
Abstract
Coronavirus disease 2019 (COVID-19) is caused by the novel SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) first discovered in Wuhan, Hubei province, China in December 2019. SARS-CoV-2 has infected several millions of people, resulting in a huge socioeconomic cost and over 2.5 million deaths worldwide. Though the pathogenesis of COVID-19 is not fully understood, data have consistently shown that SARS-CoV-2 mainly affects the respiratory and gastrointestinal tracts. Nevertheless, accumulating evidence has implicated the central nervous system in the pathogenesis of SARS-CoV-2 infection. Unfortunately, however, the mechanisms of SARS-CoV-2 induced impairment of the central nervous system are not completely known. Here, we review the literature on possible neuropathogenic mechanisms of SARS-CoV-2 induced cerebral damage. The results suggest that downregulation of angiotensin converting enzyme 2 (ACE2) with increased activity of the transmembrane protease serine 2 (TMPRSS2) and cathepsin L in SARS-CoV-2 neuroinvasion may result in upregulation of proinflammatory mediators and reactive species that trigger neuroinflammatory response and blood brain barrier disruption. Furthermore, dysregulation of hormone and neurotransmitter signalling may constitute a fundamental mechanism involved in the neuropathogenic sequelae of SARS-CoV-2 infection. The viral RNA or antigenic peptides also activate or interact with molecular signalling pathways mediated by pattern recognition receptors (e.g., toll-like receptors), nuclear factor kappa B, Janus kinase/signal transducer and activator of transcription, complement cascades, and cell suicide molecules. Potential molecular targets and therapeutics of SARS-CoV-2 induced neurologic damage are also discussed.
Similar content being viewed by others
Introduction
Coronavirus (CoV) (from “corona”—Latin for “crown”) is a family of enveloped, single-stranded, positive-sense RNA viruses with protruding spike-like appearance on the membrane that gives rise to a crown-like shape (McBride et al. 2014; Li 2015; Chen et al. 2020a). CoV is one of the two virus genera within the family Coronaviridae of the order Nidovirales that is widely distributed in humans and animals, including bats, dogs, mice, cats etc. (Ksiazek et al. 2003; Zaki et al. 2012; McBride et al. 2014). To date, 7 species belonging to the α- and β-CoV genera of the Coronaviridae family have been identified to infect humans (Lefkowitz et al. 2018). The species of α-CoV genus are 229E and NL63, while those of β-CoV genus include OC43, HKU1, Middle East respiratory syndrome coronavirus (MERS-CoV), severe acute respiratory syndrome coronavirus (SARS-CoV) (Esper et al. 2006; Li 2015; Wu et al. 2020a; Yeo et al. 2020) and SARS-CoV-2 (novel CoV 2019) (Chen et al. 2020a).
This review is focused on the recently discovered SARS-CoV-2 that causes CoV disease 2019 (COVID-19), first identified in Wuhan, the capital of Hubei province, China in December 2019 (Wu et al. 2020a; Yeo et al. 2020). CoV is a frequent cause of upper and lower respiratory tract infections, affecting all age groups. Symptoms of COVID-19 include fever, cough, fatigue (Yeo et al. 2020; Zhu et al. 2020), sore throat, breathlessness, malaise (Singhal 2020), diarrhea (Yeo et al. 2020), and vomiting (Yeo et al. 2020). Notably, data have shown that a subset of patients with positive SARS-CoV-2 RNA test initially experience gastrointestinal symptoms such as diarrhea and nausea about 24–48 h prior to development of fever and respiratory symptoms (Yeo et al. 2020). Evidence also indicates that intestinal epithelial inflammation and degeneration precede pulmonary infection in CoV disease, suggesting that respiratory impairment, at least in part, may be due to dysfunction of the gut-lung axis (Yeo et al. 2020). Though the majority of reported CoV infections are mild (Conti et al. 2020), CoV-19 may cause potentially serious damage to the respiratory system (Ksiazek et al. 2003; Zaki et al. 2012) and other organs of the body (Zhu et al. 2020). As of March 4, 2021, more than 115 million people have been infected with several disabilities and over 2.5 million deaths worldwide (John Hopkins University and Medicine 2020). Alas, SARS-CoV-2 infection has also resulted to huge global economic losses (Baldwin and Mauro 2020; Fernandes 2020).
Though the respiratory and gastrointestinal tracts have been frequently implicated in the pathogenesis of SARS-CoV-2 infection, available evidence also indicates a critical role of central nervous system (CNS) dysfunction in COVID-19 pathogenesis (Yeo et al. 2020). Unfortunately, however, the mechanisms of SARS-CoV-2 induced cerebral damage are not completely known. Here, we review the literature on possible neuropathogenic mechanisms of SARS-CoV-2 induced cerebral damage. Potential molecular targets and emerging therapeutics for SARS-CoV-2 induced neurologic damage are also discussed.
Neurological manifestations of SARS-COV-2 neuroinvasion
The SARS-CoV-2 exhibits neurotropic properties (Wu et al. 2020b) and has been found to cause a range of neurological diseases including Guillain–Barré syndrome, Miller Fisher syndrome, polyneuritis cranialis and epilepsy (Gutiérrez-Ortiz et al. 2020; Zhao et al. 2020b) as well as cerebral stroke (Al Saiegh et al. 2020; Helms et al. 2020; Mao et al. 2020a). Similarly, neurologic injury was reported in previous CoV outbreak involving the MERS-CoV (Arabi et al. 2015; Kim et al. 2017), human CoV-OC43 (Salmi et al. 1982; Morfopoulou et al. 2016), and human CoV-229E (Salmi et al. 1982) among others (Li et al. 2017).
Interestingly, biomolecular analysis by polymerase-chain-reaction (PCR) reportedly revealed the presence of CoV RNA in the cerebrospinal fluid (CSF) or cerebral tissues of CoV infected patients. So, Gutiérrez-Ortiz et al. (2020) reported the presence of SARS-CoV-2 RNA in the CSF of SARS-CoV-2 infected patients (Gutiérrez-Ortiz et al. 2020). Previous studies have reported the identification of human CoV-229E (Fazzini et al. 1992; Dessau et al. 2001, 2009), human CoV-OC43 (Fazzini et al. 1992; Yeh et al. 2004; Morfopoulou et al. 2016), SARS-CoV (Hung et al. 2003; Yeh et al. 2004) in human CSF or postmortem brain tissues. In contrast, Al Saiegh et al. (2020) reported the absence of SARS-CoV-2 RNA in CSF despite the presence of neurological problems such as headache, loss of consciousness and ischaemic/haemorrhagic cerebral injury in SARS-CoV-2 infected patients (Al Saiegh et al. 2020). Consistent with these findings, Helms et al. (2020) also demonstrated the absence of SARS-CoV-2 RNA in CSF of patients admitted for SARS-CoV-2 infection, confirmed by reverse-transcriptase PCR assay of the nasopharyngeal tissue (Helms et al. 2020). Though these inconsistencies remain to be clarified, neurological symptoms of possible SARS-CoV-2 neuroinvasion reported across studies (Gutiérrez-Ortiz et al. 2020; Mao et al. 2020b; Al Saiegh et al. 2020; Helms et al. 2020) should not be ignored.
The SARS-CoV-2 invasion of the brain may cause cerebral vascular/endothelial dysfunctions, which may result in cerebral circulatory impairments that characterise the acute cerebrovascular problems of SARS-CoV-2 infection as reported by Mao et al. (2020b), Al Saiegh et al. (2020), and Helms et al. (2020). Indeed, Helms et al. (2020), using perfusion imaging, demonstrated in COVID-19 patients that SARS-CoV-2 neuroinvasion causes bilateral frontotemporal hypoperfusion, indicating cerebral circulatory impairment (Helms et al. 2020).
The neurological problems of COVID-19 may be due to the ensuing inflammatory or immune system responses to SARS-CoV-2 neuroinvasion. Gutiérrez-Ortiz et al. (2020) reported a range of neurological damages, concomitantly with inflammatory/immune dysfunction in two patients diagnosed with COVID-19 in Madrid, Spain. The patients had ageusia, anosmia, bilateral abducens palsy (or right internuclear ophthalmoparesis, right fascicular oculomotor palsy), ataxia and areflexia (Gutiérrez-Ortiz et al. 2020). Furthermore, a CSF positive test was demonstrated for albuminocytologic dissociation, indicating a hallmark of inflammatory demyelinating polyneuropathies, while immunoglobulin (Ig)G antibodies to GD1b ganglioside was also positive, which suggests acute immune-mediated ataxic neuropathies (Gutiérrez-Ortiz et al. 2020). Interestingly, for both patients, these neurological damages occurred 3–5 days before onset of cough, malaise, diarrhea, fever, headache, and low back pain, after which COVID-19 was confirmed by PCR using oropharyngeal tissue (Gutiérrez-Ortiz et al. 2020). These findings suggest that neurological manifestations may be the initial symptoms of SARS-CoV-2 infection or neuroinvasion and may be responsible for peripheral organ dysfunctions in some COVID-19 patients.
Consistent with the data of Gutiérrez-Ortiz et al. (2020), other clinicians have shown that SARS-CoV-2 infection adversely affects the neuromuscular system (Guidon and Amato 2020). In addition to ataxia and areflexia reported by Gutiérrez-Ortiz et al. (2020), Zhao et al. (2020b), Guidon and Amato (2020), and Mao et al. (2020b) described associated muscle weakness in COVID-19 patients. At the same time, Zhao et al. (2020b) showed reduced tendon reflexes in both legs and feet in COVID-19 patients. In contrast, Helms et al. (2020) reported enhanced tendon reflexes among 67% of the 58 patients admitted for SARS-CoV-2 infection with positive reverse-transcriptase PCR test using nasopharyngeal samples. These patients also had disorganized movement in response to command and ankle clonus (Helms et al. 2020).
In a retrospective study, conducted among 214 patients with laboratory confirmed diagnosis of COVID-19 in Wuhan, China, Mao et al. (2020b) reported central and peripheral dysfunctions such as headache, impaired consciousness, ataxia, generalized tonic–clonic seizures, hyposmia and hypogeusia in 36% patients. Indeed, headache was one of the most frequently reported symptoms of SARS-CoV-2 neuroinvasion (Gutiérrez-Ortiz et al. 2020; Zhu et al. 2020). In addition, neurological manifestations of COVID-19 included fatigue (Wang et al. 2020a), nausea, vomiting (Guan et al. 2020; Wang et al. 2020a), disorientation, inattention (Helms et al. 2020), delirium tremens, and coma (Gutiérrez-Ortiz et al. 2020; Helms et al. 2020), which are similar to data reported by other authors (Mao et al. 2020b). Neuropathic pain as a neurological symptom was frequently reported by the majority of studies (Guidon and Amato 2020; Gutiérrez-Ortiz et al. 2020; Mao et al. 2020b; Wang et al. 2020a). For instance, Guidon and Amato (2020) and Wang et al. (2020a) described an association between SARS-CoV-2 infection and myalgia or low back pain (Gutiérrez-Ortiz et al. 2020). Wang et al. (2020a) reported abdominal pain in COVID-19. However, pain arising from the gastrointestinal tract may be due to local injury caused by the virus or SARS-CoV-2 induced neurogenic injury that generates pain sensation.
Though the precise mechanisms of neurological manifestations of SARS-CoV-2 infection are not clearly known, neuroinflammation and blood brain barrier disruption appear to be critical pathophysiological processes responsible for the central and peripheral nervous system damages, and thus provide important information for the prevention and management of patients with initial symptoms of the disease. The next section will be concerned with the receptors and enzymes required for SARS-CoV-2 entry into the host cells. In subsequent sections, the key pathways involved in SARS-CoV-2 neuroinvasion, neuroimmune dysregulation by SARS-COV-2 neuroinvasion, including inflammatory and oxidative stress, and cytokine storm as well as the molecular signalling cascades implicated in these processes will be discussed.
Receptors and enzymes required for SARS-CoV-2 entry into the host cells
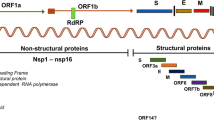
SARS-CoV-2 infection occurs via the fecal–oral route of transmission (Yeo et al. 2020). The virus can be transmitted via direct droplet transmission, excrements, contact, fomites, and environmental contamination (Yeo et al. 2020). SARS-CoV-2 enters the host cells by binding to the angiotensin converting enzyme 2 (ACE2) with its spike (S) protein, followed by priming of the S protein through the activities of the transmembrane protease, serine 2 (TMPRSS2) (Fig. 1) (Sungnak et al. 2020) and cathepsin L (Ou et al. 2020).

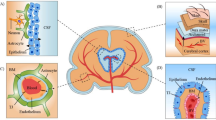
Schematic representation of SARS-CoV-2 invasion of olfactory/nasopharyngeal, pulmonary, intestinal and tongue or oropharyngeal epithelial cells. ACE2 angiotensin converting enzyme 2, ER endoplasmic reticulum, SARS-CoV-2 severe acute respiratory syndrome coronavirus 2, TMPRSS2 transmembrane protease, serine 2 (see explanation in text)
The serine protease, TMPRSS2, is a transmembrane enzyme required for SARS-CoV-2 invasion of the host cells. TMPRSS2 is responsible for cleaving and activating the S protein of SARS-CoV-2, thereby enhancing the fusion of the virus with the host cell membrane (Hoffmann et al. 2020). TMPRSS2 is primarily expressed in nasal epithelial cells and alveolar epithelial type II cells of subsegmental bronchial branches (Chen et al. 2020c; Sungnak et al. 2020), pancreatic and renal tissues (Glowacka et al. 2011). TMPRSS2 is highly expressed in the fetal brain (Ubuka et al. 2018), but available data suggest that expression of the protease is low in the adult brain (Glowacka et al. 2011; Ubuka et al. 2018). It is, therefore, important for future investigation to identify key aspects of TMPRSS2 ontogenesis, and the implication of age-dependent expression of this protease in neuroinvasiveness of CoV.
It is widely acknowledged that ACE2 receptor binding affinity constitutes a key determinant of viral infectivity (Yeo et al. 2020). ACE2 is a zinc-dependent carboxy-peptidase, comprising an integral component of the central renin–angiotensin system that has been consistently implicated in the regulation of cardiovascular/cerebrovascular functions (Doobay et al. 2007; Xia and Lazartigues 2008). Interestingly, ACE2 downregulation has been associated with neuroinflammation and cerebral endothelial disruption (vide infra). ACE2 is expressed on vascular endothelial cells, nasal epithelial and type II alveolar cells of the lungs (Oudit et al. 2009; Chen et al. 2020c; Sungnak et al. 2020), cardiac cells (Oudit et al. 2009), corneal epithelial cells (Sungnak et al. 2020), intestinal epithelial cells, vascular endothelial cells and smooth muscle cells of lungs, kidney and heart (Hamming et al. 2004; Sungnak et al. 2020). The widespread expression of ACE2 points to the severe complications that arise from SARS-CoV-2 infection: acute respiratory distress syndrome, acute kidney injury, acute cardiac injury, shock, and disseminated intravascular coagulopathy (Deng et al. 2020).
