be normal. Direct Sanger sequencing of all the coding exons and splice sites of the STS gene revealed a hemizygous variant in exon 4 (chromosome X: 7,175,519; c.287G > A) in the index case (Fig. 1B). The identified variant was validated in all the included family members and all the affected males were found to be hemizygotes (Fig. 1A and B). The two affected females (V:7 and V:9, Fig. 1A) who were born to affected father (IV:23) and carrier mother (IV:22) in a consanguineous union, were found to be homozygous for the identified variant. The transition mutation c. 287G > A leads to a substitution of tryptophan at position 96 by a stop codon (p.W96*), producing a loss of 487 amino acids downstream (Fig. 1C). The sequence alteration was not found in 200 ethnically matched controls, dbSNP database build 151 (http://browser.1000genomes.org), 1000 Genome project and ExAC; discarding any possibility of polymorphism. In addition, the identified variant has not been described in the literature or Human Gene Mutation Database (HGMD®; http://www.hgmd.cf.ac.uk) as mutation. The identified variant p.W96* was predicted as “disease causing” by Mutation Taster. Hence the factors that p.W96* being a null variant, their absence in population data/controls, segregation analysis and relevance to the patients phenotype, led us to classify this allelic variants as “likely pathogenic” according to the recommendations of ACMG guidelines [20] for the interpretation of sequence variants. The identified variant in this study has been submitted to the ClinVar database at NCBI archives (ID: SUB5568796; (https://www.ncbi.nlm.nih.gov/clinvar/).
Discussion
We performed molecular investigation of STS gene in a highly inbred Pakistani consanguineous family associated with XLI. The patients showed clinical features of dark brown scales especially prominent on the skin of the abdomen, legs and arms, segregated STS and a lack of any other phenotypic abnormalities. All the affected male members were found to carry a novel hemizygous mutation c.287G > A resulting in p.W96* premature termination of translation. Structural change predictions of the mutant form of STS with Swiss Model revealed that the p.W96* mutant is predicted to lack transmembrane, cytoplasmic and luminal domains thus resulting in loss of STS enzyme activity (Fig. 1D). Other possibility is the presence of non-sense-mediated mRNA decay (NMD) mechanism, which selectively degrades mRNAs harboring premature termination codons. However, in both instances it would result in absence or null activity of the STS enzyme.
Globally about 90% of XLI patients have complete deletions of the STS gene and flanking sequences, one of the highest ratios of chromosomal deletions among all genetic disorders [16, 21, 22]. A small number of point mutations with functional implications and partial deletions have also been reported (Human Gene Mutation Database, HGMD; (http://www.hgmd.cf.ac.uk, July 2019) without any evidence of genotype/phenotype correlation [18, 23]. So far, only 5 nonsense mutations, 2 in the N-terminal, 1 in the catalytic site and 2 in the C-terminal had been reported (Fig. 1E). The premature stop codon at codon 96 is expected to have serious consequences on the formation of mature protein and ultimately a deleterious effect on protein structure and function. This is consistent with other studies with nonsense hemizygous mutations detected in the sporadic subjects [24,25,26,27].
STS converts dehydroepiandrosterone sulfate in dehydroepiandrosterone, both steroids being involved in neurological processes [28]. Deletions encompassing STS and contiguous gene deletion syndromes have been associated with multiple behavioral, cognitive and neurological phenotypes notably: attention deficit hyperactivity disorder (ADHD), autism, mental retardation, and seizures [29, 30]. ADHD is also reported in XLI patients with partial STS deletions, suggesting that STS deficiency plays a direct role in the pathogenesis of inattentive or hyperactivity symptoms [31, 32]. None of our affected patients presented any neuropsychiatric symptoms; including ADHD, motor disabilities, behavioral abnormalities or social communication deficits. In addition, no other extra-cutaneous features or corneal opacities were detected in our family. The young family members have no learning disability and have normal academic performance in school.
The whole family is highly inbred and resides in a remote village in Pakistan; the elders and mothers are familiar with the disease and almost unmistakably notice the thin translucent membrane over the entire body for the affected newborns or a fine whitish scaling in the first days of life, confirming that XLI manifests around birth in a significant proportion of cases. However, dark polygonal scaling appears from late infancy to early childhood. The patients had not received any topical or systemic medical treatment, although the use of emollients helped with temporary remission of the cutaneous lesions. Clinical improvement during summer time is a common feature in ichthyosis patients. Family in our study belongs to a village in Pakistan and the male members work as farmers thus having a lot of sun-exposure, resulting in complete disease resolution during summer. Hypohidrosis is also known to be present due to keratotic plugs filling in sweat glands orifices [33]. The focus of treatment is lubrication, cutaneous hydration, and keratolysis, it should also include topical moisturizers and topical retinoids.
