
Abstract
Severe acute respiratory syndrome coronavirus (SARS-CoV) encodes a highly basic nucleocapsid (N) protein which can inhibit the synthesis of type I interferon (IFN), but the molecular mechanism of this antagonism remains to be identified. In this study, we demonstrated that the N protein of SARS-CoV could inhibit IFN-beta (IFN-β) induced by poly(I:C) or Sendai virus. However, we found that N protein could not inhibit IFN-β production induced by overexpression of downstream signaling molecules of two important IFN-β induction pathways, toll-like receptor 3 (TLR3)- and RIG-I-like receptors (RLR)-dependent pathways. These results indicate that SARS-CoV N protein targets the initial step, probably the cellular PRRs (pattern recognition receptors)-RNAs-recognition step in the innate immune pathways, to suppress IFN expression responses. In addition, co-immunoprecipitation assays revealed that N protein did not interact with RIG-I or MDA5. Further, an assay using truncated mutants revealed that the C-terminal domain of N protein was critical for its antagonism of IFN induction, and the N deletion mutant impaired for RNA-binding almost completely lost the IFN-β antagonist activity. These results contribute to our further understanding of the pathogenesis of SARS-CoV.
Similar content being viewed by others


Introduction
Severe acute respiratory syndrome coronavirus (SARS-CoV) is highly pathogenic in humans, with an overall mortality rate of 9.6%. Its 29.7 kb single-stranded RNA genome, which may be recognized by cytosolic RNA sensors, is wrapped in a helical nucleocapsid composed of multiple copies of N protein. The high pathogenicity suggests that SARS-CoV has developed mechanisms to overcome the host innate immune response.
Production of type I interferon (IFN) is a key step in the innate immune response to viral infection. Two major pathways are employed for the activation of type I interferon following virus infection. The first one is toll-like receptor (TLR)-dependent pathway, for example, TLR3 can detect viral dsRNAs in endosomal compartments. The second one is RIG-I-like receptors (RLR)-dependent pathway, in which cytoplasmic CARD domain-containing RNA helicases, RIG-I and MDA5, can sense viral RNA in the cytoplasm. Both pathways are based on sensor interactions with pathogen associated molecular patterns (PAMPs), such as double stranded RNA accumulated during replication or released upon lysis of infected cells, or structured single-stranded RNAs of the viral genome. Two different groups of adaptors are utilized in the two pathways. The TLRs signal through TRIF and/or MyD88, whereas RLRs signal through MAVS/IPS-1/VISA/CARDIF. Downstream of the different adaptors, IKKε and TBK1, are recruited and ultimately IRF-3, NFκB, and AP-1 are activated to initiate type I IFN gene transcription. Once interferon is synthesized and released from cells, it binds to interferon receptors, initiating a signaling cascade of JAK/STAT pathway that results in the expression of interferon-stimulated genes (ISGs) which enable the cell to combat viral infection and interfere with viral replication.
Until now, many proteins of SARS-CoV have been identified as effective interferon antagonists including nsp1 [1–3], PLP domain of nsp3 [3–5], nsp7 and nsp15 [3], M [6], and ORF3b, ORF6, and N [7, 8], which can suppress IFN responses by different mechanisms at multiple levels of IFN production pathways.
In SARS-CoV, nucleocapsid protein (N) is one of the most crucial structural components with the primary function of encapsidating the viral genome. In addition, it was demonstrated that N protein interfered with different cellular pathways [8–13], thus implying it to be a key regulatory component of the virus. Recently, N protein has been reported to inhibit IFN-β production by Sendai virus [8]. However, the underlying molecular mechanism of this inhibition is not clear.
In this study, we found that N could inhibit IFN-β production induced by poly(I:C) in 293T cells. Interestingly, N could not inhibit IFN-β production induced by RIG-I-2CARD, MAVS, TRIF, TBK-1, or IKKε, which are important molecules of TLR3- or RLR-dependent pathways. These results indicate that N blocks a very early step for IFN-β production, most probably at the RNA-sensor recognition step. The direct association of N with RIG-I or MDA5 could not be detected in vitro, and the RNA-binding region in C-terminal domain was essential for IFN expression inhibition, thus implying that the inhibition of IFN-β production by N protein may probably due to its ability in binding to and shielding viral RNAs from the recognition by cellular PRRs.
