
Abstract
NETosis is a type of regulated cell death dependent on the formation of neutrophil extracellular traps (NET), where net-like structures of decondensed chromatin and proteases are produced by polymorphonuclear (PMN) granulocytes. These structures immobilise pathogens and restrict them with antimicrobial molecules, thus preventing their spread. Whilst NETs possess a fundamental anti-microbial function within the innate immune system under physiological circumstances, increasing evidence also indicates that NETosis occurs in the pathogenic process of other disease type, including but not limited to atherosclerosis, airway inflammation, Alzheimer’s and stroke. Here, we reviewed the role of NETosis in the development of organ injury, including injury to the brain, lung, heart, kidney, musculoskeletal system, gut and reproductive system, whilst therapeutic agents in blocking injuries induced by NETosis in its primitive stages were also discussed. This review provides novel insights into the involvement of NETosis in different organ injuries, and whilst potential therapeutic measures targeting NETosis remain a largely unexplored area, these warrant further investigation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
BACKGROUND
Cell death is commonly segregated into necrosis and apoptosis; apoptosis being programmed cell death, for instance during development and physiological cellular turnover, whilst necrosis predominantly takes place in an unregulated manner [1]. NETosis, like necrosis, is a mode of cell death that involves the loss of membrane integrity. During NETosis, decondensation of chromatin is thought to be initiated by peptidyl arginine deiminase 4 (PAD4) [2]; its subsequent release together with granule contents is vital in the innate immune response to infection and inflammation. Recent studies suggest that NET formation is of central to pathogenesis of organ injury. This review will summarise the current understanding of the molecular mechanisms of NETosis and the therapeutic approaches under development targeting NET-induced organ injury.
MOLECULAR MECHANISM OF NET FORMATION
Although NETosis is closely associated with NET formation, not all NET formation requires the process of cell death to take place beforehand. According to Nomenclature Committee on Cell Death, the term ‘NETosis’ should only be used in the context of cell death, and not just based on the presence of NET formation [3].
Two main pathways of NET formation have been described and categorised according to their dependence on the activity of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase pathway (Fig. 1) [5].

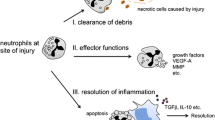
Type of cell death for neutrophil in organ injury. During solid organ injury, neutrophils could be prompted to undergo caspase-dependent apoptosis, which results in controlled dissolution of cell into apoptotic bodies containing cellular debris to prevent immune and inflammatory responses. Neutrophil extracellular traps (NETs) form via two pathways. The first is through lytic NETosis, a cell death pathway characterised by nuclear de-lobulation and disintegration of the nuclear envelope, which precedes loss of cellular polarisation, chromatin de-condensation and plasma membrane rupture. The second mechanism involves the non-lytic form of NETosis, which is not associated with cellular death but prompts expulsion of nuclear chromatin together with release of granule proteins through degranulation. These components can assemble in the extracellular space into NETs, leaving behind enucleated cytoplasts that continue to ingest microorganisms [4]. In addition, neutrophils could undergo unregulated necrosis that does not involve specific molecular pathways, with uncontrolled release of cellular debris acting as danger-associated molecular patterns (DAMPs) to trigger pro-inflammatory response.
NADPH Oxidase-Dependent NET Formation
The NADPH oxidase-dependent molecular pathway of NET formation begins with activation of neutrophil surface receptors by stimuli derived from pathogenic or non-pathogenic sources, such as cholesterol or urate, and ends with cellular lysis. These stimuli trigger calcium release from the endoplasmic reticulum (ER), resulting in the activation of protein kinase C (PKC) and the assembly of the NADPH oxidase complex generating reactive oxygen species (ROS) [6]. Following this, neutrophil elastase (NE), a protease stored in the cytoplasmic granules, migrates to the nucleus in a myeloperoxidase (MPO)-dependent manner and cleaves histones to initiate chromatin decondensation [7]. This is promoted by the citrullination of histone arginine residues by peptidylarginine deiminase IV (PAD4) [8]. Finally, mixing of the chromatin and granule proteins takes place as cellular membranes are broken down. Interestingly, there have been reports of mitochondrial DNA (mtDNA), instead of nuclear DNA, being the source of the DNA fibres in NETs, with observations of mtDNA being released from granulocytes in response to disease states such as trauma and systemic lupus erythematosus (SLE) [9, 10]. Moreover, it seems that histone citrullination is not always required for NET formation, as observed by Kenny and colleagues in their study of neutrophils activated by the PKC agonist phorbol 12-myristate 13-acetate (PMA); this highlights the diversity of pathways for NET formation following their induction [11].
Degradation of NETs can take place through several pathways, for example, by DNases [12], or endocytosed by macrophages [13].
Factors that influence NET formation include pH, CO2 and HCO3− levels, which modulate neutrophil activation. This explains why NETs form more readily in the periphery of an inflammatory site, where the pH is more alkaline [14]. This may influence treatment efficacy as NETs can seal off the affected area. An acidic environment has been hypothesised to reduce NADPH oxidase-dependent NET formation by reducing neutrophil glycolysis [15].
NADPH oxidase-dependent NET formation also requires neutrophils to be in the cell cycle, necessitating the activation of cyclin-dependent kinases (CDK) [16], phosphorylating the retinoblastoma protein.
NADPH Oxidase-Independent NET Formation
This mechanism of NET formation is more relevant in the context of infection, as inducers of NETosis here are calcium ionophore A23187 and the potassium ionophore nigericin, which are products of Streptomyces chartreusensis and Streptomyces hygroscopicus, respectively [11]. How this pathway leads to NET release is poorly understood.
NETosis AND INFLAMMATION
NETs under physiological conditions are central to pathogen clearance. When there is excessive formation or suboptimal, NETs are able to initiate further destructive signalling through interaction with other tissue constituents and the immune system. Moreover, the antimicrobial histones and peptides within the NET structure impose a direct cytotoxic effect on tissues [17]. To date, there have been numerous accounts of NETosis being present in diseases of major organs. Understanding of NETosis in pathophysiology may offer unique opportunities for clinical management.
NETosis IN ORGAN INJURY
There is an expanding body of research describing NETosis in infectious and non-infectious organ injury (summarised in Fig. 2). Although it is valid that in these scenarios, nuclear DNA released during necrotic cell death can contribute to tissue injury and exacerbate the extent of organ damage, here we focus on the contribution by aberrant NET formation and the implication of understanding its underlying pathogenesis for therapeutic interventions.