Our findings on lesion appearance and their distribution pattern in XLI patients are in keeping with literature survey [11, 33]. Scalp was affected in all the children and also mildly affected in few adults in the study family. The presence of fold involvement in about half of the reported cases limits its usefulness in differentiating XLI from ichthyosis vulgaris where folds are regularly spared. Because STS seems to play an important role in testosterone metabolism, the male-pattern baldness seems to be common in men with XLI [34]. Nevertheless, affected members of our family did not have any significant baldness pattern. Phenotypic variability of symptoms, even for patients with same STS mutations is likely due to effect of an individual’s genetic background, genetic modifiers and or other environmental factors. In addition the skin phenotype varies with age, older patients having dark brown scales than the younger individuals.
Recessive XLI is almost exclusively manifested in males. Female carriers of STS gene do not exhibit XLI symptoms, they may occasionally exhibit mild associated phenotypes such as dry skin [8, 23, 35, 36]. Interestingly, we observed no clinical signs of the any manifestations in the mothers of an affected male child, in agreement with previous reports and with the concept that STS gene is localized to a region of the X-chromosome that does not undergo X-inactivation [5, 16]. The presence of two XLI affected females with a homozygous STS mutation is an uncommon finding which could be explained by the high number of consanguineous unions in our family. So far, very few female patients have been reported in the literature [8, 37,38,39,40,41,42].
Diagnosis of XLI at or after birth is usually made clinically, but confirmation may be obtained through biochemically and the dissimilarities between XLI and regular skin and/or ichthyosis vulgaris can be quantified using protein electrophoresis [43]. Nevertheless, genetic diagnosis of affected individuals remains the most reliable method to identify the causative mutation leading to more accurate and effective genetic counseling, diagnosis of female carriers in the family allowing a correct evaluation of the risk of recurrence, especially in cultures where consanguineous marriages are preferred.
Conclusions
In conclusion, we define a unique nonsense alteration in the STS gene which expands the spectrum of known STS mutations in XLI; although the molecular mechanism underlying pathogenesis requires further functional studies and research. We also report two affected female siblings from this highly consanguineous family; carrier testing for at-risk relatives in extended family members is offered for genetic counseling purposes.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- 1000G:
-
1000 Genomes Project
- ABI:
-
Applied Biosystems
- aCGH:
-
Comparative Genomic Hybridization array
- ACMG:
-
American College of Medical Genetics and Genomics
- ADHD:
-
Attention Deficit Hyperactivity Disorder
- CNV:
-
Copy Number Variations
- ExAC:
-
The Exome Aggregation Consortium
- HGMD:
-
Human Gene Mutation Database
- OMIM:
-
Online Mendelenian Inheritance in Man
- PCR:
-
Polymerase Chain Reaction
- STS :
-
Steroid sulfatase
- XLI:
-
X-linked ichthyosis
References
Ballabio A, Carrozzo R, Parenti G, Gil A, Zollo M, Persico MG, et al. Molecular heterogeneity of steroid sulfatase deficiency: a multicenter study on 57 unrelated patients, at DNA and protein levels. Genomics. 1989;4(1):36–40.
Wang X, Tan L, Shen N, Lu Y, Zhang Y. Exacerbation of ichthyosis vulgaris phenotype by co-inheritance of STS and FLG mutations in a Chinese family with ichthyosis: a case report. BMC Med Genet. 2018;19(1):120.
Traupe H, Happle R. Clinical spectrum of steroid sulfatase deficiency: X-linked recessive ichthyosis, birth complications and cryptorchidism. Eur J Pediatr. 1983;140(1):19–21.
Hung C, Ayabe RI, Wang C, Frausto RF, Aldave AJ. Pre-Descemet corneal dystrophy and X-linked ichthyosis associated with deletion of Xp22.31 containing the STS gene. Cornea. 2013;32(9):1283–7.
Crane JS, Wu B, Paller AS. Ichthyosis X-linked. StatPearls. Treasure Island: StatPearls Publishing LLC; 2019.
Hobkirk R. Steroid sulfotransferases and steroid sulfate sulfatases: characteristics and biological roles. Canadian journal of biochemistry and cell biology =. Rev Can Biochim Biol Cell. 1985;63(11):1127–44.
Dibbelt L, Kuss E. Human placental sterylsulfatase. Interaction of the isolated enzyme with substrates, products, transition-state analogues, amino-acid modifiers and anion transport inhibitors. Biol Chem Hoppe Seyler. 1991;372(3):173–85.
Fernandes NF, Janniger CK, Schwartz RA. X-linked ichthyosis: an oculocutaneous genodermatosis. J Am Acad Dermatol. 2010;62(3):480–5.