ACE2 is highly expressed in the brain of animals (Doobay et al. 2007; Xia and Lazartigues 2008; Chen et al. 2020c) and humans with comparative distribution in these organisms (Chen et al. 2020c). ACE2 is richly expressed in the cerebral ventricles, substantia nigra, middle temporal gyrus, posterior cingulate cortex, olfactory bulb (Chen et al. 2020c), subfornical organ, dorsal motor nucleus of the vagus nerve and the ventrolateral medulla (Doobay et al. 2007). ACE2 expression has been documented in multiple cerebral neurons (Doobay et al. 2007; Chen et al. 2020c), and oligodendrocytes (Chen et al. 2020c). The high expression of ACE2 in cellular components of the blood brain barrier [vascular endothelial cells, pericytes, and astrocytes (Chen et al. 2020c)] suggests that SARS-CoV-2 invasion of the brain may cause blood brain barrier disruption. Scientific evidence (Welcome 2020a, b) indicates that these components of the blood brain barrier also represent principal mediators of neuroinflammatory processes in several CNS diseases. Consequently, inflammatory responses to SARS-CoV-2 invasion of the brain can potentially cause serious CNS dysfunctions in COVID-19 (Chen et al. 2020c). So, Al Saiegh et al. (2020) reported ischaemic and haemorrhagic cerebral injury [(which is often associated with neuroinflammation and blood brain barrier disruption (Welcome 2020a, b)] accompanied by headache and loss of consciousness in COVID-19 patients, suggesting a role of SARS-CoV-2 infection in blood brain barrier impairment.
Cathepsin L is a cysteine protease that is primarily expressed in endolysosomal vesicles. This protease mediates proteolytic cleavage of multiple proteins, including receptors, enzymes, and transcription factors to regulate several aspects of cell physiology (Rawlings and Salvesen 2013). Ou et al. (2020) showed that treatment of 293/hACE2 cells with the cathepsin L inhibitor, SID 26681509, resulted in over 76% decrease in the entry of SARS-CoV-2 S virions into the cells (Ou et al. 2020). Interestingly, cathepsin L (Hook et al. 2012) is expressed in the brain, suggesting a potential role of this protease as a target for the treatment of SARS-CoV-2 infection. Zhang and co-researchers reported that teicoplanin, a glycopeptide antibiotic, suppressed the activation of the SARS‐CoV‐2 S protein inside endosomes by inhibiting cathepsin L (Zhang et al. 2020a). Indeed, Zhou et al. (2016) previously showed that teicoplanin, which is recommended for use in the clinic, effectively blocked both CoVs and Ebola virus from invading the host cells (Zhou et al. 2016).
Though the mechanisms are not well understood, phosphatidylinositol 3-phosphate 5-kinase (PIKfyve)/FAB1 (formation of aploid and binucleate cells) and two pore channel subtype 2 (TPC2) may also play a role in SARS-CoV-2 invasion of the host cells (Ou et al. 2020). PIKfyve is a lipid kinase involved in the regulation of endosome biogenesis and vesicle transport. It is the main enzyme responsible for the synthesis of phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2) (a lipid molecule regulating intracellular trafficking of vesicles and membrane recycling to the Golgi network) in early endosome (Nelson et al. 2017; Qiu et al. 2018). TPC2 is an intracellular voltage- and ligand-gated ion channel and a key downstream effector of PI(3,5)P2 signalling in lysosomes (Patel 2015). Interestingly, PIKfyve (Martin et al. 2013) and TPC2 (Kelu et al. 2018) are expressed in the brain, suggesting a possible role of these molecules in SARS-CoV-2 neuroinfection. Ou et al. (2020) demonstrated that the PIKfyve inhibitor, YM201636 or apilimod, and the TPC2 inhibitor, tetrandrine, effectively inhibited the entry of CoV virion into 293/hACE2 cells (Ou et al. 2020). Notably, previous studies have shown that both CoVs and Ebola virus require the PIKfyve for effective entry into the host cells (Nelson et al. 2017; Qiu et al. 2018). These data suggest that PIKfyve and TPC2 may serve as critical targets for the treatment of SARS-CoV-2 infection.
The molecular architecture that regulates SARS-CoV-2 entry into the host cells are not limited to ACE2, TMPRSS2, cathepsin L, PIKfyve, and TPC2, but also, T cell cluster of differentiation (CD) 32/CD64, CD147/besigin, and bradykinin B2 receptor. CD147 is expressed on the surface of T lymphocytes (Wang et al. 2020b). Bradykinin B2 receptor (van de Veerdonk et al. 2020) and CD32/CD64 (Yip et al. 2016; Quinlan et al. 2020) are expressed in neurons, glia and endothelial cells (Curtin et al. 2007; Zhu et al. 2014; Sharma et al. 2019). These receptors have been reportedly shown to play a role in SARS-CoV-2 invasion of the host cells (Wang et al. 2020b; van de Veerdonk et al. 2020; Quinlan et al. 2020). Interestingly, these receptors, especially CD147 and bradykinin B2 receptor, are involved in neuroinflammatory response and blood brain barrier breakdown (Li et al. 2013a; Zhu et al. 2014; Sharma et al. 2019). Further research is required to ascertain the contribution of each of these receptors and proteases to SARS-CoV-2 invasion of the host cells.
Pathways mediating SARS-COV-2 induced cerebral infection
Literature data indicate that the humoral and neural pathways are the main routes through which the neurotropic SARS-COV-2 infects the CNS (vide infra). However, due to the paucity of information on SARS-CoV-2 neuroinvasiveness, data on similar viruses will be used at strategic point of the discussion to ultimately achieve the main goals of this review.
Humoral pathway
The humoral pathway mediates the transport of viral components (including viral RNA), cytokines, toxic metabolites, certain hormones, and peptide molecules produced as a result of SARS-CoV-2 activities (Fig. 1) (Zhu et al. 2020). These molecules may be produced in the respiratory, gastrointestinal tract or other regions of the body, and are then transported via the circulatory system to the brain (Zhu et al. 2020). In the brain, the molecules first interact with the cerebral vascular endothelium to mediate a range of pathological processes that promote neuroinflammation and blood–brain barrier disruption (Fig. 2) (Peña-Silva et al. 2012).

Mechanisms involved in SARS-CoV-2 induced neuroinfection, neuroinflammation and neurodegeneration. ACE2 angiotensin converting enzyme 2, AJ adherens junction, BBB blood brain barrier, CCL-2 C–C motif chemokine ligand, cJNK c-Jun N-terminal kinase, CXCL-8 C-X-C motif chemokine ligand, Fas FS-7-associated surface antigen, IFN-γ interferon gamma, IL interleukin, iNOS inducible nitric oxide synthase, IP-10 IFNγ-induced protein 10, MAPK mitogen-activated protein kinase, MCP-1 monocyte chemoattractant protein-1, MIP-1A macrophage inflammatory protein-1A, NADPH nicotinamide adenine dinucleotide phosphate, NF-κB nuclear factor kappa-light-chain-enhancer of activated B cells, NLRP3 nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3, NO nitric oxide, P2X7R P2X purinergic receptor 7, PRR pattern recognition receptor, ROS reactive oxygen species, SARS-CoV-2 severe acute respiratory syndrome coronavirus 2, TGF-β transforming growth factor, TJ tight junction, TLR toll-like receptor, TMPRSS2 transmembrane protease, serine 2, TNF-α tumor necrosis factor alpha (see explanation in text)
Neural pathway
In the neural pathway, viral components or molecules released during SARS-CoV-2 invasion interact with enteric neurons and glia, vagus, facial, glossopharyngeal and olfactory nerve fibers to initiate neural signalling or modulate neurotransmission that may lead to cerebral dysfunctions. Indeed, activation of nerve fibers, enteric neurons or glia can stimulate or inhibit the activities of their control centers (enteric ganglia, brainstem nuclei or olfactory nuclei) (Fig. 1) (Mengeling et al. 1972; Andries and Pensaert 1980; Butowt and Bilinska 2020).
CoV can transmigrate via peripheral nerve fibers into the brain (Fig. 1) (Andries and Pensaert 1980; Butowt and Bilinska 2020; Li et al. 2020d). Niu et al. (2020), using non-invasive bioluminescence imaging, investigated the spatio-temporal progression of human CoV-OC43 in the CNS of mice after intranasal or intracerebral inoculation and showed substantial expression of the viral RNA in the brain before the infection spread to the spinal cord of the laboratory animals (Niu et al. 2020). Niu et al. (2020) also demonstrated that the viral replication was effectively inhibited with chloroquine/hydroxychloroquine in live mice (Niu et al. 2020) or humans (Liu et al. 2020a) due to its ability to inhibit toll-like receptors and high mobility group box 1 (HMBG1), thereby attenuating the proinflammatory responses (Liu et al. 2020a). In addition, the inhibitory action of chloroquine and hydroxychloroquine on CoV is also due to their ability to cause impairment in glycosylation of ACE2 receptor and thus binding of the S protein. Again, these quinolones can increase the intraorganellar pH of endosomes and lysosomes—organelles critical to SARS-CoV-2 replication and accumulation in the host cells (Liu et al. 2020b).
Previous studies conducted by independent groups of researchers also reported the transmigration of CoV via peripheral nerve fibers into the brain. So, experimental data have shown that HEV 67 N, the CoV that causes hemagglutinating encephalomyelitis, infects the respiratory and intestinal epithelial cells, transmigrating in a retrograde direction through peripheral nerves to the brainstem neurons (Mengeling et al. 1972; Andries and Pensaert 1980). Though the mechanism of transmigration of CoV from the peripheral nerves into the central neurons are not completely understood, Li et al. (2013a, b) showed that trans-synaptic communication of HEV 67 N occurs via a special membranous-coating that facilitates migration of the virus in nerve fibers or neurons (Li et al. 2013b). Importantly, HEV 67 N shares over 90% amino acid sequence homology with human CoV‐OC43 (González et al. 2003) and also shares about 90% homology with SARS-CoV (Li et al. 2020d). The SARS-CoV in turn shares about 75% homology with SARS-CoV-2 (Xie et al. 2020). Indeed these CoVs also have other properties in common. For example, SARS-CoV and SARS-CoV-2 use the same receptor to gain access into the host cells (Xie et al. 2020). Interestingly, interneuronal and synaptic transfer of similar viruses and subsequent neuroinfection has been reported (Matsuda et al. 2004). Identification of infectivity of the SARS-CoV-2 in different regions of the brain confirms the neuroinvasiveness of CoV. Previous data have demonstrated the presence of viral antigens in regions of the brain, where SARS-CoV receptors are widely expressed (Kalia and Mesulam 1980; Hadziefendic and Haxhiu 1999; Raux et al. 2000). Data also indicate that the olfactory epithelium, where ACE2 and TMPRSS2 are highly expressed, can facilitate SARS-CoV-2 binding, replication, and accumulation, suggesting that the nasal cavity may be a better site from collecting samples for SARS-CoV-2 detection at the early stages of invasion (Butowt and Bilinska 2020). Again, the olfactory pathway represents a relatively fast pathway through which the virus can cause neuroinfection/neuroinflammation, in addition to smell impairment in COVID-19 patients (Butowt and Bilinska 2020). Previous studies have consistently shown the involvement of CoVs in cerebral neuroinfection: 229E (Dessau et al. 2009), SARS-CoV (Lau et al. 2004) and human CoV-HKU1 (Esper et al. 2006).
Neuroimmune dysregulation by SARS-CoV-2 neuroinvasion
SARS-CoV-2 invasion of host cells, replication and accumulation
SARS-CoV-2 invades the brain cells by binding to the ACE2 with its S protein S1 subunit, triggering conformational change of the S2 subunit, which enables membrane fusion with the host cell (Fig. 2). SARS-CoV-2 initially interacts with the cerebral vascular endothelial cells through the humoral pathway to initiate endothelial dysfunction, thereby allowing direct access of the virus and its components to the elements of the blood brain barrier such as pericytes, perivascular astrocytes, neurons and microglia (Fig. 2) (Xia et al. 2020). Some proteases (vide supra) also facilitate the entry of the virions into the host cells. In addition to membrane fusion, SARS-CoV-2 may enter the host cell through clathrin-dependent mechanism, which has been previously reported for SARS-CoV (Wang et al. 2008; Kuba et al. 2010). Experimental investigation by Li and coworkers revealed that HEV 67 N infects cerebral neurons by clathrin-mediated endocytosis and exocytosis (Li et al. 2020d). Furthermore, viruses may enter the cell via cell adhesion molecules (Li 2015; Mateo et al. 2015). Research findings reported by Kubo and colleagues showed that the murine CoV, mouse hepatitis virus, invades the host cells via the cell–cell adhesion molecule, CEACAM1 (carcinoembryonic antigen related cell adhesion molecule 1) (Kubo et al. 1994). Frankly, upregulation of CEACAM8 by activated monocytes and granulocytes has been reported in SARS-CoV infection (Reghunathan et al. 2005).
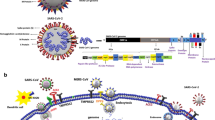
The virus enters the host cell through an endocytic pathway, hijacking the endosomes for delivery of its RNA molecule into the cytosol. The viral RNA is then translated into several structural proteins required for viral assembly, non-structural proteins (produced from the cleavage of two large polyproteins) required for viral RNA synthesis, and accessory proteins required to overcome the immunity of the host (Fig. 1) (Bartlam et al. 2005; Perlman and Netland 2009; Liu et al. 2014; Valencia 2020). Thereafter, the virus starts to replicate, and subsequently assembled to form similar viruses, which may be slightly different from the original viral RNA genome. However, the viral RNA genome may be transcribed to the corresponding mRNA, which is then translated in the cytoplasm to form envelope glycoproteins (Fig. 1) (Bartlam et al. 2005; Perlman and Netland 2009; Liu et al. 2014; Valencia 2020). These glycoproteins are inserted into the membrane of the endoplasmic reticulum or Golgi network, and together with the nucleocapsid protein, germinate in the endoplasmic reticulum-Golgi intermediate compartment. The products are assembled; bud off to form new viral replicates (Fig. 1) (Bartlam et al. 2005; Perlman and Netland 2009; McBride et al. 2014; De Wit et al. 2016; Valencia 2020). The viral vesicles then fuse with the plasma membrane, releasing the virus to infect other cells (Fig. 1) (De Wit et al. 2016).
Some pharmacological agents acting as viral RNA polymerase inhibitors [(Clevudine, NCT04347915; Favipiravir, NCT04336904 & NCT04359615; Lopinavir/ritonavir, NCT04321993; and Remdesivir, NCT04292899 (https://www.clinicaltrials.gov)] that specifically target the SARS-CoV-2 replication machinery have shown promise for potential application in the treatment of COVID-19.
Immune responses from SARS-CoV-2 invasion
Some viruses in the endosomal compartment are channeled into the lysosomal degradation pathway, which produces viral components (i.e., CoV antigenic peptides) that are subsequently loaded into major histocompatibility complex (MHC I & II molecules) or human leukocyte antigen (HLA) and presented on the surface of the brain perivascular macrophages, their tissue specific subtype—microglia, dendritic cells and other antigen presenting cells (APC) (Figs. 1, 2) (Jensen et al. 2018). Flanked CoV antigenic molecules by the APC are recognized by naïve T cells through viral specific T-cell receptor, thereby triggering their proliferation and differentiation with the formation of different isotypes of T cells (Fig. 3). Homing of activated T cells to the site of invasion occurs with the aid of environmental cues (Jensen et al. 2018).