Materials and methods
Cells and virus
293T cells were grown and maintained in DMEM containing 10% heat-inactivated FBS. Sendai virus was obtained from Wuhan Institute of Virology, Chinese Academy of Sciences.
Reagents and antibodies
Poly(I:C) (Invitrogen), monoclonal mouse and polyclonal rabbit anti-Flag antibodies (Sigma), monoclonal mouse anti β-actin antibody (Santa Cruz), monoclonal mouse anti-Myc antibody (TIANGEN), TRITC-conjugated goat anti-mouse IgG (PIERCE), HRP-conjugated goat anti-mouse secondary antiserum (Thermo), and DAPI (Invitrogen) were obtained commercially.
Plasmids
The reporter plasmids pIFN-β-luc, pRL-TK, the plasmids expressing Flag-RIG-I, Flag-MDA5, Flag-IRF-3, HA-MAVS, HA-TRIF, HA-TBK1, HA-IKKε, Flag-RIG-I-2CARD, and Flag-NS3/4A were kindly provided by Dr. Hongbing Shu [14]. pCMV-tag2b-N was constructed as described [15]. pCMV-Myc-N was constructed by substituting the Flag-tag coding sequence of pCMV-tag2b-N with Myc-tag coding sequence. Plasmids expressing truncated N were amplified from pCMV-tag2b-N with specific primers by PCR. The PCR fragments were digested with EcoRI and SalI, and ligated into EcoRI and SalI-digested pCMV-tag2b vector. pEGFP-N1-IRF-3 was constructed by inserting IRF-3 coding sequence into pEGFP-N1 vector (Clontech), which achieves a fusion expression of IRF-3-GFP after transfection in mammalian cells. Plasmid pcDNA3.0-NS3/4A was constructed by inserting the coding sequence of NS3/4A into pcDNA3.0 vector.
Dual-luciferase reporter assays
The 293T cells (~1 × 105) were seeded on 24-well dishes and transfected the following day with pCMV-tag2b-N or an empty vector (0.3 μg), an indicated expression plasmid (0.2 μg), pIFN-β-luc (0.1 μg), pRL-TK (0.05 μg) (expressing Renilla luciferase constitutively) by standard calcium phosphate precipitation. At 24 h post-transfection, cells were analyzed by Dual-Luciferase reporter Assay System (Promega) according to the manufacturer’s instructions, or the cells were infected with SenV (MOI = 10) or transfected with poly(I:C) (1 μg/well) by Lipofectamine (Invitrogen) for 18 h prior to determining the relative luciferase activities of the cell lysate. Luciferase activity was normalized to pRL-TK signal and is depicted as RLU.
RNA analysis
RNA from 293T cells was extracted using Trizol reagent (Invitrogen) according to the manufacturer’s instructions. The final number of cells used for RNA extraction was approximately 5 × 106 . cDNA synthesized from 2 μg of RNA was diluted at 1:30 and 5 μl of each sample was subjected to real-time PCR using Power SYBR Green PCR master mix (Toyobo). The reaction was carried out for the IFN-β and GAPDH genes using specific primers (IFN-β F 5′-AGACTTACAGGTTACCTCCGAA-3′, IFN-β R 5′-CAGTACATTCGCCATCAGTCA-3′; GAPDH F 5′-GAGTCAACGGATTTGGTCGT-3′, GAPDH R 5′-GACAAGCTTCCCGTTCTCAG-3′) and an ABI 7300 real-time PCR system. 45 amplification cycles were performed with cycling conditions of 15 s at 94°C, 15 s at 55°C, and 45 s at 72°C. The specificity of the primers was confirmed by sequencing the PCR products. The threshold cycle (Ct) for IFN-β and the difference between their Ct values (ΔCt) were determined. The results were normalized by GAPDH expression to obtain the ΔΔCt value. Finally, the 2−D value was calculated to reflect the relative expression of IFN-β gene.
Fluorescence microscopy
Cells were fixed in acetone:methanol (1:1) and blocked in PBS containing 1% BSA, then incubated overnight at 4°C with mouse anti-Flag antibody. Subsequently cells were incubated with TRITC-conjugated goat anti-mouse IgG for 1 h at 37°C before stained with DAPI for 5 min. Cells were examined using a conventional fluorescent microscope.