Elias PM, Williams ML, Choi EH, Feingold KR. Role of cholesterol sulfate in epidermal structure and function: lessons from X-linked ichthyosis. Biochim Biophys Acta. 2014;1841(3):353–61.
Bonifas JM, Morley BJ, Oakey RE, Kan YW, Epstein EH Jr. Cloning of a cDNA for steroid sulfatase: frequent occurrence of gene deletions in patients with recessive X chromosome-linked ichthyosis. Proc Natl Acad Sci U S A. 1987;84(24):9248–51.
Shapiro LJ, Yen P, Pomerantz D, Martin E, Rolewic L, Mohandas T. Molecular studies of deletions at the human steroid sulfatase locus. Proc Natl Acad Sci U S A. 1989;86(21):8477–81.
Nomura K, Nakano H, Umeki K, Harada K, Kon A, Tamai K, et al. A study of the steroid sulfatase gene in families with X-linked ichthyosis using polymerase chain reaction. Acta Derm Venereol. 1995;75(5):340–2.
Cuevas-Covarrubias SA, Kofman-Alfaro SH, Maya-Nunez G, Diaz-Zagoya JC, Orozco E. X-linked ichthyosis in Mexico: high frequency of deletions in the steroid sulfatase encoding gene. Am J Med Genet. 1997;72(4):415–6.
Yen PH, Li XM, Tsai SP, Johnson C, Mohandas T, Shapiro LJ. Frequent deletions of the human X chromosome distal short arm result from recombination between low copy repetitive elements. Cell. 1990;61(4):603–10.
Ross MT, Grafham DV, Coffey AJ, Scherer S, McLay K, Muzny D, et al. The DNA sequence of the human X chromosome. Nature. 2005;434(7031):325–37.
Hernandez-Martin A, Gonzalez-Sarmiento R, De Unamuno P. X-linked ichthyosis: an update. Br J Dermatol. 1999;141(4):617–27.
Cuevas-Covarrubias SA, Gonzalez-Huerta LM. Analysis of the VCX3A, VCX2 and VCX3B genes shows that VCX3A gene deletion is not sufficient to result in mental retardation in X-linked ichthyosis. Br J Dermatol. 2008;158(3):483–6.
Valdes-Flores M, Kofman-Alfaro SH, Vaca AL, Cuevas-Covarrubias SA. Deletion of exons 1-5 of the STS gene causing X-linked ichthyosis. J Inves Dermatol. 2001;116(3):456–8.
Ramzan K, Bin-Abbas B, Al-Jomaa L, Allam R, Al-Owain M, Imtiaz F. Two novel LHX3 mutations in patients with combined pituitary hormone deficiency including cervical rigidity and sensorineural hearing loss. BMC Endocr Disord. 2017;17(1):17.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Toral-Lopez J, Gonzalez-Huerta LM, Cuevas-Covarrubias SA. Segregation analysis in X-linked ichthyosis: paternal transmission of the affected X-chromosome. Br J Dermatol. 2008;158(4):818–20.
Canueto J, Ciria S, Hernandez-Martin A, Unamuno P, Gonzalez-Sarmiento R. Analysis of the STS gene in 40 patients with recessive X-linked ichthyosis: a high frequency of partial deletions in a Spanish population. J Eur Acad Dermatol Venereol. 2010;24(10):1226–9.
Alperin ES, Shapiro LJ. Characterization of point mutations in patients with X-linked ichthyosis. Effects on the structure and function of the steroid sulfatase protein. J Biol Chem. 1997;272(33):20756–63.
Morita E, Katoh O, Shinoda S, Hiragun T, Tanaka T, Kameyoshi Y, et al. A novel point mutation in the steroid sulfatase gene in X-linked ichthyosis. J Invest Dermatol. 1997;109(2):244–5.
Winge MC, Hoppe T, Lieden A, Nordenskjold M, Vahlquist A, Wahlgren CF, et al. Novel point mutation in the STS gene in a patient with X-linked recessive ichthyosis. J Dermatol Sci. 2011;63(1):62–4.
del Refugio Rivera Vega M, Murillo-Vilches MR, Toral-Lopez J, Sanchez EG, Sanchez AT, Gonzalez-Huerta LM, et al. X-linked ichthyosis in a patient with a novel nonsense mutation in the STS gene. J Dermatol Sci. 2015;80(2):160–2.
Oyama N, Matsuda M, Hamada T, Numata S, Teye K, Hashimoto T, et al. Two novel missense mutations of STS gene underlie X-linked recessive ichthyosis: understanding of the mutational and structural spectrum. J Eur Acad Dermatol Venereol. 2016;30(9):1629–31.
Mueller JW, Gilligan LC, Idkowiak J, Arlt W, Foster PA. The regulation of steroid action by Sulfation and Desulfation. Endocr Rev. 2015;36(5):526–63.