Simplified schematic representation of SARS-CoV-2 infection of blood. MHC major histocompatibility complex, SARS-CoV-2 severe acute respiratory syndrome coronavirus 2, Th T helper, Treg T regulatory (see explanation in text)
At the point of invasion, virus-specific effector T cells produce cytokines and chemokines directed against viral replication, facilitation of antigen presentation, and recruitment of immune cells (Channappanavar et al. 2014; Chen et al. 2020b). CoV antigenic peptides-APC-T cell complex also activates CD4+ and CD8+ T cells that participate in blocking APC-CoV antigenic peptide complex. However, the T cell responses may not be effective enough to resolve SARS-CoV-2 in the majority of infected patients (Li et al. 2020a). The ability of CD4+ and CD8+ T cells to secrete cytokines such as interleukin-2 (IL-2), interferon (IFN)-γ, and tumor necrosis factor (TNF)-α has been demonstrated to be higher in severe cases of infection compared to mild cases (Peng et al. 2006; Li et al. 2008a; Fan et al. 2009). However, previous data about the effects of SARS-CoV infection showed marked decrease in CD4+ and CD8+ T cells in approximately 80–100% of patients compared to similar indices in healthy controls, which was demonstrated to be due to impaired activation of these T cells in SARS-CoV infected patients (Channappanavar et al. 2014). Similarly, data about SARS-CoV-2 infection also showed substantial decrease in CD4+ and CD8+ T cells in the peripheral blood (Diao et al. 2020; Qin et al. 2020; Wang et al. 2020c; Xu et al. 2020; Zhou et al. 2020b), indicating impaired activation of T cells (Zhou et al. 2020b). The cytotoxic T cells can directly kill the CoV, but may not efficiently execute their functions, in part, due to impaired immune responses. These data strongly suggest that T-cell responses to SARS-CoV-2 invasion may not be adequate enough to prevent or resolve the activities of the pathogen.
The generated helper T cells (Th1, Th2, and Th17) present the viral antigens to the B cells (differentiated plasma cells), which produce CoV-specific antibodies or Ig required to neutralise the virus (Fig. 3) (Li et al. 2017, 2020a; Ahmed et al. 2020; Huang et al. 2020). Several studies have shown association between higher antibody production with stronger helper T cell cytokine secretion in SARS-CoV infection (Peng et al. 2006; Li et al. 2008a; Fan et al. 2009). However, Qin et al. (2020) reported a substantial reduction in the number of serum helper T cells and regulatory T (Treg)/suppressor T cells in patients with COVID-19 of different severities (Qin et al. 2020). Again, Qin and coworker reportedly established substantial reduction in the number of B- and natural killer (NK) cells in severe cases of COVID-19 (Qin et al. 2020). Furthermore, generated memory cells may not effectively protect from reinfection (Peng et al. 2006; Chen et al. 2010; Channappanavar et al. 2014). Importantly, Qin et al. (2020) reported considerable reduction of memory cells in severe COVID-19 (Qin et al. 2020). Indeed cases of reinfection have been reported in patients who were previously diagnosed and treated of COVID-19 (Lan et al. 2020). Notwithstanding, convalescent serum has demonstrated effectiveness in treating COVID-19 patients, suggesting that viral-specific antibodies may play a critical role in preventing reinfection (Shen et al. 2020). On the contrary, however, decreased immunity of previously treated COVID-19 patients, especially in severe cases, may increase the likelihood of reinfection during a repeat exposure.
The CoV antigenic peptides and single-stranded RNA trigger cellular signalling pathways that mediate the production of inflammatory mediators/molecules in the immune cells (Li et al. 2020a). Experimental investigation by Iwata-Yoshikawa et al. (2019) showed that replication and accumulation of SARS-CoV were integral causes of elevated inflammatory chemokines observed in wild type mice (Iwata-Yoshikawa et al. 2019). The authors also suggested a likely involvement of TMPRSS2 and related proteases in development of inflammatory reactions in SARS-CoV infection (Iwata-Yoshikawa et al. 2019). Accumulation of SARS-CoV peptides and RNA due to viral replication also triggers the activation of neutrophils (Figs. 2, 3) (Li et al. 2020a). Consistent with these findings, Qin et al. (2020) demonstrated increased neutrophil count in the blood of COVID-19 patients (Qin et al. 2020). Upon activation by CoV, neutrophils release C3a, granzyme B, defensin and other alarmins that inhibit the viral activities by killing infected cells (Reghunathan et al. 2005; Chen et al. 2020b; Li et al. 2020a). However, reports by Guan et al. (2020), Liu et al. (2020c), and Deng et al. (2020) showed leukopenia in COVID-19 patients, which are comparable with previous data reported by Channappanavar et al. (2014) about the marked leukopenia in the acute phase of SARS-CoV infection in humans (Channappanavar et al. 2014). These inconsistencies in data regarding the effects of SARS-CoV-2 on blood cell indices may be due to the different stages of progression of the disease as well as sequestering of both lymphocytes and granulocytes into the sites of inflammation.
The serum monocyte and basophil (Diao et al. 2020; Qin et al. 2020; Wang et al. 2020c; Xu et al. 2020; Zhou et al. 2020b), eosinophil and lymphocyte (Deng et al. 2020; Guan et al. 2020; Liu et al. 2020c; Qin et al. 2020) and thrombocyte (Deng et al. 2020; Guan et al. 2020; Liu et al. 2020c) counts have been consistently shown to decrease in COVID-19 patients. In COVID-19, abnormal blood cell counts suggest atypical immune responses that prime the blood brain barrier for subsequent disruptive processes, resulting to neuroimmune dysregulation and neuroinflammation that worsen the course of progression of SARS-CoV-2 infection (Fig. 2).
Therefore, modulators of the immune system such as cyclosporine A (Liu et al. 2020a), intravenous IgG (Ye et al. 2020), fingolimod (NCT04280588, https://www.clinicaltrials.gov), and anti-CoV-19 antibody from recovered COVID-19 patients (ChiCTR2000030012, http://www.chictr.org) may provide benefits in SARS-CoV-2 infection. Though the effects of several immunomodulators are yet to be reported, preliminary evidence indicates that intravenous IgG, for instance, improves COVID-19 symptoms (Fu et al. 2020), at least in part, by downregulating proinflammatory cytokines, complement proteins and promoting T suppressor cell functions (vide infra) (Katz et al. 2000; Aschermann et al. 2010; Kaufman et al. 2015). In addition, Fu et al. (2020) demonstrated that the anti-inflammatory effects of intravenous IgG against SARS-CoV-2 occur via inhibition of Fc receptor activation.
Immuno-inflammatory stress, oxidative stress, and cytokine storm in SARS-CoV-2 infection
The immune dysregulation in COVID-19 (Ye et al. 2020) favors upregulation of multiple genes that enhance proinflammatory and oxidative stress responses, resulting to inflammatory stress (Reghunathan et al. 2005), oxidative stress (Horowitz et al. 2020; Zhao et al. 2020a), hormonal and neuro-peptide/-transmitter signaling disorder (Zhang et al. 2020a, b), as well as cytokine storm (Huang et al. 2020; Li et al. 2020b; Liu et al. 2020a) in severe cases of infection.
Oxidative stress
Though there is a scarcity of data, available information indicates the involvement of oxidative stress in the pathogenetic mechanisms of the SARS-CoV-2 infection (Horowitz et al. 2020; Zhao et al. 2020a). Increased expression of oxidative stress genes due to SARS-CoV invasion was reportedly demonstrated to promote downregulation of antioxidant enzymes with elevated expression of prooxidants (Reghunathan et al. 2005). An abnormal level of lactate dehydrogenase was observed in SARS-CoV-2 infected patients (Zhao et al. 2020a). A recent report by Horowitz et al. (2020) also revealed that COVID-19 may cause substantial decrease in antioxidant enzymes such as glutathione, and promote pro-oxidative stress enzymes (Horowitz et al. 2020). Interestingly, high dose (2000 mg) glutathione (per os or intravenous) showed improvement in symptoms one hour after treatment, indicating that antioxidants may provide substantial benefits for use in the treatment of SARS-CoV-2 infection (Horowitz et al. 2020). Importantly, several antioxidants (including vitamin A, C, D, and E) have shown promise for potential use as adjuncts in the treatment of COVID-19 (NCT04323228, NCT04323514, NCT04351490, NCT04363840, NCT04334005, and NCT04344041, https://www.clinicaltrials.gov).
Immuno-inflammatory stress predisposes to hormonal, neuropeptide and neurotransmitter dysregulation in SARS-CoV-2 infection
Up-regulation of alarmins (including the 55–60 kDa microbicidal permeability increasing protein) by SARS-CoV activated monocytes and granulocytes along with increased immune cell chemotaxis (Reghunathan et al. 2005) not only primes the cerebral microenvironment for inflammatory processes, but also, favors disruptive processes in the blood brain barrier by downregulating the expression of junctional proteins that seal the cerebral endothelium (Welcome 2020a, b). Indeed increased leucocyte chemotaxis has been consistently demonstrated in blood brain barrier disruption or neuroinflammation (Takeshita 2012; Welcome 2020c).
Though the mechanisms are not clearly understood, immune dysregulation and inflammatory stress can potentially cause disordered signaling of hormones, neuro-peptide and -transmitters such as VEGF and melatonin (Zhang et al. 2020a, b). VEGF dysregulation due to inflammatory stress has been recently associated with SARS-CoV-2 infection and may underlie the development of neuroinflammation and blood brain barrier disruption in COVID-19 (Zhang et al. 2020a, b). Experimental data indicate that VEGF can trigger cerebral inflammation (Argaw et al. 2009) and blood brain barrier leakage (Zhang et al. 2000) in several brain pathologies. Argaw et al. (2009) showed that upregulation of VEGF-A in brain disorders downregulates the endothelial tight junction proteins, occludin and claudin-5, in animal model of autoimmune encephalomyelitis (Argaw et al. 2009). Furthermore, intracerebral injection of VEGF-A in mice resulted to corresponding loss of occludin and claudin-5 with concomitant breakdown of the blood brain barrier (Argaw et al. 2009). Thus, VEGF dysregulation in SARS-CoV-2 infection (Zhang et al. 2020a, b) can predispose to blood brain barrier breakdown. The neurotransmitter, melatonin, was demonstrated to effectively attenuate the proinflammatory and oxidative responses in animal models of COVID-19, suggesting that neurotransmitter dysregulation may play a critical role in the pathogenesis of the disease (Zhang et al. 2020a, b). There is also a possible role of glucocorticoid signalling disorder caused by immuno-inflammatory stress in SARS-CoV-2 infection. Indeed, impaired glucocorticoid signalling has been consistently implicated in viral infection (Ng et al. 2013; Biron 2018). A recent analysis showed that impaired glucocorticoid signalling can result from inflammatory/immune dysregulation (Welcome 2020c). Though the mechanisms are not exactly clear, data indicate that impaired glucocorticoid signalling can also prime neuroinflammatory responses especially in stress responsive brain regions (Welcome 2020c). Though glucocorticoids exert pleiotropic effects on the cell, short term treatment in severe cases of COVID-19 was reportedly associated with some benefits (Ye et al. 2020). Clinical trials on short term use of corticosteroids or glucocorticoids (e.g., NCT04329650, NCT04244591, https://www.clinicaltrials.gov) as adjunct therapy in severe cases of SARS-CoV-2 infection also demonstrated evidence of health benefits.
Excessive inflammatory and oxidative stress due to immune dysregulation is responsible for triggering the cytokine storm in SARS-CoV-2 infection
Excessive activation of the immune effector cells by SARS-CoV-2 causes continued release of proinflammatory cytokines such as IL-1β, IL-6, IL-8, IL-12, IL-18, IL-33, granulocyte colony-stimulating factor (G-CSF), monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-1A (MIP-1A), TNF-α, transforming growth factor (TGF)-β, IFN-α, IFN-β, IFN-γ, etc. (Li et al. 2017, 2020b; Conti et al. 2020; Ye et al. 2020; Zhang et al. 2020a, b) and chemokines such as C–C motif chemokine ligand (CCL)-2, CCL3, CCL5, CCL7, CCL12, C-X-C motif chemokine ligand (CXCL)-8, CXCL9, CXCL10, IFNγ-induced protein 10 (IP-10), etc. (Li et al. 2020b; Ye et al. 2020). These proinflammatory cytokines and chemokines along with elevated reactive oxygen species (Figs. 2, 3) (Conti et al. 2020; Zhang et al. 2020a, b) trigger cytokine release syndrome, also known as cytokine storm, a fatal irrepressible systemic inflammatory response (Huang et al. 2020; Li et al. 2020b; Liu et al. 2020a) that progresses to acute respiratory distress syndrome/acute respiratory injury, acute kidney injury, acute cardiac injury, multiple organ failure, coma and death in severe cases of SARS-CoV-2 infection (Xu et al. 2020).
The expression of the pro-inflammatory cytokines/chemokines largely depends on the severity of the illness. Multiple lines of evidence indicate that mild to moderate cases of COVID-19 may be associated with such plasma pro-inflammatory cytokines/chemokines as IL-1β, IFN-γ, MCP-1 and IP-10. In a severe form of the disease, significantly higher levels of TNF-α, IL-8, IL-10, G-CSF, MCP-1, MIP-1A and IP-10 have been reported (Huang et al. 2020; Qin et al. 2020). Furthermore, several studies (Deng et al. 2020; Guan et al. 2020; Liu et al. 2020c) have identified elevation of C-reactive protein (CRP) in COVID-19. Notably, sustained increase of inflammatory molecules in circulation has been associated with blood brain barrier inflammation and leakage (Haruwaka et al. 2019), as well as neuroinflammation in the affected brain regions (Fig. 2) (Biesmans et al. 2013; Zhang et al. 2018). Li et al. (2017) investigated 183 and 236 paediatric patients with acute encephalitis-like syndrome and respiratory tract infection, respectively. The researchers documented increased level of cytokines not only in the serum, but also, significantly higher IL-6, IL-8, MCP-1, and granulocyte macrophage colony-stimulating factor (GM-CSF) levels in CSF of patients with CoV-induced CNS infection than in matched serum samples, indicating a greater vulnerability of the brain to SARS-CoV infection than other organs (Li et al. 2017).
Other inflammatory molecules that may be associated with SARS-CoV infection include lipocalin-2, oligoclonal bands and IgG. Lipocalin 2 is a glycoprotein that is secreted by activated neutrophils, cerebral endothelial cells, and perivascular astrocytes in response to pathological (e.g., inflammatory) stimuli (Ni et al. 2013; Jin et al. 2014; Ferreira et al. 2015). Serum lipocalin-2 is thought to be a crucial biomarker of peripheral inflammation (Ni et al. 2013; Moschen et al. 2017). More importantly, due to its high expression in the CNS and involvement in innate immunity, cerebrospinal lipocalin-2 (Llorens et al. 2020) is currently considered as a potential biomarker of cerebral inflammation or damage (Llorens et al. 2020). Increased expression of lipocalin-2 is associated with cerebral inflammation and blood brain barrier hyperpermeability (Jin et al. 2014; Du et al. 2019). Indeed deficiency of lipocalin 2 expression in the brain reportedly attenuates neuroinflammatory responses and blood brain barrier disruption in animal models of cerebral stroke (Jin et al. 2014). Reghunathan et al. (2005) previously established up-regulation of genes coding for lipocalin 2 in SARS-CoV-infected patients (Reghunathan et al. 2005). These data strongly suggest that SARS-CoV-2 induced upregulation of lipocalin genes can predispose to blood brain barrier damage or neuroinflammation. In recent study, Helms et al. (2020) reported identification of oligoclonal bands and elevated IgG level in the serum of SARS-CoV-2 infected patients (Helms et al. 2020). These oligoclonal bands and IgG indicate B-cell responses resulting from CNS inflammation (Link and Huang 2006; Franciotta et al. 2008). Thus, CNS dysfunctions in SARS-COV-2 infection may be triggered by neuroinflammation and blood brain barrier disruption.