Native PAGE and immunoblotting
Native PAGE and immunoblotting were performed as described [16, 17].
Co-immunoprecipitation assays
Co-immunoprecipitation was performed as described [18].
Statistical analyses
Statistical analyses were performed using an unpaired, two-tailed Student’s t-test. P values of less than 0.05 were considered to be statistically significant.
Results
N inhibits IFN-β promoter activation induced by poly(I:C) and Sendai virus
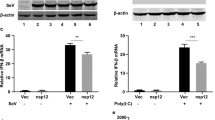
To determine whether N can interfere with the transcriptional activation of IFN-β promoter induced by poly(I:C), 293T cells were transfected with a plasmid (pIFN-β-luc) expressing firefly luciferase under the control of IFN-β promoter. The cells were co-transfected with plasmid expressing SARS-CoV N protein, or with an empty vector. A renilla luciferase reporter plasmid (pRL-TK) was also co-transfected to normalize for transfection efficiency. At 24 h post-transfection, cells were transfected with poly(I:C) or infected with SenV to induce interferon synthesis, and 18 h later, cells were subjected to dual-luciferase reporter assays. As shown in Fig. 1a and b, IFN-β-luc activities were markedly induced by poly(I:C) and SenV in 293T cells, and N could significantly inhibit poly(I:C)-or SenV-induced activation of IFN-β promoter compared to the empty vector. To further determine that whether N could interfere with poly(I:C)- and SenV-induced IFN-β expression in MDA5 or RIG-I transfected cells, an increasing amount of pCMV-tag2b-N (0.05, 0.2, and 0.4 μg) and plasmid expressing full-length MDA5 or RIG-I was transfected into 293T cells. At 24 h post-transfection, cells were transfected with poly(I:C) or infected with SenV, and 18 h later, cells were subjected to dual-luciferase reporter assays. As shown in Fig. 1c and d, N could significantly inhibit the IFN-β-luc activities, and the inhibition effect was dose-dependent.

SARS-CoV N protein inhibits IFN-β production induced by poly(I:C) and SenV. 293T cells (1 × 105) were transfected with a plasmid expressing Flag-N or the empty vector and pIFN-β-luc, pRL-TK. At 24 h post-transfection, a cells were transfected with poly(I:C) or b infected with SenV. 18 h later, reporter assays were performed. c, d Reporter assays were performed similarly as in (a) and (b) except that an increasing amount of Flag-N-expressing plasmids and MDA5- or RIG-I-expressing plasmid were transfected. The data represent the mean ± S.D. of three independent experiments. * P < 0.05
N does not inhibit IFN-β promoter activation induced by RIG-I-2CARD, MAVS, TRIF, TBK1, or IKKε
MAVS, TRIF, TBK-1, IKKε, and RIG-I-2CARD (the CARD domains of RIG-I, a constitutively active form of RIG-I), when expressed exogenously in cells, can induce high level expression of IFN-β. To assess whether N protein has an inhibitory effect in these two pathways, 293T cells were co-transfected with pIFN-β-luc, pRL-TK, pCMV-tag2b-N, and the expression constructs of RIG-I-2CARD, MAVS, TRIF, TBK-1, or IKKε. At 24 h post-transfection, cells were subjected to dual-luciferase reporter assays. Upon overexpression of RIG-I-2CARD, MAVS, TRIF, TBK-1, or IKKε, luciferase expression driven by IFN-β promoter was greatly increased. Interestingly, overexpression of N protein did not inhibit but even stimulated IFN-β promoter activation induced by these signaling molecules (Fig. 2). These results suggest that N’s inhibition of IFN-β production might occur at RIG-I or upstream of RIG-I and TRIF in the signal transduction pathway.