Involvement of NETosis in organ injury. Accumulating evidences now point to an important role of NETosis in infectious and non-infectious solid organ injury. Neutrophil invasion into brain parenchyma and release of neutrophil extracellular traps (NETs) have been established in the pathophysiology of Alzheimer’s disease to cause destruction to neural cells and blood-brain barrier. Abnormal NETosis activity and reactive oxygen species (ROS) response, a key element to NETosis initiation, were observed in stroke patients. The degree of neutrophil infiltration, NET formation/component (e.g. cell-free nucleosomes) and NETosis have been found to correlate with the severity of a range of lung diseases, including cystic fibrosis, acute lung injury (ALI)/acute respiratory distress syndrome (ARDS) and lung infection. NETosis was also shown to be involved in allergic asthma, chronic obstructive pulmonary disease and pulmonary hypertension, wherein degree of NET formation correlates with disease severity. During liver ischaemia-reperfusion, toll-like receptor-dependent NET release has been suggested to mediate liver inflammation and injury. Conversely, deficiency in NET release was reported in decompensated cirrhotic liver disease and could explain susceptibility to bacterial peritonitis infection in those patients. NET formation and NETosis have further been implicated in atherosclerosis and myocardial infarction, wherein NET was found in thrombi and infarct lesion and correlate with disease severity. In rheumatoid arthritis, enhanced NET release and NETosis are observed in synovial tissue, rheumatoid nodules and skin, whilst pro-inflammatory cytokines and autoantibodies further aggravate neutrophil infiltration and NETosis. Neutrophils could also be potently activated by monosodiumurate (MSU) crystals in gout joints and point to a potential role of NET/NETosis in gout pathogenesis. Moreover, neutrophil activation and NET deposition were also observed in colon mucosa of ulcerative colitis. Excessive neutrophil activation, NET formation and NETosis could also be responsible different pregnancy-related disorders, including pre-eclampsia wherein NET deposition and NETosis in the intervillous space may damage maternal endothelium and impair foetal oxygen exchange.
Brain
Alzheimer’s Disease
Alzheimer’s disease (AD) is a common disorder of neurodegeneration characterised by gradual loss of cognitive functions. In AD patients, neutrophils have been observed to invade the brain parenchyma and release NETs, causing destruction of neural cells and the blood-brain barrier [18].
Stroke
It is well known that the adaptive immune system is altered after a stroke, predisposing patients to infections [19,20,21]. Interestingly, NETosis has also been described as significantly impaired up until on day 5 in those with an ischaemic stroke [22]. Though NETosis in haemorrhagic stroke patients has yet to be documented, it has been noted that the generation of ROS, a key requirement for chromatin decondensation, is suppressed in these patients for up to 20 days [23].
Lung
Cystic Fibrosis
It has been well established that chronic infections in cystic fibrosis (CF) patients are due to the highly viscous mucus production harbouring microbial growth. Although impaired clearance of mucus has been principally named responsible, there is increasing evidence that the high viscosity is also due vast amounts of free DNA, found in sputum samples [24], which was in concordance with the high concentration of neutrophil and NETs found in CF lungs. The source was believed to be from necrotic neutrophils [25] but is now considered to be secondary to NETosis [26]. Additionally, NET production was found to be promoted by bacterial infection in CF airways and was defective in clearance of the bacteria Pseudomonas aeruginosa. NETs are also named as a facilitative factor for biofilm formation. There is evidence that surfactant protein D (SP-D), responsible for opsonising pathogens and dying cells for clearance by alveolar macrophages [27], is essential for NET clearance, through binding directly to the chromatin within the NETs. SP-D levels are decreased in CF patients, and the level of decrease is proportional to the degree of inflammation, through accumulation of NETs [28].
Lung Infection
Neutrophils migrated into the affected site and initiate the cascade of antimicrobial mechanisms, including NET generation [29] to combat microorganisms. This happens more readily in the lungs compared with in other tissues, with neutrophils found to exist in higher concentrations in pulmonary vasculature compared with systemic blood vessels [30]. A prominent pathway leading to NET formation in infection is through the interaction of lipopolysaccharide (LPS), with toll-like receptor 4 (TLR4) found on neutrophils [31].
In patients with community-acquired pneumonia (CAP), increased levels of cell-free nucleosomes in serum samples, used as surrogate markers of NETosis, were found. This was associated with prolonged hospitalisation and a greater 30-day all-cause mortality [32]. This finding suggests NETs could function as a novel marker of prognosis in CAP.
Acute Lung Injury and Acute Respiratory Distress Syndrome
The degree of neutrophil influx into the lungs and NET release during ALI and ARDS positively correlates with disease progression and severity [17], with neutrophil depletion conferring protection in a transfusion-related ALI animal model [33]. NETosis seems to be a key component of ventilator-induced lung injury (VILI) [34] as evidenced by detection of citrullinated histone-3, suggesting that this was a mode of cell death independent from apoptosis and necrosis [35]. The authors suggested that this may be due to increased levels of IL-1β and HMGB1, which have been both shown to be able to induce NETosis.
Allergic Asthma
Patients with neutrophilic asthma have a greater severity of disease and reduced response to corticosteroid treatment compared with the eosinophilic type [34]. The increased expression of neutrophil chemoattractant IL-8 in airway smooth muscle cells could be contributing to disease severity [35] through inducing NETosis.
Chronic Obstructive Pulmonary Disease
NETosis has been documented as an integral part of chronic obstructive pulmonary disease (COPD) pathophysiology. Unlike asthma, where neutrophils are important in certain subtypes, NETosis has been directly linked to disease severity in COPD [36, 37]; TLR-4 expression, one of the main potentiators for NET formation, is increased during COPD exacerbations [38].
Pulmonary Hypertension
NETs are also able to potentiate dysregulated angiogenesis, as seen in patients with chronic thromboembolic pulmonary hypertension and idiopathic pulmonary hypertension as plasma levels of DNA, NE and MPO levels are significantly elevated. Moreover, NETs also seem to destabilise intercellular junctions and increase endothelial cell motility. Through direct contact with endothelial cells, NETs were found to induce the activity of the pro-inflammatory transcription factor NFκB by approximately 3-fold. Moreover, NETs increase the surface expression of von Willebrand Factor and platelet adhesion, thereby producing a prothrombotic state [39].
Kidney
Glomerulonephritides
NETs have been visualised upon immunostaining renal biopsies from patients with SLE and antineutrophil cytoplasmic antibodies-associated vasculitis (AAV) [40] and may be at least partially responsible for activating complement pathways resulting in disease exacerbations [41]. These autoimmune conditions also seem to affect the patient’s ability to degrade NETs, amplifying their deleterious inflammatory effects [12]. In crescentic glomerulonephritis, neutrophil-mediated glomerular damage is worsened by addition of extracellular MPO, which could have been released during NETosis [42]. NETosis could also contribute to the loss of immune tolerance through further externalisation of crucial autoantigens during cell death [4].
Haemolytic Uraemic Syndrome (HUS)
Plasma from affected patients exhibited a greater capacity to undergo NETosis compared with healthy patients. The ensuing damage has been linked to the pro-inflammatory cytokines IL-6 and IL-8 released from glomerular epithelial cells, upon stimulation by NETs. This potentiates microvasculature inflammation and thrombosis, precipitating renal failure [43].