Kent L, Emerton J, Bhadravathi V, Weisblatt E, Pasco G, Willatt LR, et al. X-linked ichthyosis (steroid sulfatase deficiency) is associated with increased risk of attention deficit hyperactivity disorder, autism and social communication deficits. J Med Genet. 2008;45(8):519–24.
Chatterjee S, Humby T, Davies W. Behavioural and psychiatric phenotypes in men and boys with X-linked Ichthyosis: evidence from a worldwide online survey. PLoS One. 2016;11(10):e0164417.
Brookes KJ, Hawi Z, Kirley A, Barry E, Gill M, Kent L. Association of the steroid sulfatase (STS) gene with attention deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2008;147B(8):1531–5.
Rodrigo-Nicolas B, Bueno-Martinez E, Martin-Santiago A, Canueto J, Vicente A, Torrelo A, et al. Evidence of the high prevalence of neurological disorders in nonsyndromic X-linked recessive ichthyosis: a retrospective case series. Br J Dermatol. 2018;179(4):933–9.
Diociaiuti A, Angioni A, Pisaneschi E, Alesi V, Zambruno G, Novelli A, et al. X-linked ichthyosis: clinical and molecular findings in 35 Italian patients. Exp Dermatol. 2018.
Axt-Gadermann M, Schlichting M, Kuster W. Male-pattern baldness is common in men with X-linked recessive ichthyosis. Dermatology. 2003;207(3):308–9.
Ghosh D. Mutations in X-linked ichthyosis disrupt the active site structure of estrone/DHEA sulfatase. Biochim Biophys Acta (BBA) - Mol Basis Dis. 2004;1739(1):1–4.
Wu B, Paller AS. Ichthyosis, X-linked. StatPearls. Treasure Island: StatPearls Publishing LLC; 2018.
Ramesh R, Chen H, Kukula A, Wakeling EL, Rustin MH, McLean WH. Exacerbation of X-linked ichthyosis phenotype in a female by inheritance of filaggrin and steroid sulfatase mutations. J Dermatol Sci. 2011;64(3):159–62.
Nagtzaam IF, Stegmann AP, Steijlen PM, Herbergs J, Van Lent-Albrechts JA, Van Geel M, et al. Clinically manifest X-linked recessive ichthyosis in a female due to a homozygous interstitial 1.6-Mb deletion of Xp22.31. Br J Dermatol. 2012;166(4):905–7.
Murtaza G, Siddiq S, Khan S, Hussain S, Naeem M. Molecular study of X-linked ichthyosis: report of a novel 2-bp insertion mutation in the STS and a very rare case of homozygous female patient. J Dermatol Sci. 2014;74(2):165–7.
Mevorah B, Frenk E, Muller CR, Ropers HH. X-linked recessive ichthyosis in three sisters: evidence for homozygosity. Br J Dermatol. 1981;105(6):711–7.
Thauvin-Robinet C, Lambert D, Vaillant G, Caillier P, Donzel A, Cusin V, et al. X-linked recessive ichthyosis in a girl: strategy for identifying the causal mechanism. Br J Dermatol. 2005;152(1):191–3.
Diociaiuti A, Angioni A, Pisaneschi E, Margollicci M, Boldrini R, Alesi V, et al. Next generation sequencing uncovers a rare case of X-linked Ichthyosis in an adopted girl homozygous for a novel nonsense mutation in the STS gene. Acta Derm Venereol. 2019.
Thyssen JP, Godoy-Gijon E, Elias PM. Ichthyosis vulgaris: the filaggrin mutation disease. Br J Dermatol. 2013;168(6):1155–66.
Acknowledgements
We thank the patients and all the family members for their participation into the study.
Funding
Not Applicable.
Author information
Authors and Affiliations
Contributions
SA and ABW conceived and designed the analysis, SA and AJ identified the subjects and collected the blood samples from the subjects, AJ and SB verified the analytical methods and findings of this work, SA and SU contributed in data analysis, SA, KR and SMW performed the analysis, SA, SB, and KR wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the guidelines of the Ethics Committee of Faculty of Allied & Health Sciences, Imperial College of Business Studies, Lahore, Pakistan. Written informed consent was obtained from all subjects or their parents/legal guardians prior to their participation.
Consent for publication
Written informed consent for publication of individual/genetic data and clinical images was obtained from the participants or parents/legal guardians of any participant under the age of 18.
Competing interests
The authors declare that they have no competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Questionnaire for recruitment of the patients with Ichthyosis for genetic studies.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Afzal, S., Ramzan, K., Ullah, S. et al. A novel nonsense mutation in the STS gene in a Pakistani family with X-linked recessive ichthyosis: including a very rare case of two homozygous female patients. BMC Med Genet 21, 20 (2020). https://doi.org/10.1186/s12881-020-0964-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-020-0964-y