The majority of clinical studies investigating inflammatory biomarkers in COVID-19 frequently showed elevated expression of IL-6 (Huang et al. 2020; Sun et al. 2020; Wang et al. 2020e; Zhou et al. 2020a). For instance, Chen et al. (2020b) and Qin et al. (2020) demonstrated a positive correlation between serum IL-6 levels and severity of illness in COVID-19 patients, suggesting that approved monoclonal antibodies can be explored for potential treatment of COVID-19. Indeed, clinical trials have shown that some monoclonal antibodies such as siltuximab (anti-IL-6 antibody) (NCT04329650, https://www.clinicaltrials.gov) may be beneficial in attenuating the proinflammatory responses in SARS-CoV-2 infection (Table 1). However, due to the relatively long period required in getting approval for these drugs (Table 1), already approved monoclonal antibodies, especially those that inhibit IL-6 and TNF, as well as Bevacizumab (Food and Drug Administration, FDA, 2018), Ruxolitinib (FDA, 2019), Baricitinib (FDA, 2018), and Emapalumab (FDA, 2019) may be considered for possible use in the treatment of SARS-CoV-2 infection with associated inflammatory stress or cytokine storm. Even though the effects of these antibodies on neuroinflammation are not known, previous studies have demonstrated a beneficial role of monoclonal antibodies in psychopathological illnesses (Essali et al. 2019) and multiple sclerosis (Sirbu and Budisteanu 2020), which are characterized by neuroinflammatory response and blood brain barrier disruption (Welcome 2020a, b).
Molecular signalling in SARS-CoV-2 induced immuno-inflammatory stress, oxidative stress, and cytokine storm
Research data suggest the involvement of critical molecular signalling pathways (vide infra) in SARS-CoV-2 infection along with immuno-inflammatory stress, oxidative stress, and cytokine storm. Identification of key molecular mechanisms mediating SARS-CoV-2 invasion of the host cells may provide useful information for COVID-19 therapeutics.
JAK/STAT signalling
JAK/STAT (Janus kinase/signal transducer and activator of transcription) signalling is an immune signalling pathway that regulates the processes of proliferation, differentiation, migration, cell survival, and antiviral immunity depending on the cell/tissue type and the physiological/pathophysiological state of the host (Harrison 2012; Jupatanakul et al. 2017). The JAK/STAT pathway also interacts with mitogen-activated protein (MAP) kinase and PI-3-kinase signalling pathways to mediate a range of physiological processes (Fig. 4) (Harrison 2012). The JAK/STAT pathway is activated by cytokines (IFN-α/β/γ, IL1-7, GM-CSF etc.) and growth factors (platelet derived growth factor, growth hormone, epidermal growth factor etc.) (Fig. 4) (Li et al. 2020a; Xin et al. 2020). IFN mediated activation of the JAK/STAT pathway is used by the host cells as a defense mechanism against viral infection by enhancing the expression of IFN‐regulated genes (Ma and Suthar 2015; Nelemans and Kikkert 2019). Unfortunately, however, some viruses have acquired properties that are able to overpower the IFN-antiviral effects (Fig. 4). For instance, Lin et al. (2004) reported inhibition of IFN-α-activation of Stat-1, -2, and -3 by the Japanese encephalitis virus (Lin et al. 2004). This virus inhibits the tyrosine phosphorylation of Tyk2 (tyrosine kinase 2), which is associated with IFN-induced JAK activation (Lin et al. 2004). Similarly, SARS‐CoV and other CoVs can block IFN‐α/β-induced JAK/STAT signalling using their N protein to antagonise the responses initiated by IFN (Spiegel et al. 2005; Kopecky-Bromberg et al. 2007; Lu et al. 2011).

JAK-STAT signalling in SARS-CoV-2 infection. IFN interferon, IL interleukin, JAK Janus kinase, MAP kinase mitogen activated protein kinase, PI-3-kinase phosphoinositide 3-kinase, Raf Rapidly Accelerated Fibrosarcoma (protein that interacts with small GTPases), STAT signal transducers and activators of transcription (see explanation in text)
Evidence indicates that impaired JAK/STAT signalling affects the infectivity of viruses, suggesting that this pathway may serve as a critical target for potential treatment of SARS-CoV infection (Schneider et al. 2014). Interestingly, the JAK/STAT signalling has been implicated in neuroinflammatory and neurodegenerative disorders (Copf et al. 2011). JAK/STAT signalling disorder primes T and myeloid cells to pathogenic phenotypes that cause neuroinflammation (Yang et al. 2010; Qin et al. 2016), in part, by activation of microglia (Qin et al. 2016). In an in vivo study, Qin et al. (2016) demonstrated that AZD1480, a JAK1/2 inhibitor, effectively inhibited the α-synuclein-induced MCH II molecule and inflammatory gene expression in both macrophages and microglia by blocking the activation of Stat-1 and -3 (Qin et al. 2016). Baricitinib, a selective JAK inhibitor with potentially powerful anti-inflammatory action, effectively reduced the levels of the proinflammatory cytokines reported in COVID-19 patients (Richardson et al. 2020; Stebbing et al. 2020). This agent also inhibited clathrin-mediated endocytosis to suppress SARS-CoV-2 invasion of the host cells (Stebbing et al. 2020). The results of a completed clinical trial (NCT04358614, https://www.clinicaltrials.gov) conducted in Italy showed considerable effectiveness of baricitinib against proinflammatory cytokines (Richardson et al. 2020). Another clinical trial in Germany is evaluating the immunomodulatory and inflammatory role of the JAK 1/2 inhibitor, ruxolitinib (NCT04338958) in SARS-CoV-2 infected patients.
TLR signalling
TLR (toll like receptor) is a type of pattern recognition receptors (PRRs) involved in translating pathogenic signals into cellular responses that are initially aimed at counteracting the invasion but may also lead to collateral tissue damage. There are three main groups of PRRs, which include the membrane type (TLR2, TLR4, mannose receptor, scavenger receptor), the cytoplasmic type (melanoma differentiation‐associated gene 5, MDA5; retinoid/inducible gene 1, RIG-1; TLR3, TLR7, TLR8, TLR9, and NLRs—nucleotide-binding oligomerization domain-like receptors) and the secretory type (CRP and mannose/binding lectin, MBL) (Gordon 2002; Kato et al. 2006; Phadwal et al. 2012; De Wit et al. 2016). Interestingly, different PRR types have been implicated in SARS-CoV infection. However, emphasis is made on TLR due to its widespread implication in viral infection (Fig. 5) (De Wit et al. 2016), neuroinflammation and blood brain barrier disruption (Fig. 2) (Welcome , 2019,2020a; b).

TLR signalling in SARS-CoV-2 infection. ACE2 angiotensin converting enzyme 2, IFN interferon gamma, IPS-1 interferon-β promotor stimulator-1, IRE interferon regulatory element, IRF interferon regulatory factor, ISRE Interferon-sensitive response element, MAVS mitochondrial antiviral signalling protein, MDA5 melanoma differentiation-association gene 5, MyD88 myeloid differentiation primary response 88, NF-κB nuclear factor kappa-light-chain-enhancer of activated B cells, NLRP3 nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3, ORF open reading frame, P2X7R P2X purinergic receptor 7, PPRγ peroxisome proliferator-activated receptor gamma, PRR pattern recognition receptor, RIG-1 retinoic acid inducible gene-I, ROS reactive oxygen species, SARS-CoV-2 severe acute respiratory syndrome coronavirus 2, TLR toll-like receptor, TMPRSS2 transmembrane protease, serine 2 (see explanation in text)
The singled stranded RNA and viral proteins constitute the viral pathogen-associated molecular patterns (PAMPs), which are recognized by the innate immune system (Hopcraft and Damania 2017). Following endocytosis of the virus in dendritic, macrophages and vascular endothelial cells, SARS-CoV-2 PAMPs (single stranded RNA) are recognized by the endosomal receptors, TLR7/8/9, and the cytosolic sensors, MDA5 and RIG-1 to trigger the synthesis of cytokines/chemokines and type I IFN such as IFN-α/β (Fig. 5) (Phadwal et al. 2012; De Wit et al. 2016; Li et al. 2016, 2020a; Channappanavar and Perlman 2017). Activation of membrane TLRs (e.g., TLR-2) (Law et al. 2009) by viral components or molecules resulting from the activities of the virus (e.g., HMGB1) can also trigger such reactions (Chen et al. 2004; Wang et al. 2006). Type I IFN synthesis is triggered by interferon response factor (IRF)-3/7 (Fig. 5) (Yang et al. 2015; Deng et al. 2019). The initiation of cytokine/chemokine synthesis is triggered by downstream stimulation of the nuclear factor κB (NF‐κB), a transcription factor that is currently known as a master regulator of inflammation (Fig. 5) (Yang et al. 2015; Deng et al. 2019; Conti et al. 2020). The stimulation of NF‐κB causes its nuclear translocation, activating gene expression with subsequent release of pro-IL-1β and other inflammatory mediators. The pro-IL-1β for instance, undergoes cleavage by caspase-1, then inflammasome activation, accompanied by generation and secretion of active mature IL-1β along with other alarmins that mediate destructive processes in the brain microenvironment (Fig. 5) (Conti et al. 2020). Data indicate that MAVS (mitochondrial antiviral signalling), an adapter protein localized at the outer membrane of the mitochondria, physically interacts with the cytoplasmic viral RNA sensors, RIG-I and MDA5 receptors, to trigger antiviral response through the induction of IFN type I and NF-κB inflammatory signalling (Fig. 5) (Belgnaoui et al. 2011; Sun et al. 2016).
Recent experimental results have implicated the nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3) inflammasome in CoV induced immune response (Fig. 5). So, Shi et al. (2019a) showed that SARS-CoV open reading frame (ORF) 8b, which is a type of accessory proteins, activates the NLRP3 inflammasome through the Leucine Rich Repeat domain of NLRP3 and ASC (apoptosis-associated speck-like protein) to mediate endoplasmic stress and dysfunctional innate immune response in macrophages. Consistently with the functions of NLRP3 inflammasome activation (Welcome 2020c), ORF8b reportedly induced pyroptosis (a form of cell death) in the macrophages (Shi et al. 2019a).
Data also indicate that SARS-CoV moderately upregulates NF-κB and B-cell lymphoma 3-encoded protein (bcl-3) (Reghunathan et al. 2005), a proto-oncogene involved in regulation of cell cycle and granulopoiesis (Kreisel et al. 2011). Interestingly, bcl-3 also interacts with NF-κB (Mansour et al. 2015), Stat3 (Zhao et al. 2016) and peroxisome proliferator-activated receptor gamma (Yang et al. 2009), which are implicated in the regulation of neuroinflammatory responses (Mrak and Landreth 2004; Borlongan et al. 2012; Bai et al. 2016) or blood brain barrier disruption (Borlongan et al. 2012; Bai et al. 2016; Nishikawa et al. 2018). Recent investigation indicates that SARS-CoV may share similar inflammatory mechanisms with SARS-CoV-2, suggesting that these inflammatory and apoptotic signalling pathways may serve as critical treatment targets in COVID-19 (Fu et al. 2020). Thus, anti-inflammatory agents that inhibit the activation of NF-κB or other key inflammatory signalling pathways may be harnessed for potential treatment of SARS-CoV-2 infection. These anti-inflammatory agents include, but are not limited to, doxycycline (NCT04371952, NCT04407130, https://www.clinicaltrials.gov), minocycline (IRCT20081019001369N4, https://www.irct.ir) (Sun et al. 2015), azithromycin (NCT04332107, NCT04359316) (Stellari et al. 2014; Haydar et al. 2019), fluoxetine (NCT04377308), vinpocetine (NCT02878772), ezetimibe (NCT01370603), atorvastatin (NCT04380402), simvastatin (NCT01424891, NCT04348695), statin (NCT04407273), lenalidomide (NCT04361643), zinc and vitamin D3 (NCT03005496), N-acetylcysteine (NCT04374461), and dexamethasone (NCT04347980, NCT04327401, NCT04395105) (https://www.clinicaltrials.gov).
Complement protein hyperactivation may drive the pathological sequelae of SARS-CoV-2 induced CNS damage
The complement system is a critical part of the host defense system that functions in concert with PRRs to neutralize microbial (including viral) invasion through the activation of adaptive immune response (Gralinski et al. 2018). In a study conducted by Gralinski et al. (2018), it was demonstrated that intranasal SARS-CoV infection of C57BL/6J mice resulted in considerable activation of the complement system beginning from day 1 of inoculation along with inflammatory cytokines/chemokines and immune system dysregulation in the lung (Gralinski et al. 2018). In contrast, C3 deficient mice infected with SARS-CoV had substantially reduced pulmonary impairment with decreased expression of proinflammatory cytokines/chemokines, infiltration of the lungs by neutrophils and monocytes despite similar pulmonary viral load compared with the wild type mice (Gralinski et al. 2018). Previous investigation has also implicated this central component of the complement cascade in SARS-CoV induced innate immune dysregulation (Reghunathan et al. 2005). Accumulating evidence indicates that disordered activation of the complement proteins can trigger neuroinflammation and prime the blood brain barrier for destructive processes (Orsini et al. 2014; Magdalon et al. 2020). Some pathogens can induce the stimulation of the G-protein coupled receptor, C5a receptor 1 (C5aR1), to initiate inflammatory processes in brain (Herrmann et al. 2018). Indeed, the complement protein C5a activates its cognate receptor (C5aR1) to cause hyperpermeability of the blood brain barrier in neuroinflammatory disorders, in part, by interacting with the NF-κB and microRNA (Jacob and Alexander 2014). Interestingly, inhibition of C5aR1 was reportedly shown to confer a considerable level of protection in brain disorders involving neuroinflammatory responses and blood brain barrier disruption (Jacob and Alexander 2014). Jiang et al. (2019) reported complement overactivation in macrophages, which resulted in immunopathology with overexpression of caspase-1 and IL-1β levels in the serum, and subsequently, pyroptosis of these cells in a mouse model of MERS-CoV infection (Jiang et al. 2019). Notwithstanding, treatment with anti-C5aR1 antibody effectively suppressed the expression of caspase-1 and IL-1β, to abrogate the immunopathology and pyroptosis of the macrophages, suggesting that C5a/C5aR1 signalling pathway constitutes a potential target for the treatment of SARS-CoV-2 infection (Jiang et al. 2019). Comparable findings were reported by Jiang et al. (2018). Inhibition of the complement proteins or their receptors is currently considered as a novel treatment option in many neuropathological diseases (Brennan et al. 2016; Carpanini et al. 2019). Importantly, some clinical trials on SARS-CoV-2 also target the complement proteins (Table 1).