Liver
Decompensated Cirrhotic Liver Disease
A deficiency in NET release has been demonstrated to play a role in the onset of end-stage liver disease [44] as neutrophils in cirrhotic patients are found to have defective ROS production [45], which commonly triggers NET release [46]. This may also partially explain why these have a predisposition to recurrent bacterial infections [47] and increased rates of decompensatory complications such as spontaneous bacterial peritonitis (SBP). This is corroborated by defective NET release from ascitic fluid neutrophils in cirrhotic patients compared with controls in vitro [48]. Cirrhotic patients with SBP were also found to have an increase in pro- and anti-inflammatory cytokines in peripheral blood and ascitic fluid [49].
Ischaemia-Reperfusion Injury
Ischaemia-reperfusion injury (IRI) is an inherent consequence of liver transplantation, hypovolaemia or trauma and results in the release of damage-associated molecular patterns (DAMPs) which, in turn, cause NET formation in a TLR-dependent manner, exacerbating inflammation [50]. Treatment with a peptidyl-arginine-deiminase 4 (PAD4) inhibitor [51] or DNase has been shown to be significantly hepatoprotective following liver IRI [50].
Cardiovascular System
Atherosclerosis
NETs are a well-known constituent of atherosclerotic lesions [52]. MPO has been strongly associated with diminishing the cardioprotective effects of high-density lipoprotein cholesterol (HDL-C) through oxidation reactions, and an increased enzymatic activity is linked to increased plaque rupture [53]. Other proteins found in NETs, such as cathelin-related antimicrobial peptide (CRAMP), have also been shown to contribute to disease progression [54]. Moreover, in vitro studies show that hypercholesterolemia triggers NET formation [55]. A large-scale study in patients with suspected coronary artery disease revealed that the markers of NETosis, such as extracellular DNA, are independently associated with disease severity [56]. Coronary specimens from patients after an acute myocardial infarction (MI) showed the presence of NETs in both fresh and lytic thrombi, therefore suggesting NETosis happens in the early stages of thrombus evolution [57]. Furthermore, NET burden was shown to be positively correlated with the infarct size in patients undergoing primary percutaneous coronary interventions post-MI. This is supported by increased levels of MPO, DNA and NE in the lesion site [58]. Therefore, NETs could potentially be considered as a novel biomarker in atherosclerosis [59].
Diabetes Mellitus-Induced Vasculopathy
It has been shown that neutrophils form peripheral blood of diabetes mellitus (DM) patients showed increased spontaneous NETosis [60]. Interestingly, metformin reduces the deleterious effects of NETosis in a mechanism independently from glucose control. One recent study showed that 2-month treatment with metformin in pre-DM patients reduced levels of components of NETs, whereas glycaemic control with other medication such as insulin saw no difference when compared with placebo controls. This has been attributed to a direct effect of metformin on inhibiting the activation of NADPH oxidase [61].
Musculoskeletal System
Rheumatoid Arthritis
Neutrophils from the peripheral blood and synovial fluid of patients with rheumatoid arthritis (RA) exhibit increased NETosis compared with healthy controls and patients with osteoarthritis. The externalisation of citrullinated proteins during the process of NETosis was found to initiate and perpetuate the aberrant immune response in RA. Moreover, the autoantibodies and inflammatory cytokines commonly seen in RA promote NETosis, resulting in a vicious cycle of tissue destruction [62].
Gout
Gout is an inflammatory disease that involves the deposition of monosodiumurate (MSU) crystals in joints. During acute gout, there is increased movement of neutrophils into the synovial fluid. MSU is a known neutrophil stimulus and it has been shown that acute gout is associated with an increase in IL-1β levels [63], a key player in NET formation.
Gut
Ulcerative Colitis
There is prominent neutrophil infiltration in the colon mucosa in ulcerative colitis (UC) [64] and this correlates with disease severity. In UC, the inflammatory environment promotes neutrophil activation and IL-1β expression [65]. In contrast, NETs do not seem to play a key role in Crohn’s disease. This may explain why mesalazine, a known inhibitor of IL-1β production and the first-line treatment for UC flare-ups, is not therapeutic in Crohn’s patients per se [66].
Reproductive System
Pre-eclampsia
Placentas from women diagnosed with pre-eclampsia showed increased neutrophil infiltration and NETosis when compared with non-hypertensive pregnant controls [67, 68] and are probably involved in causing widespread damage to the maternal endothelium [69].
Placental and Endothelial Injury During Pregnancy
Aberrant neutrophil activity during pregnancy is also associated with other severe complications, including recurrent foetal loss [70, 71]. One recent study indicated neutrophils in pregnant women seem to have an increased propensity to undergo NETosis, secondary to an increase in granulocyte colony-stimulating factor during pregnancy [72]. Progesterone has been shown to attenuate neutrophil-mediated ROS production, whereas 17β-estradiol induces intracellular ROS generation in a dose-dependent manner [73].
NETosis AS A THERAPEUTIC TARGET
Targeting critical steps in NET formation and degradation is critical for developing treatment strategies for NETosis-associated organ injury associated with NETosis (Fig. 3) [4]. Examples of recent publications on potential therapeutic compounds targeting NETosis are summarised in Table 1.

Therapeutic strategies targeting NET formation. Stimulation of neutrophil receptors (e.g. FC γ receptor, toll-like receptor) by microorganisms or sterile signals leads to release of calcium (Ca2+) from the endoplasmic reticulum (ER). Cellular Ca2+ overload results in activation of protein kinase C (PKC), assembly of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex, and/or mitochondrial activation, thus stimulating reactive oxygen species (ROS) production. Oxidative stress promotes myeloperoxidase (MPO)-dependent migration of granular neutrophil elastase (NE) into the nucleus to cleave histones. Subsequent activation of peptidylarginine deiminase (PAD) 4 induces histone citrullination to cause chromatin decondensation. The last step involves nuclear membrane degradation and extrusion of a mixture of chromatin and granular proteins into extracellular space, whereby extracellular DNase eventually digests and removes neutrophil extracellular traps (NETs). In this regard, modulation of critical steps in NET formation and degradation (shown by blocking arrows) might be beneficial for the treatment of inflammatory disorders (figure modified and reproduced with permission) (42). FcγR, Fc γ receptor; TLR, toll-like receptor.
TLR Inhibitors
Dexamethasone (Dex) has been shown in vitro to reduce NETosis in neutrophils that are stimulated with Staphylococcus aureus but not in those stimulated with PMA. The TLRs involved in S. aureus-induced NET formation seem to be TLR2 and TLR4, as agonists of these receptors rescued Dex inhibition. Interestingly, although Dex reduced NET formation, it did not significantly affect ROS production [74].