N cannot inhibit IFN-β promoter activation induced by overexpressing signaling molecules in innate immune pathways. 293T cells (1 × 105) were co-transfected with Flag-N or empty vector, pIFN-β-luc, pRL-TK and indicated plasmid expressing RIG-I-2CARD (a), MAVS, TRIF (b), TBK1, or IKKε (c). At 24 h post-transfection, reporter assays were performed. The data represent the mean ± S.D. of three independent experiments. * P < 0.05
N reduces IFN-β mRNA synthesis induced by Sendai virus and poly(I:C), but not by RIG-I-2CARD, MAVS, TRIF, TBK1, or IKKε
To further verify the inhibitory effect of N on IFN-β transcriptional activation, real-time PCR was used to analyze IFN-β mRNA synthesis. Gene-specific mRNA was amplified by RT and real-time PCR, using primers for IFN-β, while GAPDH was used as an endogenous control. As expected, strong IFN-β mRNA signals were induced by SenV and poly(I:C) in vector-tranfected cells, while overexpression of N could downregulate the IFN-β mRNA level induced by SenV and poly(I:C) (Fig. 3a). Consistent with Figs. 1 and 2, overexpression of N cannot downregulate the IFN-β mRNA level induced by RIG-I-2CARD, MAVS, TBK1, TRIF, and IKKε (Fig. 3b, c).

N reduces IFN-β mRNA synthesis induced by Sendai virus and poly(I:C), but not by overexpressing signaling molecules in innate immune pathways. 293T cells (1 × 105) were similarly treated as in Figs. 1 and 2 without pIFN-β-luc and pRL-TK in transfections. The mRNAs for IFN-β in transfected cells were detected by real-time PCR. The data represent the mean ± S.D. of three independent experiments. * P < 0.05
N inhibits the activation of IRF-3 induced by poly(I:C) but not by MAVS, TRIF, TBK1, and IKKε
Dimer formation and nuclear translocation are two important signals for effective activation of IRF-3, which can then form a complex with CBP/p300. The holocomplex can then bind to the IRF-E sites in IFN-β promoter region to activate the transcription of IFN-β gene eventually. We next sought to define whether SARS-CoV N could inhibit IRF-3 activation induced by poly(I:C) or downstream signaling molecules.
To examine the effect of N on the nuclear translocation of GFP-IRF-3 caused by poly(I:C) and signaling molecules, 293T cells were transfected with a GFP-IRF-3 expression plasmid and poly(I:C) or plasmid expressing each of the signaling molecules (MAVS, TRIF, TBK1, and IKKε) with or without the N plasmid. As shown in Fig. 4, poly(I:C) or expression of MAVS induced the nuclear translocation of GFP-IRF-3, as expected (panels 1 and 3). The nuclear translocation of GFP-IRF-3 was impaired in cells transfected with poly(I:C) and N (Fig. 4 panel 2), but not in cells co-transfected with MAVS and N (Fig. 4 panel 4). HCV NS3/4A, which cleaves MAVS and can thus inhibit the activation of IRF-3 [19], was used as a control, and nuclear translocation of GFP-IRF-3 induced by expression of MAVS was inhibited by NS3/4A as expected (Fig. 4 panel 5).

N inhibits the nuclear translocation of IRF-3 promoted by poly(I:C), but not by MAVS. 293T cells (1 × 105) were transfected with indicated plasmids, together with a plasmid expressing the IRF-3 and GFP fusion (GFP-IRF-3) (left panels) to follow IRF-3’s localization. At 24 h post-transfection, cells were either analyzed by microscopy (b) or tranfected with poly(I:C) for another 12 h before microscopy analysis (a). N proteins were detected with monoclonal antibodies against Flag (right panels), and DAPI staining was used to show the localization of nucleus (middle panels). The experiments were repeated for three times with similar results
Then, we tested whether the dimerization of IRF-3 induced by poly(I:C) or signaling molecules could be inhibited by N. As expected, the overexpression of N could inhibit the dimer formation of IRF-3 promoted by poly(I:C) (Fig. 5a) but not by MAVS (Fig. 5b). HCV NS3/4A as a control could suppress IRF-3 dimer formation induced by MAVS (Fig. 5b). Similar results were obtained when TRIF, TBK1, or IKKε were overexpressed instead of MAVS in IRF-3 translocation and dimerization assays (data not shown).