Fas and other apoptotic signalling pathways
Fas (CD95) signalling is a pathway that involves interaction between the type II transmembrane Fas ligand (FasL) and the membrane spanning Fas receptor (FasR) that leads to apoptosis in activated cells (Fig. 6) (Marfè et al. 2011). Upon activation of FasL/FasR, the adaptor Fas-associated death domain (FADD) is recruited to activate caspases-8/10, forming the ‘death-receptor complex’, which contains procaspases (Fig. 6). The procaspases pass through a series of cleavage reactions that generate active caspases, which in turn mediate the generation of caspases-3/6/7, ultimately culminating in apoptosis (Fig. 6) (Ye et al. 2020). Apart from the FasL/FasR, the TNF/TNFR (tumor necrosis factor receptor)/TNFR-associated death domain (TRADD) signalling has also been implicated in apoptosis (Fig. 6) (Ye et al. 2020). Again, IFN-α/β/γ can induce inflammatory reactions that mobilize the Fas/FasL or TNF-related apoptosis-inducing ligand (TRAIL)/DR5 (death receptor 5) to cause apoptotic cell death (Fig. 6) (Ye et al. 2020). Furthermore, Xiong et al. (2020) reported upregulation of p53 pathway with resultant apoptotic events in monocytes and lymphocytes of COVID-19 patients, suggesting the involvement of multiple pathways in SARS-CoV-2 induced cell-death signalling (Xiong et al. 2020). Experimental results suggest that the mouse hepatitis virus (also known as murine CoV, and closely related to the human CoV-OC43) that causes hepatitis and CNS pathology in mice activates the Fas signalling pathway through the recruitment of Fas, FADD and procaspase-8 to form the ‘death-receptor complex’ (Liu and Zhang 2007). Through a series of cleavage reactions, this complex activates apoptotic cell death in rat oligodendrocytes (Liu and Zhang 2007). However, treatment with the Fas/FasL blocker, Fas/Fc chimera, effectively suppressed apoptosis by inhibiting the formation of the ‘death-receptor complex’, at least in part, via repression of caspase-8 activation in murine CoV-infected oligodendrocytes (Liu and Zhang 2007). Marfè et al. (2011) also studied the effects of canine CoV type II in A-72 canine cell line and showed that CoV induces substantial increase in TRAIL expression along with up-regulation of Fas/FasL (Marfè et al. 2011). Concomitantly, there was an increase in bax (Bcl-2-associated X) and a decrease in bcl-2 protein expression, indicating a proapoptotic response (Marfè et al. 2011). Though the mechanisms are yet to be completely understood, further research revealed that the effects of canine CoV type II infection on cell death signalling was associated with modulation of sirtuin and forkhead box O (Marfè et al. 2011). Evidence also suggests that mitochondrial metabolic dysregulation resulting to elevated ROS can trigger the cytochrome c activities to activate caspase-3/9, which in turn initiates the proapoptotic response in SARS-CoV (Wong et al. 2005; Zhang et al. 2007). In an in vitro model using SARS-CoV-infected Vero cell line, Ren et al. (2005) showed caspase 3 activation, bcl-2 downregulation, and bax upregulation, which ultimately resulted to apoptosis of the cells (Ren et al. 2005). Though the exact mechanisms are not completely known, MAVS is speculated to play a role in regulation of apoptotic events and metabolic activities by engaging multiple signalling pathways that link the mitochondria with endoplasmic reticulum, peroxisomes and autophagosomes (Belgnaoui et al. 2011). Further investigation is required to unravel specific molecular mechanisms involved in translating pathogenic signals from CoV to cytosolic metabolic and stress sensors such as the sirtuins and forkhead box O that modulate cellular responses to inflammation, immune and metabolic dysregulation.

Fas signalling in SARS-CoV-2 infection. Atg5 autophagy 5 protein, bax Bcl-2-associated X protein, bcl-2 B-cell lymphoma 2-encoded protein, CD cluster of differentiation, FADD Fas-associated death domain, Fas FS-7-associated surface antigen, FasL Fas ligand, FasR Fas receptor, NK natural killer, SARS-CoV-2 severe acute respiratory syndrome coronavirus 2, TNFR tumor necrosis factor receptor, TRAIL TNF(tumor necrosis factor)-related apoptosis-inducing ligand (see explanation in text)
Autophagy signalling
Autophagy refers to a process of self-degradation of cellular constituents, characterized by sequestration of cell organelles or cytosolic components into autophagosomes that merge with lysosomes for degradation (He and Klionsky 2009). The process is usually triggered by inflammatory, oxidative, and endoplasmic reticulum stress, as well as pathogenic invasion (Kolattukudy and Niu 2012; Trachsel-Moncho et al. 2018). Research data have implicated multiple signalling pathways in the initiation of autophagy (Vergne et al. 2009; Li et al. 2015; Zeng and Ju 2018; Rahman et al. 2020). Though autophagy is a survival mechanism essential in clearing worn out components of the cell (Glick et al. 2010), defective autophagy signalling plays an important role in the pathogenesis of several diseases including infectious and inflammatory diseases of the CNS (He and Klionsky 2009). While the mechanisms are not clearly understood, data (Maier and Britton 2012; Shi et al. 2019a) indicate that the cytotoxic effects exerted by SARS-CoV on the host cells not only cause dysfunctional activation of the innate immune system, but also, aberrant activation of the autophagy signalling pathway. Indeed Shi et al. (2019a, b) demonstrated that aggregation of SARS-CoV ORF8b activates endoplasmic reticulum stress response, lysosomal dysfunction and the transcription factor EB, a master regulator of autophagy and lysosomal biogenesis (Shi et al. 2019a). Similarly, Liu et al. (2014) showed that SARS-CoV ORF-9b interacts with the MAVS through a series of signalling reactions that trigger the degradation of MAVS and TNF receptor associated factor 3/6 by ubiquitination with the involvement of proteasomes, thereby reducing the host cell IFN responses. Importantly, the authors also demonstrated that increase in ORF-9b expression resulted in autophagy induction, which was dependent on expression of Atg5 (autophagy 5 protein) (Shi et al. 2014), a protein that plays a central role in cell death by interacting with FADD (Fig. 6) (Pyo et al. 2005). MAVS activation has been consistently known to play an integral role in autophagy (Belgnaoui et al. 2011; Sun et al. 2016; Shi et al. 2019b). Available evidence also suggests that CoV infected cells, especially during viral replication, generate autophagosomes with increased expression of the microtubule associated protein 1A/1B light chain 3 (LC3)—a central autophagy protein involved in the biogenesis of autophagosomes (Klionsky et al. 2012; Maier and Britton 2012). Importantly, Sun et al. (2016) demonstrated that MAVS directly interacts with LC3 to trigger autophagy, suggesting that MAVS may serve as a major receptor during autophagy induction (Sun et al. 2016). Interestingly, Xiong et al. (2020) found upregulation of autophagy in monocytes, T-, NK- and B cells of SARS-CoV-2 infected patients (Xiong et al. 2020). These data suggest that defective autophagy signalling due to SARS-CoV-2 infection may promote uncontrolled immune cell death, and provide information for further research that may unravel possible mechanisms involved in SARS-CoV-2 induced leukopenia and thrombocytopenia, and identification of effective treatment options for SARS-CoV-2 infection.
Effects of ACE2 downregulation by SARS-CoV-2 neuroinvasion
Downregulation of ACE2 by CoV facilitates cellular and tissue injury (Li 2015), which further worsens the neuroinflammatory responses, oxidative stress and cerebrovascular endothelial impairment (Peña-Silva et al. 2012; Zhang et al. 2020a, b). The neuroinflammatory responses are characterized by increased macrophage infiltration with continued production of proinflammatory cytokines and chemokines (Oudit et al. 2009). However, the proinflammatory cytokines and chemokines may be transported into the brain via the circulatory system (from other sites of viral invasion—lungs, gut etc.), which further worsen blood brain barrier dysfunction or inflammatory processes in the cerebral microenvironment (Welcome 2020b). SARS-CoV invasion with downregulation of ACE2 has been reportedly associated with inflammation in postmortem tissues (Oudit et al. 2009). ACE2 downregulation due to SARS-CoV infection reportedly reversed the protective role of ACE2 overexpression not only in peripheral tissues, but also, CNS (Xia and Lazartigues 2008). Cerebral ACE2 overexpression was demonstrated to substantially reduce cyclooxygenase (COX)-1 or COX-2-mediated neuroinflammation and oxidative stress, thereby resulting in an improvement in the antioxidant status and nitric oxide homeostasis (Sriramula et al. 2015). Thus, pharmacological activation of ACE2 may confer substantial protection against the neuropathological sequelae of SARS-CoV-2 infection. Pharmacological ACE2 activators, which include diminazene aceturate (Hernández Prada et al. 2008; Shenoy et al. 2013; Haber et al. 2014), resorcinolnaphthalein and xanthenone (Hernández Prada et al. 2008; Haber et al. 2014) have been demonstrated to confer anti-inflammatory and vasoprotective effects, and restoration of endothelial dysfunction in animal models. However, their roles in attenuating inflammatory responses and endothelial dysfunction have not been established in humans. Two clinical trials in Europe are assessing potential use of recombinant human ACE2 (EUCTR2020-001172-15-AT & EUCTR2020-001172-15-DK, https://www.clinicaltrialsregister.eu) as a treatment option in COVID-19 patients.
Factors determining the severity of SARS-CoV-2 infection
The infectivity of SARS-CoV-2 depends on a range of factors including genetic polymorphisms of molecules implicated in immune response (Conti et al. 2020), viral load (Liu et al. 2020d; Zheng et al. 2020), presence of chronic illnesses (Li et al. 2020c), ageing, development, and lifestyle such as smoking and diet (Li et al. 2020c).
Research has shown that certain polymorphisms of the HLA gene either promote or reduce susceptibility to SARS-CoV infection (Yuan et al. 2014). In particular, MBL gene polymorphisms have been reportedly demonstrated to be associated with risk of SARS-CoV infection (MBL is a component of the innate immune system, whose activation can lead to complement protein C3 and C4 cleavage, required for opsonization and phagocytosis) (Zhang et al. 2005). Similar data were reported by Tu et al. (2015), who revealed an association between the MBL B allele and increased risk of SARS-CoV infection in a Chinese population of 932 patients with SARS-CoV. Notably, Tu et al. (2015) also showed that the G-2518A polymorphism at the CCL2 gene can potentially increase the susceptibility to SARS-CoV infection (Tu et al. 2015). Furthermore, MBL B allele and CCL2 G-2518A polymorphisms had a cumulative effect on SARS-CoV infection (Tu et al. 2015) that may, at least in part, explain why some individuals with multiple susceptible polymorphisms may develop serious complications associated with SARS-CoV-2 infection. More so, polymorphism of CLEC4M (type II transmembrane C-type lectin domain family 4 member M), a putative receptor for CoV is also associated with risk of SARS-CoV infection (Li et al. 2008b).
Importantly, ACE2 polymorphisms have been associated with susceptibility to SARS-CoV-2 infection, multi-organ dysfunctions, and COVID-19 outcome (Devaux et al. 2020). In contrast to the results of previous research (Chiu et al. 2004), recent experimental findings indicate that certain variants of the human ACE2 can increase the susceptibility to SARS-CoV-2 infection, whereas other ACE2 variants confer protection against SARS-CoV-2 invasion (Stawiski et al. 2020). Indeed, the ACE2 receptors on the surface of endothelial cells of blood vessels (Hamming et al. 2004) provide crucial pathway for SARS-CoV-2 entry, and may be responsible for the ‘endotheliopathic’ effects of SARS-CoV-2 on organ damage at a very early stage of infection. SARS-CoV-2 invasion of endothelial cells can cause inflammation of the endothelium (endotheliitis) and may also be responsible for CoV-associated disorders such as disseminated intravascular coagulopathy, and the recently identified pediatric multisystem inflammatory syndrome (Varga et al. 2020). Interestingly, in vitro experiment using engineered human blood vessel organoids and in vivo data from COVID-19 patients have confirmed the involvement of endothelial cells in the pathogenesis of COVID-19 (Varga et al. 2020). These data suggest that COVID-19 associated cerebrovascular diseases such as cerebral thrombosis, ischaemic and haemorrhagic stroke, at least in part, may be due to endotheliitis triggered by SARS-CoV-2 invasion of the endothelial cells. Though the mechanism of dysregulation of vascular endothelial cell ACE2 that resultsin organ dysfunctions is not clearly understood, ACE2 dysregulation can potentially cause calcium dyshomeostasis and metabolic derangement, activating platelet aggregation, concomitantly with increased cell permeability and inflammatory responses as well as oxidative stress that trigger a series of pathophysiological processes, culminating in blood brain barrier disruption, neuroinflammation, and neurodegeneration (Pierce et al. 2017). Thus, endothelial cells are critical to several aspects of organ and system malfunctions, including dysregulation of immune cell trafficking across the endothelium in SARS-CoV-2 infection.
In a relatively recent report, Wang and colleagues revealed that elevated glycosylated hemoglobin (HbA1c) level is associated with higher mortality in COVID-19 patients (Wang et al. 2020d). Indeed, HbA1c is associated with systemic inflammation, hypercoagulability, and low arterial oxygen saturation in COVID-19 (Wang et al. 2020d). Furthermore, Wang et al. (2020d) reported a positive correlation between HbA1c and CRP levels (Wang et al. 2020d). Guo et al. (2020) also found that COVID-19 patients with diabetes, without evidence of comorbidity, were at higher risk of excessive inflammation (due to uncontrolled secretion of IL-6 and CRP) and hypercoagulable states. Surprisingly, the inflammatory responses in diabetes itself have been shown to also predispose to blood brain barrier impairment and neuroinflammation. Evidence from animal experimental indicates that diabetes causes blood–brain barrier dysfunction, neuroinflammation and neurodegeneration (Srodulski et al. 2014; Takechi et al. 2017). Similar findings are reported elsewhere (Brook et al. 2019). These data may explain why the severity of COVID-19 is higher in diabetic patients compared to those without diabetes. Therefore, HbA1c level on admission may be essential is determining the severity of COVID-19. Consequently, reduction in HbA1c level with pharmacological agents such as sevelamer (NCT01493050, https://www.clinicaltrials.gov) (Vlassara et al. 2012) and vitamin C (Kotb and Azzam 2015) may provide health benefits to diabetic patients with COVID-19.
The neuroimmnue dysregulation in SARS-CoV-2 infection is also dependent on the viral load and the severity of COVID-19 (Liu et al. 2020d; Zheng et al. 2020). The effect of chronic diseases, ageing, development, and lifestyle pattern (smoking and diet) on the severity of SARS-CoV-2 infection has been discussed (Li et al. 2020c).
Conclusion
In addition to the pathological impact on the respiratory and gastrointestinal tract, SARS-CoV-2 infection considerably affects CNS functions through the humoral and neural pathways due to the widespread cerebral expression of ACE2 and proteases essential for the viral invasion. SARS-CoV-2 induced cerebral damage is mainly characterized by neuroinflammation and blood brain barrier disruption due to replication and accumulation of viral RNA, its antigenic peptides, downregulation of ACE2, increased activity of the transmembrane protease serine 2, and cathepsin L. These changes favor upregulation of proinflammatory cytokines/chemokines, generation of reactive oxygen species, downregulation of antioxidant defense system, and hormonal/neurotransmitter dysregulation, which collectively underlie the neuropathologic sequelae of SARS-CoV-2 neuroinvasion. The viral RNA or antigenic peptides activate or interact with molecular signalling pathways mediated by pattern recognition receptors (e.g., toll-like receptors), nuclear factor kappa B, Janus kinase/signal transducer and activator of transcription, and complement cascades, as well as cell suicide molecules, which constitute key molecular targets for potential treatment of COVID-19. The spate of emerging therapeutics for SARS-CoV-2 infection (including associated CNS impairment) provides hope in the fight against COVID-19.