Calcineurin Inhibitors
Calcineurin is a calcium-dependent serine/threonine protein phosphatase that is important for neutrophil activity. Many stages of NETosis induction depend upon calcium mobilisation. Hence, modulators of the calcineurin pathway are potential pharmacological inhibitors of NET formation. Cyclosporine A (CsA), an antagonist of the calcineurin pathway, has been shown to reduce the effect of key physiological activators of neutrophils. This effect on NETosis may in part explain CsA’s efficacy in RA [75] and steroid-resistant UC patients [76]. PMA-induced NETosis seems to be calcium-independent, as this was not inhibited by CsA [77].
ROS Scavengers
The mitochondria are a powerful source of ROS [78]. ROS scavengers such as N-acetyl cysteine (NAC) reduce NET formation and severity of SLE in patients [79]. Trolox and Tempol are two antioxidants which have also been shown to prevent NETosis of PMA-stimulated human neutrophils in a dose-dependent manner and have been recommended for treatment of autoimmune and inflammatory pathologies [80].
PAD Inhibitors
Using a murine model of atherosclerosis, Knight and colleagues have shown that pharmacological inhibition of PAD4 using 11 weeks of daily Cl-amidine injections reduced NET-induced vascular damage, with delayed plaque progression in the carotid arteries [81]. The same group also showed that PAD inhibitors reduce disease activity in murine models of SLE, by reducing endothelial dysfunction [82]. It is worth mentioning that the possibility of PAD inhibition as a therapeutic avenue to be pursued in NET-induced organ damage in glomerulonephritis has been recently challenged by the work of Gordon and colleagues on murine models on SLE with PAD4 deletion. They showed that this did not reduce end-organ damage as measured by proteinuria [83], suggesting that mechanisms other than NET formation are implicated in this complex autoimmune condition.
DNase Therapy
DNase therapy has been proposed to improve outcomes in CF patients through reducing mucous viscosity. However, it appears that recombinant DNAse does not reduce the load of DNA/protein complexes seen in NETosis. One solution to this is to combine elastase with DNAse, in order to degrade histones and provide DNAse access to chromatin. This combination has been shown to enhance solubilisation of sputum [84].
Tetrahydroisoquinolines
In contrary to the aforementioned mechanisms of NETosis inhibitors, tetrahydroisoquinolines (THIQs) are a new class of NET formation inhibitors that do not target ROS formation or granular protein activity. As functional neutrophils are paramount to maintaining immune reactivity, this difference offers an advantage to selectively target NET overproduction without impairing normal function. The exact molecular mechanisms of THIQs are yet to be determined; however, it is known that THIQ inhibition of NETosis take place at micromolar concentrations and possibly at different stages of NET formation [85].
CONCLUSION
When regulated as part of normal physiology, NETs are anti-microbial and fundamental to the innate immune system. Dysregulated NET formation contributes to the pathogenesis of a plethora of diseases. This review has summarised the role of NETosis in pathologies of multiple body systems, as well as highlighted the stages of NETosis that has so far been investigated as emerging pharmacological targets. These putative strategies seem to hold therapeutic potential and warrant further investigation.
Abbreviations
- NET:
-
neutrophil extracellular traps
- PMN:
-
polymorphonuclear
- PAD4:
-
peptidyl arginine deiminase 4
- NADPH:
-
nicotinamide adenine dinucleotide phosphate
- ER:
-
endoplasmic reticulum
- PKC:
-
protein kinase C
- ROS:
-
reactive oxygen species
- NE:
-
neutrophil elastase
- MPO:
-
myeloperoxidase
- mtDNA:
-
mitochondrial DNA
- SLE:
-
systemic lupus erythematosus
- PMA:
-
phorbol 12-myristate 13-acetate
- CDK:
-
cyclin-dependent kinases
- AD:
-
Alzheimer’s disease
- CF :
-
cystic fibrosis
- SP-D:
-
surfactant protein D
- LPS:
-
lipopolysaccharide
- TLR4:
-
toll-like receptor 4
- CAP:
-
community-acquired pneumonia
- ALI:
-
acute lung injury
- ARDS:
-
acute respiratory distress syndrome
- VILI:
-
ventilator-induced lung injury
- HMGBI:
-
High Mobility Group Box 1
- COPD:
-
chronic obstructive pulmonary disease
- AAV:
-
antineutrophil cytoplasmic antibodies-associated vasculitis
- HUS:
-
haemolytic uraemic syndrome
- SBP:
-
spontaneous bacterial peritonitis
- IRI:
-
ischaemia-reperfusion injury
- DAMP:
-
damage-associated molecular patterns
- PAD4:
-
peptidyl-arginine-deiminase 4
- HDL-C:
-
high density lipoprotein cholesterol
- CRAMP:
-
cathelin-related antimicrobial peptide
- MI:
-
myocardial infarction
- DM:
-
diabetes mellitus
- RA:
-
rheumatoid arthritis
- MSU:
-
monosodiumurate
- UC:
-
ulcerative colitis
- CsA:
-
cyclosporine
- NAC:
-
N-acetyl cysteine
- THIQ:
-
tetrahydroisoquinolines
References
Vandenabeele, P., L. Galluzzi, T. Vanden Berghe, and G. Kroemer. 2010. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nature Reviews. Molecular Cell Biology 11 (10): 700–714. https://doi.org/10.1038/nrm2970.
Lewis, H.D., J. Liddle, J.E. Coote, S.J. Atkinson, M.D. Barker, B.D. Bax, K.L. Bicker, R.P. Bingham, M. Campbell, Y.H. Chen, C.W. Chung, P.D. Craggs, R.P. Davis, D. Eberhard, G. Joberty, K.E. Lind, K. Locke, C. Maller, K. Martinod, C. Patten, O. Polyakova, C.E. Rise, M. Rüdiger, R.J. Sheppard, D.J. Slade, P. Thomas, J. Thorpe, G. Yao, G. Drewes, D.D. Wagner, P.R. Thompson, R.K. Prinjha, and D.M. Wilson. 2015. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nature Chemical Biology 11 (3): 189–191. https://doi.org/10.1038/nchembio.1735.
Galluzzi, Lorenzo, Ilio Vitale, Stuart A. Aaronson, John M. Abrams, Dieter Adam, Patrizia Agostinis, Emad S. Alnemri, et al. 2018. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death and Differentiation 25 (3): 486–541. https://doi.org/10.1038/s41418-017-0012-4.
Gupta, S., and M.J. Kaplan. 2016. The role of neutrophils and NETosis in autoimmune and renal diseases. Nature Reviews. Nephrology 12 (7): 402–413. https://doi.org/10.1038/nrneph.2016.71.
Papayannopoulos, V. 2018. Neutrophil extracellular traps in immunity and disease. Nature Reviews. Immunology 18 (2): 134–147. https://doi.org/10.1038/nri.2017.105.