N inhibits the dimer formation of IRF-3 promoted by poly(I:C), but not by MAVS. a 293T cells (4 × 105) were transfected with plasmid expressing Flag-IRF-3 (1 μg) and indicated plasmids (2 μg). At 24 h post-transfection, cells were mock treated or transfected with poly(I:C) (4 μg). 12 h later, cell extracts were prepared and detected by Western blot analysis. IRF-3 dimers and monomers were separated by native PAGE and then detected with monoclonal antibodies against Flag. b 293T cells (4 × 105) were transfected with plasmids expressing Flag-IRF-3 (1 μg) and HA-MAVS (1 μg) and indicated plasmids (2 μg), and 18 h later, cell extracts were prepared and detected by Western blot analysis similarly as in (a). pcDNA3.0-NS3/4A and its empty vector pcDNA3.0 (V-3) were set as a control. V-M stands for the empty vector pCMV-Myc, which was used for expression of N. The experiments were repeated for three times with similar results
Taken together, the results of IRF-3 activation assays were consistent with the results in Figs. 1, 2 and 3. These results suggest that SARS-CoV N only suppresses IFN-β production induced by SenV or poly(I:C), but not by downstream signaling molecules; therefore, SARS-CoV N must suppress the IFN-β induction at a very early step, most probably at the PRRs-RNA-recognition step.
N does not interact with RIG-I or MDA5
Since N could not inhibit IFN-β production induced by RIG-I-2CARD, we assumed that N may block the beginning PRRs-RNA-recognition step. Many virus proteins can inhibit IFN-β production through binding and thus inactivate cytosolic PRRs [20, 21]. Therefore, we sought to investigate whether N can interact with RIG-I or MDA5. However, such proposed interactions could not be detected in co-immunoprecipitation assays (Fig. 6a). RIG-I and MDA5 could be efficiently precipitated in the same experiment, which indicated the precipitation condition was suitable (Fig. 6a). Since N has the ability to self-associate, the interaction between the Flag-tagged and Myc-tagged N was set as a positive control, and Flag-N interacted with Myc-N as expected (Fig. 6a, right). Considering that RIG-I and MDA5 will undergo conformation change upon exogenous stimuli, the same IP analysis was performed upon Sendai virus infection or poly(I:C) transfection. However, the interaction between N and RIG-I or MDA5 still cannot be detected (Fig. 6b). These results demonstrate that N does not inhibit IFN-β production through binding cytosolic PRRs.

N does not interact with RIG-I or MDA5. a 293T cells (2 × 106) were co-transfected with indicated expression plasmids (8 μg each). Cell lysates were immunoprecipitated with control rabbit IgG or rabbit anti-Flag (αFlag) antibody as indicated. The immunoprecipitates were analyzed by Western blots with mouse anti-Myc antibody (top panels) and mouse anti-FLAG antibody (middle panels). The expression levels of transfected proteins in the lysates were analyzed by Western blots with indicated antibodies (bottom panel). The experiments were repeated for three times with similar results. b 293T cells (2 × 106) were co-transfected with indicated expression plasmids (8 μg each). At 24 h post-transfection, cells were transfected with poly (I:C) (left panels) or infected with Sendai virus (right panels) for 6 h. Then, co-immunoprecipitation assays were performed similarly as in (a)
The N protein C-terminus domain is critical in inhibiting SenV-induced IFN-β promoter activation, and the RNA-binding ability is indispensible for this inhibition
Given the results that N protein suppressed the IFN-β production induced by Sendai virus, we then investigated which domain of N was responsible. We constructed plasmids expressing truncated N by the regions of the structural domains [22] (Fig. 7a). Expression of N-truncated proteins was confirmed by Western blot (Fig. 7b). Dual-luciferase reporter assays were similarly performed as mentioned above. As shown in Fig. 7c, SenV-induced IFN-β promoter activation was significantly suppressed by co-transfection of CTD and C′, as well as full-length N protein, while transfection of NTD, N′, and Linker did not significantly suppress SenV-induced IFN-β promoter activation. The C-CTD (aa 281–365), which has been demonstrated to have no RNA-binding ability [23], almost lost the ability to reduce IFN-β promoter activity. These results suggested that the C-terminal domain of N protein may have an important role in suppressing SenV-induced IFN expression responses, and RNA-binding ability is essential for this inhibition effect.