References
Ahmed SF, Quadeer AA, McKay MR (2020) Preliminary identification of potential vaccine targets for the COVID-19 Coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses. https://doi.org/10.3390/v12030254
Al Saiegh F, Ghosh R, Leibold A, Avery MB, Schmidt RF, Theofanis T, Mouchtouris N, Philipp L, Peiper SC, Wang Z-X, Rincon F, Tjoumakaris SI, Jabbour P, Rosenwasser RH (2020) Status of SARS-CoV-2 in cerebrospinal fluid of patients with COVID-19 and stroke. J Neurol Neurosurg Psychiatry. https://doi.org/10.1136/jnnp-2020-323522
Andries K, Pensaert MB (1980) Immunofluorescence studies on the pathogenesis of hemagglutinating encephalomyelitis virus infection in pigs after oronasal inoculation. Am J Vet Res 41:1372–1378
Arabi YM, Harthi A, Hussein J et al (2015) Severe neurologic syndrome associated with Middle East respiratory syndrome corona virus (MERS-CoV). Infection. https://doi.org/10.1007/s15010-015-0720-y
Argaw AT, Gurfein BT, Zhang Y et al (2009) VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.0808698106
Aschermann S, Lux A, Baerenwaldt A et al (2010) The other side of immunoglobulin G: suppressor of inflammation. Clin Exp Immunol 160:161–167
Bai Y, Zhang Y, Hua J et al (2016) Silencing microRNA-143 protects the integrity of the blood-brain barrier: implications for methamphetamine abuse. Sci Rep. https://doi.org/10.1038/srep35642
Baldwin R, di Mauro BW (2020) Economics in the time of COVID-19. CEPR Press, London
Bartlam M, Yang H, Rao Z (2005) Structural insights into SARS coronavirus proteins. Curr Opin Struct Biol 15:664–672
Belgnaoui SM, Paz S, Hiscott J (2011) Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (MAVS) adapter. Curr Opin Immunol 23:564–572
Biesmans S, Meert TF, Bouwknecht JA et al (2013) Systemic immune activation leads to neuroinflammation and sickness behavior in mice. Mediat Inflamm. https://doi.org/10.1155/2013/271359
Biron CA (2018) Glucocorticoids and NK cell PD-1. Nat Immunol 19(9):908–910
Borlongan VC, Glover EL, Sanberg RP, Hess CD (2012) Permeating the blood brain barrier and abrogating the inflammation in stroke: implications for stroke therapy. Curr Pharm Des. https://doi.org/10.2174/138161212802002841
Brennan FH, Lee JD, Ruitenberg MJ, Woodruff TM (2016) Therapeutic targeting of complement to modify disease course and improve outcomes in neurological conditions. Semin Immunol 28:292–308
Brook E, Mamo J, Wong R et al (2019) Blood–brain barrier disturbances in diabetes-associated dementia: therapeutic potential for cannabinoids. Pharmacol Res 141:291–297
Butowt R, Bilinska K (2020) SARS-CoV-2: olfaction, brain infection, and the urgent need for clinical samples allowing earlier virus detection. ACS Chem Neurosci. https://doi.org/10.1021/acschemneuro.0c00172
Carpanini SM, Torvell M, Morgan BP (2019) Therapeutic inhibition of the complement system in diseases of the central nervous system. Front Immunol 10:362
Channappanavar R, Perlman S (2017) Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol 39:529–539
Channappanavar R, Zhao J, Perlman S (2014) T cell-mediated immune response to respiratory coronaviruses. Immunol Res 59:118–128
Chen G, Chen DZ, Li J et al (2004) Pathogenic role of HMGB1 in SARS? Med Hypotheses. https://doi.org/10.1016/j.mehy.2004.01.037
Chen J, Lau YF, Lamirande EW et al (2010) Cellular immune responses to severe acute respiratory syndrome coronavirus (SARS-CoV) infection in senescent BALB/c mice: CD4+ T cells are important in control of SARS-CoV infection. J Virol. https://doi.org/10.1128/jvi.01281-09
Chen R, Wang K, Yu J et al (2020c) The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in human and mouse brain. bioRxiv. https://doi.org/10.1101/2020.04.07.030650
Chen N, Zhou M, Dong X et al (2020b) Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. https://doi.org/10.1016/S0140-6736(20)30211-7
Chen C, Zhou Y, Wang DW (2020a) SARS-CoV-2: a potential novel etiology of fulminant myocarditis. Herz 45:230–232
Chiu RWK, Tang NLS, Hui DSC et al (2004) ACE2 gene polymorphisms do not affect outcome of severe acute respiratory syndrome. Clin Chem. https://doi.org/10.1373/clinchem.2004.035436
Conti P, Ronconi G, Caraffa A et al (2020) Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by COVID-19: anti-inflammatory strategies. J Biol Regul Homeost Agents 34:1
Copf T, Goguel V, Lampin-Saint-Amaux A et al (2011) Cytokine signaling through the JAK/STAT pathway is required for long-term memory in Drosophila. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1012919108
Curtin KD, Wyman RJ, Meinertzhagen IA (2007) Basigin/EMMPRIN/CD147 mediates neuron–glia interactions in the optic lamina of Drosophila. Glia. https://doi.org/10.1002/glia.20568
De Wit E, Van Doremalen N, Falzarano D, Munster VJ (2016) SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol 14:523–534
Deng X, van Geelen A, Buckley AC et al (2019) Coronavirus endoribonuclease activity in porcine epidemic diarrhea virus suppresses Type I and Type III interferon responses. J Virol. https://doi.org/10.1128/jvi.02000-18
Deng Y, Liu W, Liu K et al (2020) Clinical characteristics of fatal and recovered cases of coronavirus disease 2019 (COVID-19) in Wuhan, China: a retrospective study. Chin Med J (Engl). https://doi.org/10.1097/CM9.0000000000000824
Dessau RB, Lisby G, Frederiksen JL (2001) Coronaviruses in brain tissue from patients with multiple sclerosis. Acta Neuropathol. https://doi.org/10.1007/s004010000331
Dessau RB, Lisby G, Frederiksen JL (2009) Coronaviruses in spinal fluid of patients with acute monosymptomatic optic neuritis. Acta Neurol Scand. https://doi.org/10.1111/j.1600-0404.1999.tb01043.x
Devaux CA, Rolain JM, Raoult D (2020) ACE2 receptor polymorphism: susceptibility to SARS-CoV-2, hypertension, multi-organ failure, and COVID-19 disease outcome. J Microbiol Immunol Infect 53:425–435
Diao B, Wang C, Tan Y et al (2020) Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). medRxiv. https://doi.org/10.1101/2020.02.18.20024364
Doobay MF, Talman LS, Obr TD et al (2007) Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin–angiotensin system. Am J Physiol Regul Integr Comp Physiol. https://doi.org/10.1152/ajpregu.00292.2006
Du Y, Li W, Lin L et al (2019) Effects of lipocalin-2 on brain endothelial adhesion and permeability. PLoS ONE. https://doi.org/10.1371/journal.pone.0218965
Esper F, Weibel C, Ferguson D et al (2006) Coronavirus HKU1 infection in the United States. Emerg Infect Dis. https://doi.org/10.3201/eid1205.051316
Essali N, Goldsmith DR, Carbone L, Miller BJ (2019) Psychosis as an adverse effect of monoclonal antibody immunotherapy. Brain Behav Immun. https://doi.org/10.1016/j.bbi.2019.06.002
Fan YY, Huang ZT, Li L et al (2009) Characterization of SARS-CoV-specific memory T cells from recovered individuals 4 years after infection. Arch Virol. https://doi.org/10.1007/s00705-009-0409-6
Fazzini E, Fleming J, Fahn S (1992) Cerebrospinal fluid antibodies to coronavirus in patients with Parkinson’s disease. Mov Disord. https://doi.org/10.1002/mds.870070210
Fernandes N (2020) Economic effects of coronavirus outbreak (COVID-19) on the world economy. SSRN Electron J. https://doi.org/10.2139/ssrn.3557504
Ferreira AC, Dá Mesquita S, Sousa JC et al (2015) From the periphery to the brain: lipocalin-2, a friend or foe? Prog. Neurobiology 131:120–136
Franciotta D, Columba-Cabezas S, Andreoni L et al (2008) Oligoclonal IgG band patterns in inflammatory demyelinating human and mouse diseases. J Neuroimmunol. https://doi.org/10.1016/j.jneuroim.2008.06.004
Fu Y, Cheng Y, Wu Y (2020) Understanding SARS-CoV-2-mediated inflammatory responses: from mechanisms to potential therapeutic tools. Virol. Sin 35:266–271
Glick D, Barth S, Macleod KF (2010) Autophagy: cellular and molecular mechanisms. J Pathol 221:3–12
Glowacka I, Bertram S, Muller MA et al (2011) Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J Virol. https://doi.org/10.1128/jvi.02232-10
González JM, Gomez-Puertas P, Cavanagh D et al (2003) A comparative sequence analysis to revise the current taxonomy of the family Coronaviridae. Arch Virol. https://doi.org/10.1007/s00705-003-0162-1
Gordon S (2002) Pattern recognition receptors: doubling up for the innate immune response. Cell 111:927–930
Gralinski LE, Sheahan TP, Morrison TE et al (2018) Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. MBio. https://doi.org/10.1128/mBio.01753-18
Guan WJ, Ni ZY, Hu Y et al (2020) Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. https://doi.org/10.1056/NEJMoa2002032
Guidon AC, Amato AA (2020) COVID-19 and neuromuscular disorders. Neurology. https://doi.org/10.1212/wnl.0000000000009566
Guo W, Li M, Dong Y et al (2020) Diabetes is a risk factor for the progression and prognosis of COVID-19. Diabetes Metab Res Rev. https://doi.org/10.1002/dmrr.3319
Gutiérrez-Ortiz C, Méndez A, Rodrigo-Rey S et al (2020) Miller Fisher Syndrome and polyneuritis cranialis in COVID-19. Neurology. https://doi.org/10.1212/wnl.0000000000009619
Haber PK, Ye M, Wysocki J et al (2014) Angiotensin-converting enzyme 2-independent action of presumed angiotensin-converting enzyme 2 activators: studies in vivo, ex vivo, and in vitro. Hypertension. https://doi.org/10.1161/HYPERTENSIONAHA.113.02856
Hadziefendic S, Haxhiu MA (1999) CNS innervation of vagal preganglionic neurons controlling peripheral airways: a transneuronal labeling study using pseudorabies virus. J Auton Nerv Syst. https://doi.org/10.1016/S0165-1838(99)00020-X
Hamming I, Timens W, Bulthuis MLC et al (2004) Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. https://doi.org/10.1002/path.1570
Harrison DA (2012) The JAK/STAT pathway. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a011205
Haruwaka K, Ikegami A, Tachibana Y et al (2019) Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat Commun. https://doi.org/10.1038/s41467-019-13812-z
Haydar D, Cory TJ, Birket SE et al (2019) Azithromycin polarizes macrophages to an M2 phenotype via inhibition of the STAT1 and NF-κB signaling pathways. J Immunol. https://doi.org/10.4049/jimmunol.1801228
He C, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. https://doi.org/10.1146/annurev-genet-102808-114910
Helms J, Kremer S, Merdji H, Clere-Jehl R, Schenck M, Kummerlen C, Collange O, Boulay C, Fafi-Kremer S, Ohana M, Anheim M, Strasbourg FM (2020) Neurologic features in severe SARS-CoV-2 infection. N Engl J Med. https://doi.org/10.1056/NEJMc2008597
Hernández Prada JA, Ferreira AJ, Katovich MJ et al (2008) Structure-based identification of small-molecule angiotensin-converting enzyme 2 activators as novel antihypertensive agents. Hypertension. https://doi.org/10.1161/HYPERTENSIONAHA.107.108944
Herrmann JB, Muenstermann M, Strobel L et al (2018) Complement c5a receptor 1 exacerbates the pathophysiology of n. Meningitidis sepsis and is a potential target for disease treatment. MBio. https://doi.org/10.1128/mBio.01755-17
Hoffmann M, Kleine-Weber H, Schroeder S et al (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. https://doi.org/10.1016/j.cell.2020.02.052
Hook V, Funkelstein L, Wegrzyn J et al (2012) Cysteine Cathepsins in the secretory vesicle produce active peptides: Cathepsin L generates peptide neurotransmitters and cathepsin B produces beta-amyloid of Alzheimer’s disease. Biochim Biophys Acta Proteins Proteomics 1824:89–104
Hopcraft SE, Damania B (2017) Tumour viruses and innate immunity. Philos Trans R Soc B Biol Sci 372:20160267
Horowitz RI, Freeman PR, Bruzzese J (2020) Efficacy of glutathione therapy in relieving dyspnea associated with COVID-19 pneumonia: a report of 2 cases. Respir Med Case Rep. https://doi.org/10.1016/j.rmcr.2020.101063
Huang C, Wang Y, Li X et al (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. https://doi.org/10.1016/S0140-6736(20)30183-5
Hung ECW, Chim SSC, Chan PKS et al (2003) Detection of SARS coronavirus RNA in the cerebrospinal fluid of a patient with severe acute respiratory syndrome. Clin Chem 49:2108–2109
Iwata-Yoshikawa N, Okamura T, Shimizu Y et al (2019) TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection. J Virol. https://doi.org/10.1128/jvi.01815-18
Jacob A, Alexander JJ (2014) Complement and blood-brain barrier integrity. Mol Immunol 61:149–152
Jensen KK, Andreatta M, Marcatili P et al (2018) Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology. https://doi.org/10.1111/imm.12889
Jiang Y, Zhao G, Song N et al (2018) Blockade of the C5a–C5aR axis alleviates lung damage in hDPP4-transgenic mice infected with MERS-CoV article. Emerg Microbes Infect. https://doi.org/10.1038/s41426-018-0063-8
Jiang Y, Li J, Teng Y et al (2019) Complement receptor c5ar1 inhibition reduces pyroptosis in hdpp4-transgenic mice infected with mers-cov. Viruses. https://doi.org/10.3390/v11010039
Jin M, Kim JH, Jang E et al (2014) Lipocalin-2 deficiency attenuates neuroinflammation and brain injury after transient middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab. https://doi.org/10.1038/jcbfm.2014.83
John Hopkins University and Medicine (2020) COVID-19 Map—Johns Hopkins Coronavirus Resource Center. John Hopkins Coronavirus Resour. Cent
Jupatanakul N, Sim S, Angleró-Rodríguez YI et al (2017) Engineered Aedes aegypti JAK/STAT pathway-mediated immunity to dengue virus. PLoS Negl Trop Dis. https://doi.org/10.1371/journal.pntd.0005187
Kalia M, Mesulam M-M (1980) Brain stem projections of sensory and motor components of the vagus complex in the cat: II. Laryngeal, tracheobronchial, pulmonary, cardiac, and gastrointestinal branches. J Comp Neurol. https://doi.org/10.1002/cne.901930211
Kato H, Takeuchi O, Sato S et al (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. https://doi.org/10.1038/nature04734
Katz SL, Frank MM, Miletic VD, Jiang H (2000) Immunoglobulin in the control of complement action. Immunol Res 22:137–146