Kobayashi, S.D., and F.R. DeLeo. 2009. Role of neutrophils in innate immunity: a systems biology-level approach. Wiley Interdisciplinary Reviews. Systems Biology and Medicine 1 (3): 309–333. https://doi.org/10.1002/wsbm.32.
Metzler, K.D., C. Goosmann, A. Lubojemska, A. Zychlinsky, and V. Papayannopoulos. 2014. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Reports 8 (3): 883–896. https://doi.org/10.1016/j.celrep.2014.06.044.
Tessarz, P., and T. Kouzarides. 2014. Histone core modifications regulating nucleosome structure and dynamics. Nature Reviews. Molecular Cell Biology 15 (11): 703–708. https://doi.org/10.1038/nrm3890.
Yousefi, S., C. Mihalache, E. Kozlowski, I. Schmid, and H.U. Simon. 2009. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death and Differentiation 16 (11): 1438–1444. https://doi.org/10.1038/cdd.2009.96.
Wang, Haiting, Ting Li, Sheng Chen, Gu Yueying, and Shuang Ye. 2015. Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthritis & Rheumatology 67 (12): 3190–3200.
Kenny, E.F., A. Herzig, R. Kruger, A. Muth, S. Mondal, P.R. Thompson, V. Brinkmann, H.V. Bernuth, and A. Zychlinsky. 2017. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife 6. https://doi.org/10.7554/eLife.24437.
Hakkim, A., B.G. Furnrohr, K. Amann, B. Laube, U.A. Abed, V. Brinkmann, M. Herrmann, R.E. Voll, and A. Zychlinsky. 2010. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proceedings of the National Academy of Sciences of the United States of America 107 (21): 9813–9818. https://doi.org/10.1073/pnas.0909927107.
Farrera, C., and B. Fadeel. 2013. Macrophage clearance of neutrophil extracellular traps is a silent process. Journal of Immunology 191 (5): 2647–2656. https://doi.org/10.4049/jimmunol.1300436.
Maueroder, C., A. Mahajan, S. Paulus, S. Gosswein, J. Hahn, D. Kienhofer, M.H. Biermann, et al. 2016. Menage-a-Trois: the ratio of bicarbonate to CO2 and the pH regulate the capacity of neutrophils to form NETs. Frontiers in Immunology 7: 583. https://doi.org/10.3389/fimmu.2016.00583.
Behnen, M., S. Moller, A. Brozek, M. Klinger, and T. Laskay. 2017. Extracellular acidification inhibits the ROS-dependent formation of neutrophil extracellular traps. Frontiers in Immunology 8: 184. https://doi.org/10.3389/fimmu.2017.00184.
Amulic, B., S.L. Knackstedt, U. Abu Abed, N. Deigendesch, C.J. Harbort, B.E. Caffrey, V. Brinkmann, F.L. Heppner, P.W. Hinds, and A. Zychlinsky. 2017. Cell-cycle proteins control production of neutrophil extracellular traps. Developmental Cell 43 (4): 449–462 e445. https://doi.org/10.1016/j.devcel.2017.10.013.
Saffarzadeh, M., C. Juenemann, M.A. Queisser, G. Lochnit, G. Barreto, S.P. Galuska, J. Lohmeyer, and K.T. Preissner. 2012. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One 7 (2): e32366. https://doi.org/10.1371/journal.pone.0032366.
Pietronigro, E.C., V. Della Bianca, E. Zenaro, and G. Constantin. 2017. NETosis in Alzheimer’s disease. Frontiers in Immunology 8: 211. https://doi.org/10.3389/fimmu.2017.00211.
Chamorro, A., X. Urra, and A.M. Planas. 2007. Infection after acute ischemic stroke: a manifestation of brain-induced immunodepression. Stroke 38 (3): 1097–1103. https://doi.org/10.1161/01.STR.0000258346.68966.9d.
Vogelgesang, A., and A. Dressel. 2011. Immunological consequences of ischemic stroke: immunosuppression and autoimmunity. Journal of Neuroimmunology 231 (1–2): 105–110. https://doi.org/10.1016/j.jneuroim.2010.09.023.
Vogelgesang, A., U. Grunwald, S. Langner, R. Jack, B.M. Broker, C. Kessler, and A. Dressel. 2008. Analysis of lymphocyte subsets in patients with stroke and their influence on infection after stroke. Stroke 39 (1): 237–241. https://doi.org/10.1161/STROKEAHA.107.493635.
Ruhnau, J., K. Schulze, B. Gaida, S. Langner, C. Kessler, B. Broker, A. Dressel, and A. Vogelgesang. 2014. Stroke alters respiratory burst in neutrophils and monocytes. Stroke 45 (3): 794–800. https://doi.org/10.1161/STROKEAHA.113.003342.
Seki, Y., Y. Sahara, E. Itoh, and T. Kawamura. 2010. Suppressed neutrophil respiratory burst in patients with haemorrhagic stroke. Journal of Clinical Neuroscience 17 (2): 187–190. https://doi.org/10.1016/j.jocn.2009.04.020.
Henke, M.O., and F. Ratjen. 2007. Mucolytics in cystic fibrosis. Paediatric Respiratory Reviews 8 (1): 24–29. https://doi.org/10.1016/j.prrv.2007.02.009.
Lethem, M.I., S.L. James, C. Marriott, and J.F. Burke. 1990. The origin of DNA associated with mucus glycoproteins in cystic fibrosis sputum. The European Respiratory Journal 3 (1): 19–23.
Manzenreiter, R., F. Kienberger, V. Marcos, K. Schilcher, W.D. Krautgartner, A. Obermayer, M. Huml, W. Stoiber, A. Hector, M. Griese, M. Hannig, M. Studnicka, L. Vitkov, and D. Hartl. 2012. Ultrastructural characterization of cystic fibrosis sputum using atomic force and scanning electron microscopy. Journal of Cystic Fibrosis 11 (2): 84–92. https://doi.org/10.1016/j.jcf.2011.09.008.
Nayak, A., E. Dodagatta-Marri, A.G. Tsolaki, and U. Kishore. 2012. An insight into the diverse roles of surfactant proteins, SP-A and SP-D in innate and adaptive immunity. Frontiers in Immunology 3: 131. https://doi.org/10.3389/fimmu.2012.00131.
Noah, T.L., P.C. Murphy, J.J. Alink, M.W. Leigh, W.M. Hull, M.T. Stahlman, and J.A. Whitsett. 2003. Bronchoalveolar lavage fluid surfactant protein-A and surfactant protein-D are inversely related to inflammation in early cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 168 (6): 685–691. https://doi.org/10.1164/rccm.200301-005OC.