Reporter assays using truncated N expression constructs. a Truncated constructs of N. The open reading frame of N was truncated in six constructs corresponding to the structural domains. b Western blot analysis was performed to determine the expression of SARS-CoV N protein and N truncation mutants. c The truncated N plasmids, pIFN-β-luc, pRL-TK were co-transfected into 293T cells (1 × 105) for 24 h before SenV infection. Reporter assays were performed 18 h after infection. The data represent the mean ± S.D. of three independent experiments. * P < 0.05. Meanwhile, cell extracts were prepared, and the expression of β-actin protein was determined by Western blot as the expression control
Discussion
Production of interferon is one of the primary host defense mechanisms against viruses. However, SARS-CoV infection does not result in IFN production in many cell cultures and pretreatment of cells with IFN blocks SARS-CoV infection [13, 24–26]. The N protein of SARS-CoV was found to inhibit the activation of IRF-3 and NF-κB induced by SenV, resulting in inhibition of IFN synthesis [8]. Sendai virus has been verified to be recognized by cytosolic sensor RIG-I to elicit type Ι IFN production, while MDA5 can recognize cytosolic poly(I:C) [27]. In this report, we found that N could inhibit IFN-β production induced not only by SenV but also by poly(I:C), and N could inhibit IFN-β production induced by SenV or poly(I:C) in MDA5- or RIG-I-transfected cells, and the inhibitory effect was dose-dependent. Further, we found that N could not inhibit IFN-β production induced by downstream signaling molecules of TLR3- and RLR-dependent pathways (RIG-I-2CARD, VISA, TRIF, TBK1, and IKKε). These results suggested that N protein suppresses the IFN-β production and N only targets a very early step in IFN induction pathway. Since N could not inhibit the IFN-β production activated by RIG-I-2CARD, it can be envisaged that N may function by blocking the beginning PRRs-RNA-recognition step of IFN-β induction. Then we sought to investigate whether N could inhibit IFN-β production through binding and thus inactivate cytosolic PRRs. However, co-immunoprecipitation assays revealed that N protein did not interact with RIG-I or MDA5, which indicates that N must have employed other mechanism to inhibit IFN-β production.
Toll-like receptors (TLRs) and cytoplasmic retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) are the two major receptor systems for detecting RNA viruses. Viruses have evolved mechanisms to evade these detections. Several virus proteins with IFNs response inhibitory activity also exhibit RNA-binding activity, such as VP35 from Ebola [28] and NS1 of influenza [21]. And it as has been demonstrated that these proteins can sequester the genomic RNA in such a way that host-sensing proteins do not detect it. SARS-CoV N protein is also an RNA-binding protein [29, 30]. Therefore, it may be possible that N protein employs a similar mechanism to inhibit the IFN response.
Surprisingly, IFN-β production was upregulated by overexpression of N in RIG-I-2CARD, MAVS, TRIF, TBK1, or IKKε-transfected cells (Figs. 2, 3b, c). The mechanism for this stimulator effect is not clear; however, due to N’s RNA-chaperone activity [31], it may help the co-transfected exogenous mRNAs of these signaling molecule to adopt their functional conformation to some extent, thus promoting the induction of IFN-β.
The N protein is a 46 kDa protein composed of 422 amino acids. N protein can be divided into two structural domains interspersed with three disordered regions [22]: the N-terminal domain(NTD, aa 45–181) which is responsible for RNA-binding; the C-terminal domain(CTD, aa 248–365) which is responsible for dimerization and have been indentified to bind to nucleic acid even stronger than the NTD [23]. A deletion mutant of CTD, which includes aa 281–365, totally lost the ability to bind to nucleic acid and retains dimerization activity [23]. Besides, it has been demonstrated that all three disordered regions of SARS-CoV N protein are involved in RNA-binding [30]. N truncation assays showed that SenV-induced IFN-β production was significantly suppressed by expression of CTD-containing constructs, while the C-CTD (aa 281–365) almost lost the ability to reduce IFN-β promoter activity (Fig. 7c). These results imply that the C-terminal domain of N protein, which is located between positions 248 and 365, may be critical for suppressing IFN expression responses in host cells. As the RNA-binding ability of CTD is indispensable for its inhibition effect, we deduce that N may function by binding to IFN-inducing RNAs and shielding them from the recognition by cellular PRRs. However, the precise mechanism of N suppression is still not clear, and further experiments are needed. In our assay, C′ exerted a stronger ability to reduce IFN-β promoter activity than CTD domain, which indicates the disorder regions may be also involved in the inhibitory function of N protein. In addition, the N-terminal domain and central linker region slightly inhibited the IFN-β promoter activation induced by SenV. As we mentioned above, all the structural domains and disordered regions have been found to have RNA-binding ability, so it may be possible that N exhibits its IFN inhibition activity through all the RNA-binding sequences. Besides, SARS-CoV N protein has been proven to undergo various post-translational modifications, such as acetylation and sumoylation in N-terminal and phosphorylation in NTD and central SR-rich motif. Whether these modifications have any effect on N’s IFN expression inhibition ability is an interesting question and deserves further exploration.
