
Abstract
Impaired intestinal mucosal integrity during colitis involves the peroxisome proliferator-activated receptor-γ (PPARγ), an important anti-inflammatory factor in intestinal mucosa homoeostasis, which is a potential target in colitis. Recurrent chronic pain is a vital pathogenetic feature of colitis. Nevertheless, potential functions of PPARγ in the colitis-associated hyperalgesia remain unclear. This study aimed to investigate biological roles of pioglitazone in relieving colitis-associated pain hypersensitivity by a PPARγ tight junction protein-dependent mechanism during the course of dextran sodium sulfate (DSS)-induced intestinal inflammation. The DSS-induced colitis model was generated in C57BL/6 mice. Changes in colitis induced the injury of intestinal mucosal barrier and hyperalgesia after a 6-day treatment of pioglitazone (25 mg/kg, IP injection) were assessed through immunofluorescent, hematoxylin and eosin (H&E) staining, western blot analysis, and determination of paw withdrawal mechanical threshold. A significant reduction of paw withdrawal mechanical threshold occurred after DSS treatment. Follow-up data showed that systematic administration of PPARγ agonist pioglitazone ameliorated the DSS-induced colitis and the development of colitis-associated hyperalgesia by repairing the intestinal mucosal barrier. The tight junction proteins ZO-1 and Claudin-5 were upregulated by PPARγ signaling, which in turn promoted the improvement of intestinal barrier function. Moreover, pioglitazone inhibited phosphorylation of ERK and NF-κB in the colon and decreased the levels of inflammatory cytokines in both colon spine tissues. Furthermore, systemically pioglitazone treatment inhibited the activation of microglia and astrocytes, as well as DSS-induced phosphorylation of NR2B subunit in spinal cord, which was correspondingly consistent with the pain behavior. Pioglitazone ameliorates DSS-induced colitis and attenuates colitis-associated mechanical hyperalgesia, with improving integrity of the intestinal mucosal barrier by directly upregulating tight junction proteins. The PPARγ-tight junction protein signaling might be a potential therapeutic target for the treatment of colitis-associated chronic pain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Inflammatory bowel diseases (IBD), including Crohn’s disease and ulcerative colitis, are characterized by diarrhea, weight loss, and chronic pain. IBD result in debilitating illness [1], among which chronic pain [2] emerges from the hyperresponsiveness of neuronal, immune, and endocrine signaling pathways within the intestines, the peripheral [3], and the central nervous system [4]. However, the mechanisms underlying IBD-associated chronic pain are largely unresolved and treatment options are limited. For the nervous system, the mechanisms involved in IBD-associated chronic pain encompass neuronal synaptic changes couple with increased neurotransmitter release [5]. The mechanisms in the inflammatory intestinal wall include interactions of immune cells, macrophages, smooth muscles, and enteric glias [6,7,8,9]. Upon epithelial injury and intestinal inflammation in IBD, compromised intestinal barrier integrity subsequently arises, dysregulated transportation of water and ion, exposures of immune cells to bacterial antigens, and triggers reactive enteric gliosis. The above pathological changes eventually result in a profound inflammatory immune response, and in turn, worsening the damage of intestinal mucosa [10, 11]. Tight junctions are composed by a series of transmembrane proteins including the claudins, occludins, junctional adhesion molecules with immunoglobulin-like domains, and intracellular scaffold proteins (i.e., zonula occludens) [12]. Tight junctions are pivotal in regulating intestinal permeability and maintaining intestinal barrier integrity. Emerging evidences from experimental intestinal inflammation models have supported the idea that a strong connection exists between tight junction protein impairment and intestinal inflammation [11, 13].
Peroxisome proliferator-activated receptor gamma (PPARγ), a number of the nuclear hormone receptor family, exerts a crucial role in mediating inflammatory diseases (e.g., colitis [14], liver steatosis [15], and rheumatoid arthritis [16]) and inflammatory and neuropathic pain development [17].
The colon expresses a high density of PPARγ, which presents anti-inflammatory effects on inhibiting the activation of NF-κB and expressions of the pro-inflammatory cytokines IL-1 and TNF-α [14, 18]. Earlier studies found PPARγ deficiency alongside elevated activation of NF-κB in the intestinal mucosa in UC patients, suggesting that PPARγ is a potential therapeutic target for UC [14, 19]. As a nuclear receptor, PPARγ-induced sustained changes in gene expressions are widely believed to be the key mechanism of pain reduction [20,21,22]. Repeated administration of PPARγ agonists reduces neuropathic pain-like behavior and associated molecular changes in the spinal cord dorsal horn [23]. However, it is still unclear how distinct intestinal inflammation contributes to the chronic pain development. Here, we characterized the anti-inflammatory and analgesia effects of systemic PPARγ activation by pioglitazone administration in dextran sodium sulfate (DSS)-induced acute colitis mice [24].