Brinkmann, V., U. Reichard, C. Goosmann, B. Fauler, Y. Uhlemann, D.S. Weiss, Y. Weinrauch, and A. Zychlinsky. 2004. Neutrophil extracellular traps kill bacteria. Science 303 (5663): 1532–1535. https://doi.org/10.1126/science.1092385.
Cheng, O.Z., and N. Palaniyar. 2013. NET balancing: a problem in inflammatory lung diseases. Frontiers in Immunology 4: 1. https://doi.org/10.3389/fimmu.2013.00001.
Caudrillier, A., K. Kessenbrock, B.M. Gilliss, J.X. Nguyen, M.B. Marques, M. Monestier, P. Toy, Z. Werb, and M.R. Looney. 2012. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. The Journal of Clinical Investigation 122 (7): 2661–2671. https://doi.org/10.1172/JCI61303.
Ebrahimi, F., S. Giaglis, S. Hahn, C.A. Blum, C. Baumgartner, A. Kutz, S.V. van Breda, B. Mueller, P. Schuetz, M. Christ-Crain, and P. Hasler. 2018. Markers of neutrophil extracellular traps predict adverse outcome in community-acquired pneumonia: secondary analysis of a randomised controlled trial. The European Respiratory Journal 51 (4): 1701389. https://doi.org/10.1183/13993003.01389-2017.
Looney, M.R., J.X. Nguyen, Y. Hu, J.A. Van Ziffle, C.A. Lowell, and M.A. Matthay. 2009. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. The Journal of Clinical Investigation 119 (11): 3450–3461. https://doi.org/10.1172/JCI38432.
Haldar, P., and I.D. Pavord. 2007. Noneosinophilic asthma: a distinct clinical and pathologic phenotype. The Journal of Allergy and Clinical Immunology 119 (5): 1043–1052; quiz 1053-1044. https://doi.org/10.1016/j.jaci.2007.02.042.
Shannon, J., P. Ernst, Y. Yamauchi, R. Olivenstein, C. Lemiere, S. Foley, L. Cicora, M. Ludwig, Q. Hamid, and J.G. Martin. 2008. Differences in airway cytokine profile in severe asthma compared to moderate asthma. Chest 133 (2): 420–426. https://doi.org/10.1378/chest.07-1881.
Grabcanovic-Musija, F., A. Obermayer, W. Stoiber, W.D. Krautgartner, P. Steinbacher, N. Winterberg, A.C. Bathke, M. Klappacher, and M. Studnicka. 2015. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respiratory Research 16: 59. https://doi.org/10.1186/s12931-015-0221-7.
Meijer, M., G.T. Rijkers, and F.J. van Overveld. 2013. Neutrophils and emerging targets for treatment in chronic obstructive pulmonary disease. Expert Review of Clinical Immunology 9 (11): 1055–1068. https://doi.org/10.1586/1744666X.2013.851347.
Sabroe, I., S.K. Dower, and M.K. Whyte. 2005. The role of toll-like receptors in the regulation of neutrophil migration, activation, and apoptosis. Clinical Infectious Diseases 41 (Suppl 7): S421–S426. https://doi.org/10.1086/431992.
Aldabbous, L., V. Abdul-Salam, T. McKinnon, L. Duluc, J. Pepke-Zaba, M. Southwood, A.J. Ainscough, C. Hadinnapola, M.R. Wilkins, M. Toshner, and B. Wojciak-Stothard. 2016. Neutrophil extracellular traps promote angiogenesis: evidence from vascular pathology in pulmonary hypertension. Arteriosclerosis, Thrombosis, and Vascular Biology 36 (10): 2078–2087. https://doi.org/10.1161/ATVBAHA.116.307634.
Villanueva, E., S. Yalavarthi, C.C. Berthier, J.B. Hodgin, R. Khandpur, A.M. Lin, C.J. Rubin, W. Zhao, S.H. Olsen, M. Klinker, D. Shealy, M.F. Denny, J. Plumas, L. Chaperot, M. Kretzler, A.T. Bruce, and M.J. Kaplan. 2011. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. Journal of Immunology 187 (1): 538–552. https://doi.org/10.4049/jimmunol.1100450.
Leffler, J., M. Martin, B. Gullstrand, H. Tyden, C. Lood, L. Truedsson, A.A. Bengtsson, and A.M. Blom. 2012. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. Journal of Immunology 188 (7): 3522–3531. https://doi.org/10.4049/jimmunol.1102404.
Odobasic, D., A.R. Kitching, T.J. Semple, and S.R. Holdsworth. 2007. Endogenous myeloperoxidase promotes neutrophil-mediated renal injury, but attenuates T cell immunity inducing crescentic glomerulonephritis. Journal of the American Society of Nephrology 18 (3): 760–770. https://doi.org/10.1681/ASN.2006040375.
Ramos, M.V., M.P. Mejias, F. Sabbione, R.J. Fernandez-Brando, A.P. Santiago, M.M. Amaral, R. Exeni, A.S. Trevani, and M.S. Palermo. 2016. Induction of neutrophil extracellular traps in Shiga toxin-associated hemolytic uremic syndrome. Journal of Innate Immunity 8 (4): 400–411. https://doi.org/10.1159/000445770.
Agraz-Cibrian, J.M., J.E. Segura-Ortega, V. Delgado-Rizo, and M. Fafutis-Morris. 2016. Alterations in neutrophil extracellular traps is associated with the degree of decompensation of liver cirrhosis. Journal of Infection in Developing Countries 10 (5): 512–517. https://doi.org/10.3855/jidc.7165.
Rolas, L., N. Makhezer, S. Hadjoudj, J. El-Benna, B. Djerdjouri, L. Elkrief, R. Moreau, and A. Perianin. 2013. Inhibition of mammalian target of rapamycin aggravates the respiratory burst defect of neutrophils from decompensated patients with cirrhosis. Hepatology 57 (3): 1163–1171. https://doi.org/10.1002/hep.26109.
Hosseinzadeh, A., P.K. Messer, and C.F. Urban. 2012. Stable redox-cycling nitroxide Tempol inhibits NET formation. Frontiers in Immunology 3: 391. https://doi.org/10.3389/fimmu.2012.00391.
Albillos, A., M. Lario, and M. Alvarez-Mon. 2014. Cirrhosis-associated immune dysfunction: distinctive features and clinical relevance. Journal of Hepatology 61 (6): 1385–1396. https://doi.org/10.1016/j.jhep.2014.08.010.
Agraz-Cibrian, J.M., V. Delgado-Rizo, J.E. Segura-Ortega, H.A. Maldonado-Gomez, J.F. Zambrano-Zaragoza, M. de Jesus Duran-Avelar, N. Vibanco-Perez, and M. Fafutis-Morris. 2018. Impaired neutrophil extracellular traps and inflammatory responses in the peritoneal fluid of patients with liver cirrhosis. Scandinavian Journal of Immunology: e12714. https://doi.org/10.1111/sji.12714.