Kaufman GN, Massoud AH, Dembele M et al (2015) Induction of regulatory T cells by intravenous immunoglobulin: a bridge between adaptive and innate immunity. Front Immunol. https://doi.org/10.3389/fimmu.2015.00469
Kelu JJ, Webb SE, Galione A, Miller AL (2018) TPC2-mediated Ca2+ signaling is required for the establishment of synchronized activity in developing zebrafish primary motor neurons. Dev Biol. https://doi.org/10.1016/j.ydbio.2018.02.011
Kim JE, Heo JH, Kim HO et al (2017) Neurological complications during treatment of middle east respiratory syndrome. J Clin Neurol. https://doi.org/10.3988/jcn.2017.13.3.227
Klionsky DJ, Abdalla FC, Abeliovich H et al (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy
Kolattukudy PE, Niu J (2012) Inflammation, endoplasmic reticulum stress, autophagy, and the monocyte chemoattractant protein-1/CCR2 pathway. Circ Res 110:174–189
Kopecky-Bromberg SA, Martínez-Sobrido L, Frieman M et al (2007) Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J Virol. https://doi.org/10.1128/jvi.01782-06
Kotb A, Al Azzam KM (2015) Effect of vitamin C on blood glucose and glycosylated hemoglobin in type II diabetes mellitus. World J Anal Chem 3:6–8. https://doi.org/https://doi.org/10.12691/WJAC-3-1A-2
Kreisel D, Sugimoto S, Tietjens J et al (2011) Bcl3 prevents acute inflammatory lung injury in mice by restraining emergency granulopoiesis. J Clin Investig. https://doi.org/10.1172/JCI42596
Ksiazek TG, Erdman D, Goldsmith CS et al (2003) A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. https://doi.org/10.1056/NEJMoa030781
Kuba K, Imai Y, Ohto-Nakanishi T, Penninger JM (2010) Trilogy of ACE2: a peptidase in the renin-angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol Ther 128:119–128
Kubo H, Yamada YK, Taguchi F (1994) Localization of neutralizing epitopes and the receptor-binding site within the amino-terminal 330 amino acids of the murine coronavirus spike protein. J Virol. https://doi.org/10.1128/jvi.68.9.5403-5410.1994
Lan L, Xu D, Ye G et al (2020) Positive RT-PCR test results in patients recovered from COVID-19. JAMA J Am Med Assoc 323:1502
Lau KK, Yu WC, Chu CM et al (2004) Possible central nervous system infection by SARS coronavirus. Emerg Infect Dis. https://doi.org/10.3201/eid1002.030638
Law HKW, Cheung CY, Sia SF et al (2009) Toll-like receptors, chemokine receptors and death receptor ligands responses in SARS coronavirus infected human monocyte derived dendritic cells. BMC Immunol. https://doi.org/10.1186/1471-2172-10-35
Lefkowitz EJ, Dempsey DM, Hendrickson RC et al (2018) Virus taxonomy: the database of the international committee on taxonomy of viruses (ICTV). Nucleic Acids Res. https://doi.org/10.1093/nar/gkx932
Li F (2015) Receptor recognition mechanisms of coronaviruses: a decade of structural studies. J Virol. https://doi.org/10.1128/jvi.02615-14
Li CK, Wu H, Yan H et al (2008a) T cell responses to whole SARS coronavirus in humans. J Immunol. https://doi.org/10.4049/jimmunol.181.8.5490
Li H, Tang NLS, Chan PKS et al (2008b) Polymorphisms in the C-type lectin genes cluster in chromosome 19 and predisposition to severe acute respiratory syndrome coronavirus (SARS-CoV) infection. J Med Genet. https://doi.org/10.1136/jmg.2008.058966
Li H, Wei M, Li S et al (2013a) Increased CD147 and MMP-9 expression in the normal rat brain after gamma irradiation. J Radiat Res. https://doi.org/10.1093/jrr/rrs072
Li YC, Bai WZ, Hirano N et al (2013b) Neurotropic virus tracing suggests a membranous-coating-mediated mechanism for transsynaptic communication. J Comp Neurol. https://doi.org/10.1002/cne.23171
Li Y, Zhang L, Li K et al (2015) ZNF32 inhibits autophagy through the mTOR pathway and protects MCF-7 cells from stimulus-induced cell death. Sci Rep. https://doi.org/10.1038/srep09288
Li SW, Wang CY, Jou YJ et al (2016) SARS coronavirus papain-like protease inhibits the TLR7 signaling pathway through removing Lys63-linked polyubiquitination of TRAF3 and TRAF6. Int J Mol Sci. https://doi.org/10.3390/ijms17050678
Li Y, Li H, Fan R et al (2017) Coronavirus infections in the central nervous system and respiratory tract show distinct features in hospitalized children. Intervirology. https://doi.org/10.1159/000453066
Li YC, Bai WZ, Hashikawa T (2020d) The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J Med Virol 92:552–555
Li G, Fan Y, Lai Y et al (2020a) Coronavirus infections and immune responses. J Med Virol 92:424–432
Li X, Geng M, Peng Y et al (2020b) Molecular immune pathogenesis and diagnosis of COVID-19. J Pharm Anal 10:102–108
Li Y, Zhou W, Yang L, You R (2020c) Physiological and pathological regulation of ACE2, the SARS-CoV-2 receptor. Pharmacol Res 157:104833
Lin R-J, Liao C-L, Lin E, Lin Y-L (2004) Blocking of the alpha interferon-induced Jak-Stat signaling pathway by Japanese encephalitis virus infection. J Virol. https://doi.org/10.1128/jvi.78.17.9285-9294.2004
Link H, Huang YM (2006) Oligoclonal bands in multiple sclerosis cerebrospinal fluid: an update on methodology and clinical usefulness. J Neuroimmunol 180:17–28
Liu Y, Zhang X (2007) Murine coronavirus-induced oligodendrocyte apoptosis is mediated through the activation of the Fas signaling pathway. Virology. https://doi.org/10.1016/j.virol.2006.10.044
Liu DX, Fung TS, Chong KKL et al (2014) Accessory proteins of SARS-CoV and other coronaviruses. Antiviral Res 109:97–109
Liu Y, Yan LM, Wan L et al (2020d) Viral dynamics in mild and severe cases of COVID-19. Lancet Infect Dis 20:656–657
Liu W, Tao ZW, Lei W et al (2020c) Analysis of factors associated with disease outcomes in hospitalized patients with 2019 novel coronavirus disease. Chin Med J (Engl). https://doi.org/10.1097/CM9.0000000000000775
Liu J, Cao R, Xu M et al (2020b) Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov 6:1–4
Liu B, Li M, Zhou Z et al (2020a) Can we use interleukin-6 (IL-6) blockade for coronavirus disease 2019 (COVID-19)-induced cytokine release syndrome (CRS)? J Autoimmun
Llorens F, Hermann P, Villar-Piqué A et al (2020) Cerebrospinal fluid lipocalin 2 as a novel biomarker for the differential diagnosis of vascular dementia. Nat Commun. https://doi.org/10.1038/s41467-020-14373-2
Lu X, Pan J, Tao J, Guo D (2011) SARS-CoV nucleocapsid protein antagonizes IFN-β response by targeting initial step of IFN-β induction pathway, and its C-terminal region is critical for the antagonism. Virus Genes. https://doi.org/10.1007/s11262-010-0544-x
Ma DY, Suthar MS (2015) Mechanisms of innate immune evasion in re-emerging RNA viruses. Curr Opin Virol 12:26–37
Magdalon J, Mansur F, Teles e Silva AL et al (2020) Complement system in brain architecture and neurodevelopmental disorders. Front Neurosci
Maier HJ, Britton P (2012) Involvement of autophagy in coronavirus replication. Viruses 4:3440–3451
Mansour NM, Bernal GM, Wu L et al (2015) Decoy receptor DcR1 is induced in a p50/Bcl3-dependent manner and attenuates the efficacy of temozolomide. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-14-2144
Mao L, Jin H, Wang M et al (2020) Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. https://doi.org/10.1001/jamaneurol.2020.1127
Mao L, Wang M, Chen S et al (2020) Neurological manifestations of hospitalized patients with COVID-19 in Wuhan, China: a retrospective case series study. SSRN Electron J. https://doi.org/10.2139/ssrn.3544840
Marfè G, Tafani M, Fiorito F et al (2011) Involvement of FOXO transcription factors, TRAIL-FasL/Fas, and Sirtuin proteins family in canine coronavirus type II-induced apoptosis. PLoS ONE. https://doi.org/10.1371/journal.pone.0027313
Martin S, Harper CB, May LM et al (2013) Inhibition of PIKfyve by YM-201636 dysregulates autophagy and leads to apoptosis-independent neuronal cell death. PLoS ONE. https://doi.org/10.1371/journal.pone.0060152
Mateo M, Generous A, Sinn PL, Cattaneo R (2015) Connections matter—how viruses use cell–cell adhesion components. J Cell Sci 128:431–439
Matsuda K, Park CH, Sunden Y et al (2004) The vagus nerve is one route of transneural invasion for intranasally inoculated influenza A virus in mice. Vet Pathol. https://doi.org/10.1354/vp.41-2-101
McBride R, van Zyl M, Fielding BC (2014) The coronavirus nucleocapsid is a multifunctional protein. Viruses 6:2991–3018
Mengeling WL, Boothe AD, Ritchie AE (1972) Characteristics of a coronavirus (strain 67N) of pigs. Am J Vet Res
Menizibeya Osain Welcome NEM (2020) Stress-induced blood brain barrier disruption: molecular mechanisms and signaling pathways. Pharmacol Res 157:104769. https://doi.org/10.1016/j.phrs.2020.104769
Morfopoulou S, Brown JR, Davies EG et al (2016) Human coronavirus OC43 associated with fatal encephalitis. N Engl J Med 375:497–498
Moschen AR, Adolph TE, Gerner RR et al (2017) Lipocalin-2: a master mediator of intestinal and metabolic inflammation. Trends Endocrinol Metab 28:388–397
Mrak RE, Landreth GE (2004) PPARγ, neuroinflammation, and disease. J Neuroinflamm. https://doi.org/10.1186/1742-2094-1-5
Nelemans T, Kikkert M (2019) Viral innate immune evasion and the pathogenesis of emerging RNA virus infections. Viruses 11:961
Nelson EA, Dyall J, Hoenen T et al (2017) The phosphatidylinositol-3-phosphate 5-kinase inhibitor apilimod blocks filoviral entry and infection. PLoS Negl Trop Dis. https://doi.org/10.1371/journal.pntd.0005540
Ng SSM, Li A, Pavlakis GN et al (2013) Viral infection increases glucocorticoid-induced interleukin-10 production through ERK-mediated phosphorylation of the glucocorticoid receptor in dendritic cells: potential clinical implications. PLoS ONE. https://doi.org/10.1371/journal.pone.0063587
Ni J, Ma X, Zhou M et al (2013) Serum lipocalin-2 levels positively correlate with coronary artery disease and metabolic syndrome. Cardiovasc Diabetol. https://doi.org/10.1186/1475-2840-12-176
Nishikawa H, Liu L, Nakano F et al (2018) Modified citrus pectin prevents blood–brain barrier disruption in mouse Subarachnoid hemorrhage by inhibiting Galectin-3. Stroke. https://doi.org/10.1161/STROKEAHA.118.021757
Niu J, Shen L, Huang B et al (2020) Non-invasive bioluminescence imaging of HCoV-OC43 infection and therapy in the central nervous system of live mice. Antiviral Res. https://doi.org/10.1016/j.antiviral.2019.104646
Orsini F, De Blasio D, Zangari R et al (2014) Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front Cell Neurosci 8:380
Ou X, Liu Y, Lei X et al (2020) Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. https://doi.org/10.1038/s41467-020-15562-9
Oudit GY, Kassiri Z, Jiang C et al (2009) SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur J Clin Investig. https://doi.org/10.1111/j.1365-2362.2009.02153.x
Patel S (2015) Function and dysfunction of two-pore channels. Sci Signal 8:re7
Peña-Silva RA, Chu Y, Miller JD et al (2012) Impact of ACE2 deficiency and oxidative stress on cerebrovascular function with aging. Stroke. https://doi.org/10.1161/STROKEAHA.112.667063
Peng H, Yang LT, Wang LY et al (2006) Long-lived memory T lymphocyte responses against SARS coronavirus nucleocapsid protein in SARS-recovered patients. Virology. https://doi.org/10.1016/j.virol.2006.03.036
Perlman S, Netland J (2009) Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol 7:439–450
Phadwal K, Alegre-Abarrategui J, Watson AS et al (2012) A novel method for autophagy detection in primary cells: impaired levels of macroautophagy in immunosenescent T cells. Autophagy. https://doi.org/10.4161/auto.18935
Pierce RW, Giuliano JS, Pober JS (2017) Endothelial cell function and dysfunction in critically ill children. Pediatrics. https://doi.org/10.1542/peds.2017-0355
Pyo JO, Jang MH, Kwon YK et al (2005) Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem. https://doi.org/10.1074/jbc.M413934200
Qin H, Buckley JA, Li X et al (2016) Inhibition of the JAK/STAT pathway protects against α-synuclein-induced neuroinflammation and dopaminergic neurodegeneration. J Neurosci. https://doi.org/10.1523/JNEUROSCI.4658-15.2016
Qin C, Zhou L, Hu Z et al (2020) Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin Infect Dis. https://doi.org/10.1093/cid/ciaa248
Qiu S, Leung A, Bo Y et al (2018) Ebola virus requires phosphatidylinositol (3,5) bisphosphate production for efficient viral entry. Virology. https://doi.org/10.1016/j.virol.2017.09.028
Quinlan BD, Mou H, Zhang L et al (2020) The SARS-CoV-2 receptor-binding domain elicits a potent neutralizing response without antibody-dependent enhancement. SSRN Electron J. https://doi.org/10.2139/ssrn.3575134
Rahman MA, Saha SK, Rahman MS et al (2020) Molecular insights into therapeutic potential of autophagy modulation by natural products for cancer stem cells. Front Cell Dev Biol. https://doi.org/10.3389/fcell.2020.00283
Raux H, Flamand A, Blondel D (2000) Interaction of the rabies virus P protein with the LC8 dynein light chain. J Virol. https://doi.org/10.1128/jvi.74.21.10212-10216.2000
Rawlings ND, Salvesen G (2013) Handbook of proteolytic enzymes
Reghunathan R, Jayapal M, Hsu LY et al (2005) Expression profile of immune response genes in patients with severe acute respiratory syndrome. BMC Immunol. https://doi.org/10.1186/1471-2172-6-2
Ren L, Yang R, Guo L et al (2005) Apoptosis induced by the SARS-associated coronavirus in vero cells is replication-dependent and involves caspase. DNA Cell Biol. https://doi.org/10.1089/dna.2005.24.496
Richardson P, Griffin I, Tucker C et al (2020) Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 395:e30–e31
Salmi A, Ziola B, Hovi T, Reunanen M (1982) Antibodies to coronaviruses OC43 and 229E in multiple sclerosis patients. Neurology. https://doi.org/10.1212/wnl.32.3.292
Schneider WM, Chevillotte MD, Rice CM (2014) Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. https://doi.org/10.1146/annurev-immunol-032713-120231