In conclusion, we have shown that dsRNA-induced IFN-β expression was suppressed by SARS-CoV N protein. N protein antagonized IFN-β induction by inhibiting the initial step for IFN induction in innate immune pathways. The C-terminal domain of N is critical, and RNA-binding ability of CTD is indispensable for this antagonism.
References
M.G. Wathelet, M. Orr, M.B. Frieman, R.S. Baric, J. Virol. 81, 11620–11633 (2007)
K. Narayanan, C. Huang, K. Lokugamage, W. Kamitani, T. Ikegami, C.T. Tseng, S. Makino, J. Virol. 82, 4471–4479 (2008)
M. Frieman, K. Ratia, R.E. Johnston, A.D. Mesecar, R.S. Baric, J. Virol. 83, 6689–6705 (2009)
M.A. Clementz, Z. Chen, B.S. Banach, Y. Wang, L. Sun, K. Ratia, Y.M. Baez-Santos, J. Wang, J. Takayama, A.K. Ghosh, K. Li, A.D. Mesecar, S.C. Baker, J. Virol. 84, 4619–4629 (2010)
S.G. Devaraj, N. Wang, Z. Chen, Z. Chen, M. Tseng, N. Barretto, R. Lin, C.J. Peters, C.T. Tseng, S.C. Baker, K. Li, J. Biol. Chem. 282, 32208–32221 (2007)
K.L. Siu, K.H. Kok, M.H. Ng, V.K. Poon, K.Y. Yuen, B.J. Zheng, D.Y. Jin, J. Biol. Chem. 284(24), 16202–16209 (2009)
M. Frieman, B. Yount, M. Heise, S.A. Kopecky-Bromberg, P. Palese, R.S. Baric, J. Virol. 81, 9812–9824 (2007)
S.A. Kopecky-Bromberg, L. Martinez-Sobrido, M. Frieman, R.A. Baric, P. Palese, J. Virol. 81, 548–557 (2007)
F.Q. Li, H. Xiao, J.P. Tam, D.X. Liu, FEBS Lett. 579, 2387–2396 (2005)
M. Surjit, B. Liu, V.T. Chow, S.K. Lal, J. Biol. Chem. 281, 10669–10681 (2006)
X. Yan, Q. Hao, Y. Mu, K.A. Timani, L. Ye, Y. Zhu, J. Wu, J. Int, Biochem. Cell Biol. 38, 1417–1428 (2006)
R. He, A. Leeson, A. Andonov, Y. Li, N. Bastien, J. Cao, C. Osiowy, F. Dobie, T. Cutts, M. Ballantine, X. Li, Biochem. Biophys. Res. Commun. 311, 870–876 (2003)
M. Spiegel, A. Pichlmair, L. Martinez-Sobrido, J. Cros, A. Garcia-Sastre, O. Haller, F. Weber, J. Virol. 79, 2079–2086 (2005)
L.G. Xu, Y.Y. Wang, K.J. Han, L.Y. Li, Z. Zhai, H.B. Shu, Mol. Cell 19, 727–740 (2005)
J. Pan, J. Pan, X. Peng, Y. Gao, Z. Li, X. Lu, Y. Chen, M. Ishaq, D. Liu, M.L. Dediego, L. Enjuanes, D. Guo, ONE PLoS 3, e3299 (2008)
T. Iwamura, M. Yoneyama, K. Yamaguchi, W. Suhara, W. Mori, K. Shiota, Y. Okabe, H. Namiki, T. Fujita, Genes Cells 6, 375–388 (2001)
W. Suhara, M. Yoneyama, T. Iwamura, S. Yoshimura, K. Tamura, H. Namiki, S. Aimoto, T. Fujita, J. Biochem. 128, 301–307 (2000)
X. Wu, Y. Zhou, K. Zhang, Q. Liu, D. Guo, FEBS Lett. 582, 2155–2160 (2008)
E. Meylan, J. Curran, K. Hofmann, D. Moradpour, M. Binder, R. Bartenschlager, J. Tschopp, Nature 437, 1167–1172 (2005)