Branzk, N., A. Lubojemska, S.E. Hardison, Q. Wang, M.G. Gutierrez, G.D. Brown, and V. Papayannopoulos. 2014. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nature Immunology 15 (11): 1017–1025. https://doi.org/10.1038/ni.2987.
Huang, H., S. Tohme, A.B. Al-Khafaji, S. Tai, P. Loughran, L. Chen, S. Wang, et al. 2015. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology 62 (2): 600–614. https://doi.org/10.1002/hep.27841.
Wang, Y., P. Li, S. Wang, J. Hu, X.A. Chen, J. Wu, M. Fisher, K. Oshaben, N. Zhao, Y. Gu, D. Wang, G. Chen, and Y. Wang. 2012. Anticancer peptidylarginine deiminase (PAD) inhibitors regulate the autophagy flux and the mammalian target of rapamycin complex 1 activity. The Journal of Biological Chemistry 287 (31): 25941–25953. https://doi.org/10.1074/jbc.M112.375725.
Megens, R.T., S. Vijayan, D. Lievens, Y. Doring, M.A. van Zandvoort, J. Grommes, C. Weber, and O. Soehnlein. 2012. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thrombosis and Haemostasis 107 (3): 597–598. https://doi.org/10.1160/TH11-09-0650.
Shao, B., S. Pennathur, and J.W. Heinecke. 2012. Myeloperoxidase targets apolipoprotein A-I, the major high density lipoprotein protein, for site-specific oxidation in human atherosclerotic lesions. The Journal of Biological Chemistry 287 (9): 6375–6386. https://doi.org/10.1074/jbc.M111.337345.
Doring, Y., M. Drechsler, S. Wantha, K. Kemmerich, D. Lievens, S. Vijayan, R.L. Gallo, C. Weber, and O. Soehnlein. 2012. Lack of neutrophil-derived CRAMP reduces atherosclerosis in mice. Circulation Research 110 (8): 1052–1056. https://doi.org/10.1161/CIRCRESAHA.112.265868.
Liu, M.L., M. Bashir, K. Williams, and V. Werth. 2014. Cholesterol loading induces neutrophil extracellular traps, and atorvastatin attenuates this effect. Arthritis & Rheumatology 66: S530–S530.
Borissoff, J.I., I.A. Joosen, M.O. Versteylen, A. Brill, T.A. Fuchs, A.S. Savchenko, M. Gallant, K. Martinod, H. ten Cate, L. Hofstra, H.J. Crijns, D.D. Wagner, and B.L.J.H. Kietselaer. 2013. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arteriosclerosis, Thrombosis, and Vascular Biology 33 (8): 2032–2040. https://doi.org/10.1161/ATVBAHA.113.301627.
de Boer, O.J., X. Li, P. Teeling, C. Mackaay, H.J. Ploegmakers, C.M. van der Loos, M.J. Daemen, R.J. de Winter, and A.C. van der Wal. 2013. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thrombosis and Haemostasis 109 (2): 290–297. https://doi.org/10.1160/TH12-06-0425.
Mangold, A., S. Alias, T. Scherz, T. Hofbauer, J. Jakowitsch, A. Panzenbock, D. Simon, et al. 2015. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circulation Research 116 (7): 1182–1192. https://doi.org/10.1161/CIRCRESAHA.116.304944.
Doring, Y., C. Weber, and O. Soehnlein. 2013. Footprints of neutrophil extracellular traps as predictors of cardiovascular risk. Arteriosclerosis, Thrombosis, and Vascular Biology 33 (8): 1735–1736. https://doi.org/10.1161/ATVBAHA.113.301889.
Fadini, G.P., L. Menegazzo, M. Rigato, V. Scattolini, N. Poncina, A. Bruttocao, S. Ciciliot, F. Mammano, C.D. Ciubotaru, E. Brocco, M.C. Marescotti, R. Cappellari, G. Arrigoni, R. Millioni, S. Vigili de Kreutzenberg, M. Albiero, and A. Avogaro. 2016. NETosis delays diabetic wound healing in mice and humans. Diabetes 65 (4): 1061–1071. https://doi.org/10.2337/db15-0863.
Menegazzo, L., V. Scattolini, R. Cappellari, B.M. Bonora, M. Albiero, M. Bortolozzi, F. Romanato, G. Ceolotto, S. Vigili de Kreutzeberg, A. Avogaro, and G.P. Fadini. 2018. The antidiabetic drug metformin blunts NETosis in vitro and reduces circulating NETosis biomarkers in vivo. Acta Diabetologica 55 (6): 593–601. https://doi.org/10.1007/s00592-018-1129-8.
Khandpur, R., C. Carmona-Rivera, A. Vivekanandan-Giri, A. Gizinski, S. Yalavarthi, J.S. Knight, S. Friday, et al. 2013. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Science Translational Medicine 5 (178): 178ra140. https://doi.org/10.1126/scitranslmed.3005580.
Mitroulis, I., K. Kambas, A. Chrysanthopoulou, P. Skendros, E. Apostolidou, I. Kourtzelis, G.I. Drosos, D.T. Boumpas, and K. Ritis. 2011. Neutrophil extracellular trap formation is associated with IL-1beta and autophagy-related signaling in gout. PLoS One 6 (12): e29318. https://doi.org/10.1371/journal.pone.0029318.
Brazil, J.C., N.A. Louis, and C.A. Parkos. 2013. The role of polymorphonuclear leukocyte trafficking in the perpetuation of inflammation during inflammatory bowel disease. Inflammatory Bowel Diseases 19 (7): 1556–1565. https://doi.org/10.1097/MIB.0b013e318281f54e.
Angelidou, I., A. Chrysanthopoulou, A. Mitsios, S. Arelaki, A. Arampatzioglou, K. Kambas, D. Ritis, V. Tsironidou, I. Moschos, V. Dalla, D. Stakos, G. Kouklakis, I. Mitroulis, K. Ritis, and P. Skendros. 2018. REDD1/autophagy pathway is associated with neutrophil-driven IL-1beta inflammatory response in active ulcerative colitis. Journal of Immunology 200 (12): 3950–3961. https://doi.org/10.4049/jimmunol.1701643.
Lim, W.C., Y. Wang, J.K. MacDonald, and S. Hanauer. 2016. Aminosalicylates for induction of remission or response in Crohn’s disease. Cochrane Database of Systematic Reviews 7: CD008870. https://doi.org/10.1002/14651858.CD008870.pub2.
Erpenbeck, L., C.S. Chowdhury, Z.K. Zsengeller, M. Gallant, S.D. Burke, S. Cifuni, S. Hahn, D.D. Wagner, and S.A. Karumanchi. 2016. PAD4 deficiency decreases inflammation and susceptibility to pregnancy loss in a mouse model. Biology of Reproduction 95 (6): 132. https://doi.org/10.1095/biolreprod.116.140293.