Sharma HS, Feng L, Muresanu DF et al (2019) Neuroprotective effects of a potent bradykinin B2 receptor antagonist HOE-140 on microvascular permeability, blood flow disturbances, edema formation, cell injury and nitric oxide synthase upregulation following trauma to the spinal cord. In: International review of neurobiology
Shen C, Wang Z, Zhao F et al (2020) Treatment of 5 critically ill patients with COVID-19 with convalescent plasma. JAMA J Am Med Assoc. https://doi.org/10.1001/jama.2020.4783
Shenoy V, Gjymishka A, Jarajapu YP et al (2013) Diminazene attenuates pulmonary hypertension and improves angiogenic progenitor cell functions in experimental models. Am J Respir Crit Care Med. https://doi.org/10.1164/rccm.201205-0880OC
Shi C-S, Qi H-Y, Boularan C et al (2014) SARS-coronavirus open reading frame-9b Suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J Immunol. https://doi.org/10.4049/jimmunol.1303196
Shi CS, Nabar NR, Huang NN, Kehrl JH (2019) SARS-Coronavirus open reading frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. https://doi.org/10.1038/s41420-019-0181-7
Shi Y, Liu Y, Zheng Y et al (2019) Autophagy triggered by MAVS inhibits Coxsackievirus A16 replication. Acta Virol. https://doi.org/10.4149/av_2019_403
Singhal T (2020) A review of coronavirus disease-2019 (COVID-19). Indian J Pediatr 87:281–286
Sirbu CA, Budisteanu M (2020) Monoclonal antibodies—a revolutionary therapy in multiple sclerosis. Neurol Neurochir Pol 54:21–27. https://doi.org/10.5603/PJNNS.a2020.0008
Spiegel M, Pichlmair A, Martínez-Sobrido L et al (2005) Inhibition of beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J Virol. https://doi.org/10.1128/jvi.79.4.2079-2086.2005
Sriramula S, Xia H, Xu P, Lazartigues E (2015) Brain-targeted angiotensin-converting enzyme 2 overexpression attenuates neurogenic hypertension by inhibiting cyclooxygenase-mediated inflammation. Hypertension. https://doi.org/10.1161/HYPERTENSIONAHA.114.04691
Srodulski S, Sharma S, Bachstetter AB et al (2014) Neuroinflammation and neurologic deficits in diabetes linked to brain accumulation of amylin. Mol Neurodegener. https://doi.org/10.1186/1750-1326-9-30
Stawiski EW, Diwanji D, Suryamohan K et al (2020) Human ACE2 receptor polymorphisms predict SARS-CoV-2 susceptibility. bioRxiv. https://doi.org/10.1101/2020.04.07.024752
Stebbing J, Phelan A, Griffin I et al (2020) COVID-19: combining antiviral and anti-inflammatory treatments. Lancet Infect Dis 20:400–402
Stellari FF, Sala A, Donofrio G et al (2014) Azithromycin inhibits nuclear factor-κB activation during lung inflammation: an in vivo imaging study. Pharmacol Res Perspect. https://doi.org/10.1002/prp2.58
Sun J, Shigemi H, Tanaka Y et al (2015) Tetracyclines downregulate the production of LPS-induced cytokines and chemokines in THP-1 cells via ERK, p38, and nuclear factor-κB signaling pathways. Biochem Biophys Rep. https://doi.org/10.1016/j.bbrep.2015.11.003
Sun X, Sun L, Zhao Y et al (2016) MAVS maintains mitochondrial homeostasis via autophagy. Cell Discov. https://doi.org/10.1038/celldisc.2016.24
Sun D, Li H, Lu XX et al (2020) Clinical features of severe pediatric patients with coronavirus disease 2019 in Wuhan: a single center’s observational study. World J Pediatr. https://doi.org/10.1007/s12519-020-00354-4
Sungnak W, Huang N, Bécavin C et al (2020) SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med. https://doi.org/10.1038/s41591-020-0868-6
Takechi R, Lam V, Brook E et al (2017) Blood–brain barrier dysfunction precedes cognitive decline and neurodegeneration in diabetic insulin resistant mouse model: an implication for causal link. Front Aging Neurosci. https://doi.org/10.3389/fnagi.2017.00399
Takeshita Y (2012) Inflammatory cell trafficking across the blood–brain barrier (BBB): Chemokine regulation and in vitro models. Immunol Rev 248:228–239. https://doi.org/10.1111/j.1600-065X.2012.01127.x
Trachsel-Moncho L, Benlloch-Navarro S, Fernández-Carbonell Á et al (2018) Oxidative stress and autophagy-related changes during retinal degeneration and development. Cell Death Dis. https://doi.org/10.1038/s41419-018-0855-8
Tu X, Chong WP, Zhai Y et al (2015) Functional polymorphisms of the CCL2 and MBL genes cumulatively increase susceptibility to severe acute respiratory syndrome coronavirus infection. J Infect. https://doi.org/10.1016/j.jinf.2015.03.006
Ubuka T, Moriya S, Soga T, Parhar I (2018) Identification of transmembrane protease serine 2 and forkhead box a1 as the potential bisphenol a responsive genes in the neonatal male rat brain. Front Endocrinol (Lausanne). https://doi.org/10.3389/fendo.2018.00139
Valencia DN (2020) Brief review on COVID-19: the 2020 pandemic caused by SARS-CoV-2. Cureus. https://doi.org/10.7759/cureus.7386
van de Veerdonk FL, Netea MG, van Deuren M et al (2020) Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome. Elife. https://doi.org/10.7554/eLife.57555
Varga Z, Flammer AJ, Steiger P et al (2020) Endothelial cell infection and endotheliitis in COVID-19. Lancet 395:1417–1418
Vergne I, Roberts E, Elmaoued RA et al (2009) Control of autophagy initiation by phosphoinositide 3-phosphatase jumpy. EMBO J. https://doi.org/10.1038/emboj.2009.159
Vlassara H, Uribarri J, Cai W et al (2012) Effects of sevelamer on HbA1c, inflammation, and advanced glycation end products in diabetic kidney disease. Clin J Am Soc Nephrol. https://doi.org/10.2215/CJN.12891211
Wang H, Ward MF, Fan XG et al (2006) Potential role of high mobility group box 1 in viral infectious diseases. Viral Immunol 19:3–9
Wang H, Yang P, Liu K et al (2008) SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. https://doi.org/10.1038/cr.2008.15
Wang Z, Yang B, Li Q et al (2020) Clinical features of 69 cases with coronavirus disease 2019 in Wuhan, China. Clin Infect Dis. https://doi.org/10.1093/cid/ciaa272
Wang W, He J, Lie P et al (2020) The definition and risks of cytokine release syndrome-like in 11 COVID-19-infected pneumonia critically ill patients: disease characteristics and retrospective analysis. medRxiv. https://doi.org/10.1101/2020.02.26.20026989
Wang Z, Du Z, Zhu F (2020) Glycosylated hemoglobin is associated with systemic inflammation, hypercoagulability, and prognosis of COVID-19 patients. Diabetes Res Clin Pract. https://doi.org/10.1016/j.diabres.2020.108214
Wang K, Chen W, Zhou Y-S et al (2020) SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. bioRxiv. https://doi.org/10.1101/2020.03.14.988345
Wang D, Hu B, Hu C et al (2020) Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA J Am Med Assoc 10:10. https://doi.org/10.1001/jama.2020.1585
Welcome MO (2019) Gut Microbiota disorder, gut epithelial and blood–brain barrier dysfunctions in etiopathogenesis of dementia: molecular mechanisms and signaling pathways. NeuroMolecular Med
Welcome MO (2020) Neuroinflammation in CNS diseases: molecular mechanisms and the therapeutic potential of plant derived bioactive molecules. PharmaNutrition. https://doi.org/10.1016/j.phanu.2020.100176
Welcome MO (2020) Cellular mechanisms and molecular signaling pathways in stress-induced anxiety, depression, and blood-brain barrier inflammation and leakage. Inflammopharmacology. https://doi.org/10.1007/s10787-020-00712-8
Welcome MO (2020) Blood brain barrier inflammation and potential therapeutic role of phytochemicals. PharmaNutrition 11:100177
Wong SLA, Chen Y, Chak MC et al (2005) In vivo functional characterization of the SARS-Coronavirus 3a protein in Drosophila. Biochem Biophys Res Commun. https://doi.org/10.1016/j.bbrc.2005.09.098
Wu JT, Leung K, Leung GM (2020) Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: a modelling study. Lancet. https://doi.org/10.1016/S0140-6736(20)30260-9
Wu Y, Xu X, Chen Z et al (2020) Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav Immun 87:18–22
Xia H, Lazartigues E (2008) Angiotensin-converting enzyme 2 in the brain: properties and future directions. J Neurochem 107:1482–1494
Xia S, Zhu Y, Liu M et al (2020) Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell Mol Immunol
Xie L, Sun C, Luo C et al (2020) SARS-CoV-2 and SARS-CoV Spike-RBD structure and receptor binding comparison and potential implications on neutralizing antibody and vaccine development. bioRxiv. https://doi.org/10.1101/2020.02.16.951723
Xin P, Xu X, Deng C, Liu S, Wang Y, Zhou X, Ma H, Wei DSS (2020) The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int Immunopharmacol 80:106210. https://doi.org/10.1016/j.intimp.2020.106210
Xiong Y, Liu Y, Cao L et al (2020) Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg Microbes Infect. https://doi.org/10.1080/22221751.2020.1747363
Xu Z, Shi L, Wang Y et al (2020) Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. https://doi.org/10.1016/S2213-2600(20)30076-X
Yang J, Williams RS, Kelly DP (2009) Bcl3 interacts cooperatively with peroxisome proliferator-activated receptor gamma (PPARγ) coactivator 1α to coactivate nuclear receptors estrogen-related receptor α and PPARα. Mol Cell Biol. https://doi.org/10.1128/mcb.01669-08
Yang X, He G, Hao Y et al (2010) The role of the JAK2-STAT3 pathway in pro-inflammatory responses of EMF-stimulated N9 microglial cells. J Neuroinflamm. https://doi.org/10.1186/1742-2094-7-54
Yang CH, Li K, Pfeffer SR, Pfeffer LM (2015) The type I IFN-induced miRNA, miR-21. Pharmaceuticals
Ye Q, Wang B, Mao J (2020) The pathogenesis and treatment of the ‘Cytokine Storm’’ in COVID-19.’ J Infect
Yeh EA, Collins A, Cohen ME et al (2004) Detection of coronavirus in the central nervous system of a child with acute disseminated encephalomyelitis. Pediatrics. https://doi.org/10.1542/peds.113.1.e73
Yeo C, Kaushal S, Yeo D (2020) Enteric involvement of coronaviruses: is faecal–oral transmission of SARS-CoV-2 possible? Lancet Gastroenterol Hepatol 5:335–337
Yip MS, Leung HL, Li PH et al (2016) Antibody-dependent enhancement of SARS coronavirus infection and its role in the pathogenesis of SARS. Hong Kong Med J Xianggang yi xue za zhi 22:25–31
Yuan FF, Velickovic Z, Ashton LJ et al (2014) Influence of HLA gene polymorphisms on susceptibility and outcome post infection with the SARS-CoV virus. Virol Sin 29:128–130
Zaki AM, Van Boheemen S, Bestebroer TM et al (2012) Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. https://doi.org/10.1056/NEJMoa1211721
Zeng X, Ju D (2018) Hedgehog signaling pathway and autophagy in cancer. Int J Mol Sci 19:2279
Zhang ZG, Zhang L, Jiang Q et al (2000) VEGF enhances angiogenesis and promotes blood–brain barrier leakage in the ischemic brain. J Clin Investig. https://doi.org/10.1172/JCI9369
Zhang H, Zhou G, Zhi L et al (2005) Association between Mannose-binding lectin gene polymorphisms and susceptibility to severe acute respiratory syndrome coronavirus infection. J Infect Dis. https://doi.org/10.1086/491479
Zhang L, Wei L, Jiang D et al (2007) SARS-CoV nucleocapsid protein induced apoptosis of COS-1 mediated by the mitochondrial pathway. Artif Cells Blood Substitutes Biotechnol. https://doi.org/10.1080/10731190601188422
Zhang X, Hartung JE, Bortsov AV et al (2018) Sustained stimulation of β 2- and β 3-adrenergic receptors leads to persistent functional pain and neuroinflammation. Brain Behav Immun. https://doi.org/10.1016/j.bbi.2018.06.017
Zhang J, Ma X, Yu F, Liu J, Zou F, Pan T, Zhang H (2020a) Teicoplanin potently blocks the cell entry of 2019-nCoV. bioRxiv. https://doi.org/10.1101/2020.02.05.935387
Zhang R, Wang X, Ni L et al (2020b) COVID-19: melatonin as a potential adjuvant treatment. Life Sci 250:117583
Zhao H, Wang W, Zhao Q et al (2016) BCL3 exerts an oncogenic function by regulating STAT3 in human cervical cancer. Onco Targets Ther. https://doi.org/10.2147/OTT.S118184
Zhao D, Yao F, Wang L et al (2020) A comparative study on the clinical features of COVID-19 pneumonia to other pneumonias. Clin Infect Dis. https://doi.org/10.1093/cid/ciaa247
Zhao H, Shen D, Zhou H et al (2020) Guillain–Barré syndrome associated with SARS-CoV-2 infection: causality or coincidence? Lancet Neurol 19:383–384
Zheng S, Fan J, Yu F et al (2020) Viral load dynamics and disease severity in patients infected with SARS-CoV-2 in Zhejiang province, China, January-March 2020: retrospective cohort study. BMJ. https://doi.org/10.1136/bmj.m1443
Zhou N, Pan T, Zhang J et al (2016) Glycopeptide antibiotics potently inhibit cathepsin l in the late endosome/lysosome and block the entry of ebola virus, middle east respiratory syndrome coronavirus (MERS-CoV), and severe acute respiratory syndrome coronavirus (SARS-CoV). J Biol Chem. https://doi.org/10.1074/jbc.M116.716100
Zhou Y, Fu B, Zheng X et al (2020) Aberrant pathogenic GM-CSF+ T cells and inflammatory CD14+CD16+ monocytes in severe pulmonary syndrome patients of a new coronavirus. bioRxiv. https://doi.org/10.1101/2020.02.12.945576
Zhou F, Yu T, Du R et al (2020) Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. https://doi.org/10.1016/S0140-6736(20)30566-3
Zhu X, Song Z, Zhang S et al (2014) CD147: a novel modulator of inflammatory and immune disorders. Curr Med Chem. https://doi.org/10.2174/0929867321666131227163352
Zhu N, Zhang D, Wang W et al (2020) A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. https://doi.org/10.1056/NEJMoa2001017
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None declared.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Welcome, M.O., Mastorakis, N.E. Neuropathophysiology of coronavirus disease 2019: neuroinflammation and blood brain barrier disruption are critical pathophysiological processes that contribute to the clinical symptoms of SARS-CoV-2 infection. Inflammopharmacol 29, 939–963 (2021). https://doi.org/10.1007/s10787-021-00806-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10787-021-00806-x