J. Andrejeva, K.S. Childs, D.F. Young, T.S. Carlos, N. Stock, S. Goodbourn, R.E. Randall, Proc. Natl. Acad. Sci. USA 101, 17264–17269 (2004)
M. Mibayashi, L. Martinez-Sobrido, Y.M. Loo, W.B. Cardenas, M. Gale Jr., A. Garcia-Sastre, J. Virol. 81, 514–524 (2007)
C.K. Chang, S.C. Sue, T.H. Yu, C.M. Hsieh, C.K. Tsai, Y.C. Chiang, S.J. Lee, H.H. Hsiao, W.J. Wu, W.L. Chang, C.H. Lin, T.H. Huang, J. Biomed. Sci. 13, 59–72 (2006)
C.Y. Chen, C.K. Chang, Y.W. Chang, S.C. Sue, H.I. Bai, L. Riang, C.D. Hsiao, T.H. Huang, J. Mol. Biol. 368, 1075–1086 (2007)
B. Zheng, M.L. He, K.L. Wong, C.T. Lum, L.L. Poon, Y. Peng, Y. Guan, M.C. Lin, H.F. Kung, J. Interferon Cytokine Res. 24, 388–390 (2004)
J. Cinatl Jr., G. Hoever, B. Morgenstern, W. Preiser, J.U. Vogel, W.K. Hofmann, G. Bauer, M. Michaelis, H.F. Rabenau, H.W. Doerr, Cell. Mol. Life Sci. 61, 2100–2112 (2004)
B.S. Tang, K.H. Chan, V.C. Cheng, P.C. Woo, S.K. Lau, C.C. Lam, T.L. Chan, A.K. Wu, I.F. Hung, S.Y. Leung, K.Y. Yuen, J. Virol. 79, 6180–6193 (2005)
H. Kato, O. Takeuchi, S. Sato, M. Yoneyama, M. Yamamoto, K. Matsui, S. Uematsu, A. Jung, T. Kawai, K.J. Ishii, O. Yamaguchi, K. Otsu, T. Tsujimura, C.S. Koh, C. Reis e Sousa, Y. Matsuura, T. Fujita, S. Akira, Nature 441, 101–105 (2006)
W.B. Cardenas, Y.M. Loo, M. Gale Jr., A.L. Hartman, C.R. Kimberlin, L. Martinez-Sobrido, E.O. Saphire, C.F. Basler, J. Virol. 80, 5168–5178 (2006)
T.K. Tang, M.P. Wu, S.T. Chen, M.H. Hou, M.H. Hong, F.M. Pan, H.M. Yu, J.H. Chen, C.W. Yao, A.H. Wang, Proteomics 5, 925–937 (2005)
C.K. Chang, Y.L. Hsu, Y.H. Chang, F.A. Chao, M.C. Wu, Y.S. Huang, C.K. Hu, T.H. Huang, J. Virol. 83, 2255–2264 (2009)
S. Zuniga, I. Sola, J.L. Moreno, P. Sabella, J. Plana-Duran, L. Enjuanes, Virology 357, 215–227 (2007)
Acknowledgments
The authors thank Dr. Hongbing Shu for providing a large panel of constructs used in this study and Yi Wang and Dan Liu for valuable discussions. This study was supported by the research grants of China National Basic Research Program (#2006CB504305, #2010CB911800) and China National Natural Science Foundation (#30925003, #30921001).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lu, X., Pan, J., Tao, J. et al. SARS-CoV nucleocapsid protein antagonizes IFN-β response by targeting initial step of IFN-β induction pathway, and its C-terminal region is critical for the antagonism. Virus Genes 42, 37–45 (2011). https://doi.org/10.1007/s11262-010-0544-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-010-0544-x