Marder, W., J.S. Knight, M.J. Kaplan, E.C. Somers, X. Zhang, A.A. O’Dell, V. Padmanabhan, and R.W. Lieberman. 2016. Placental histology and neutrophil extracellular traps in lupus and pre-eclampsia pregnancies. Lupus Science and Medicine 3 (1): e000134. https://doi.org/10.1136/lupus-2015-000134.
Powe, C.E., R.J. Levine, and S.A. Karumanchi. 2011. Preeclampsia, a disease of the maternal endothelium: the role of antiangiogenic factors and implications for later cardiovascular disease. Circulation 123 (24): 2856–2869. https://doi.org/10.1161/CIRCULATIONAHA.109.853127.
Giaglis, S., M. Stoikou, F. Grimolizzi, B.Y. Subramanian, S.V. van Breda, I. Hoesli, O. Lapaire, P. Hasler, N.G. Than, and S. Hahn. 2016. Neutrophil migration into the placenta: good, bad or deadly? Cell Adhesion & Migration 10 (1–2): 208–225. https://doi.org/10.1080/19336918.2016.1148866.
Girardi, G., J. Berman, P. Redecha, L. Spruce, J.M. Thurman, D. Kraus, T.J. Hollmann, P. Casali, M.C. Caroll, R.A. Wetsel, J.D. Lambris, V.M. Holers, and J.E. Salmon. 2003. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. The Journal of Clinical Investigation 112 (11): 1644–1654. https://doi.org/10.1172/JCI18817.
Giaglis, S., M. Stoikou, C. Sur Chowdhury, G. Schaefer, F. Grimolizzi, S.W. Rossi, I.M. Hoesli, O. Lapaire, P. Hasler, and S. Hahn. 2016. Multimodal regulation of NET formation in pregnancy: progesterone antagonizes the pro-NETotic effect of estrogen and G-CSF. Frontiers in Immunology 7: 565. https://doi.org/10.3389/fimmu.2016.00565.
Maleki, J., M. Nourbakhsh, M. Shabani, M. Korani, S.M. Nourazarian, M.R. Ostadali Dahaghi, and M.H. Moghadasi. 2015. 17beta-estradiol stimulates generation of reactive species oxygen and nitric oxide in ovarian adenocarcinoma cells (OVCAR 3). Iranian Journal of Cancer Prevention 8 (3): e2332. https://doi.org/10.17795/ijcp2332.
Wan, T., Y. Zhao, F. Fan, R. Hu, and X. Jin. 2017. Dexamethasone inhibits S. aureus-induced neutrophil extracellular pathogen-killing mechanism, possibly through toll-like receptor regulation. Frontiers in Immunology 8: 60. https://doi.org/10.3389/fimmu.2017.00060.
Gremese, E., and G.F. Ferraccioli. 2004. Benefit/risk of cyclosporine in rheumatoid arthritis. Clinical and Experimental Rheumatology 22 (5 Suppl 35): S101–S107.
Ina, K., K. Kusugami, M. Shimada, T. Tsuzuki, Y. Nishio, D.G. Binion, A. Imada, and T. Ando. 2002. Suppressive effects of cyclosporine A on neutrophils and T cells may be related to therapeutic benefits in patients with steroid-resistant ulcerative colitis. Inflammatory Bowel Diseases 8 (1): 1–9.
Gupta, A.K., S. Giaglis, P. Hasler, and S. Hahn. 2014. Efficient neutrophil extracellular trap induction requires mobilization of both intracellular and extracellular calcium pools and is modulated by cyclosporine A. PLoS One 9 (5): e97088. https://doi.org/10.1371/journal.pone.0097088.
Lood, C., L.P. Blanco, M.M. Purmalek, C. Carmona-Rivera, S.S. De Ravin, C.K. Smith, H.L. Malech, J.A. Ledbetter, K.B. Elkon, and M.J. Kaplan. 2016. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nature Medicine 22 (2): 146–153. https://doi.org/10.1038/nm.4027.
Patel, S., S. Kumar, A. Jyoti, B.S. Srinag, R.S. Keshari, R. Saluja, A. Verma, K. Mitra, M.K. Barthwal, H. Krishnamurthy, V.K. Bajpai, and M. Dikshit. 2010. Nitric oxide donors release extracellular traps from human neutrophils by augmenting free radical generation. Nitric Oxide 22 (3): 226–234. https://doi.org/10.1016/j.niox.2010.01.001.
Vorobjeva, Nina V., and Boris V. Pinegin. 2016. Effects of the antioxidants Trolox, Tiron and Tempol on neutrophil extracellular trap formation. Immunobiology 221 (2): 208–219. https://doi.org/10.1016/j.imbio.2015.09.005.
Knight, J.S., W. Luo, A.A. O’Dell, S. Yalavarthi, W. Zhao, V. Subramanian, C. Guo, et al. 2014. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circulation Research 114 (6): 947–956. https://doi.org/10.1161/CIRCRESAHA.114.303312.
Knight, J.S., W. Zhao, W. Luo, V. Subramanian, A.A. O’Dell, S. Yalavarthi, J.B. Hodgin, D.T. Eitzman, P.R. Thompson, and M.J. Kaplan. 2013. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. The Journal of Clinical Investigation 123 (7): 2981–2993. https://doi.org/10.1172/JCI67390.
Gordon, Rachael A., Jan M. Herter, Florencia Rosetti, Allison M. Campbell, Hiroshi Nishi, Michael Kashgarian, Sheldon I. Bastacky, et al. 2017. Lupus and proliferative nephritis are PAD4 independent in murine models. JCI Insight 2 (10): e92926. https://doi.org/10.1172/jci.insight.92926.
Papayannopoulos, V., D. Staab, and A. Zychlinsky. 2011. Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving DNase therapy. PLoS One 6 (12): e28526. https://doi.org/10.1371/journal.pone.0028526.
Martinez, N.E., T.J. Zimmermann, C. Goosmann, T. Alexander, C. Hedberg, S. Ziegler, A. Zychlinsky, and H. Waldmann. 2017. Tetrahydroisoquinolines: new inhibitors of neutrophil extracellular trap (NET) formation. Chembiochem 18 (10): 888–893. https://doi.org/10.1002/cbic.201600650.
Author information
Authors and Affiliations
Contributions
DM designed and reviewed the manuscript. ZC and HZ wrote the first draft of the paper. All authors read, revised and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Ethics Approval and Consent to Participate
Not applicable
Consent for Publication
Not applicable
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cahilog, Z., Zhao, H., Wu, L. et al. The Role of Neutrophil NETosis in Organ Injury: Novel Inflammatory Cell Death Mechanisms. Inflammation 43, 2021–2032 (2020). https://doi.org/10.1007/s10753-020-01294-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-020-01294-x