METHODS
Animals
Eight-week-old male C57BL/6 mice (Beijing Vital River Laboratory Animal Technology Co., Ltd.) weighing 20 to 25 g at the time of behavioral procedures were housed in a standard environment with a 12-h light/dark cycle, (20 ± 2 °C) temperature and humidity-controlled room with 4 mice per cage. Mice were given free accesses to food and water provided ad libitum. All efforts were made to minimize animal suffering, reduce the number of animals used, and use alternatives to in vivo techniques. All animal procedures were performed in accordance with the Animal Care Committee of the Institutional Animal Care and Use Committee of the Medical School of Nanjing University.
Mouse Model and Drug Treatments
For acute colitis, mice (DSS) were given one cycle of 2.5% DSS [25] (MW 36,000–50,000 Da; MP Biomedicals) for 7 days. Twenty mice (CON) and 31 mice (DSS) were used in this study. To target PPARγ in colitis, mice were IP injected with 25 mg/kg body weight [26] of the PPARγ agonist pioglitazone (HY-14601, MCE Chemicals) dissolved in PBS containing 20% DMSO (Sigma Aldrich) during the period of DSS-induced colitis. Control mice were IP injected with an equal volume of PBS containing 20% DMSO. Seven mice (CON + Veh), 21 mice (DSS + Veh), and 17 mice (DSS + Pio) were used in the study. During the course of the experiment, mice were monitored for body weight, diarrhea, and macroscopic bleeding.
Paw Withdrawal Mechanical Threshold
Paw withdrawal mechanical threshold (PWMT) determined at day 1 was recorded as the baseline. In addition, PWMT was performed every day during DSS treatment or pioglitazone injection, and every other day till day 21. All tests were performed during the light phase. Before determination, mice were allowed to acclimatize for at least 30 min. PWMT was performed in a quiet test room by the same investigator. Von Frey filaments (Stoelting, Wood Dale, IL, USA) were used to assess the mechanical allodynia as previously reported [27]. Briefly, the mice were placed into individual transparent compartments onto a metal mesh floor. Different von Frey filaments (0.16, 0.4, 0.6, 1.0, 1.4, and 2.0 g) were applied to the hind paw. The filaments were pressed vertically against the plantar surface with sufficient force to cause a slight bending against the paw for 6 to 8 s. An interval for at least 10 min was necessary between the two stimulations. Brisk withdrawal of the paw or paw flinching was regarded as a positive response. Each mouse was tested five times per stimulus strength. The lowest von Frey filament stimulus strength that produced at least 3 positive responses was recorded as the reasonable paw withdrawal mechanical threshold.
Western Blot
Total tissue or cell lysates were prepared with a detergent lysis buffer. Western blot was performed using the indicated primary antibodies: NF-κB p65 (1:1000, Cell Signaling Technology, no. 8242, MA, USA), phospho-p65 (Ser536) (1:1000, Cell Signaling Technology, no. 3031, MA, USA), ERK (1:1000, Cell Signaling Technology, no. 9102, MA, USA), phospho-ERK (p44/p42) (Thr202/Tyr204) (1:1000, Cell Signaling Technology, no. 9102, MA, USA), GFAP (1:1000, Cell Signaling Technology, no. 3670, MA, USA), MMP9 (1:1000, Abcam, no. 38898, Cambridge, UK), NR2B (1:1000, Abcam, no. 65783, Cambridge, UK), phospho-NR2B (1:1000, Abcam, no. 3856, Cambridge, UK), IL-6 (1:1000, Abcam, no. 208113, Cambridge, UK), TNF-α (1:500, Santa Cruz Biotechnology, sc52746, Santa Cruz, CA), Iba1 (1:500, Santa Cruz Biotechnology, sc32725, Santa Cruz, CA), ZO-1 (1:1000, Thermo Fisher, no. 617300, US), Claudin-5 (1:1000, Thermo Fisher, no. 35-2500, US), PPARγ (1:1000, Proteintech, 16643-1-AP, US), anti-GAPDH (1:1000, Boster Biotechnology, BM1985, Wuhan, China), and α-tubulin (1:1000, Boster Biotechnology, BM3885, Wuhan, China). Each blot was repeated three times.
Immunofluorescence
Immunofluorescence was performed as previously reported. Briefly, the lumbar segments of the spinal cords were extracted and post fixed in 4% paraformaldehyde, followed by dehydration in 30% sucrose at 4 °C. Serial frozen sections were cut in a freezing microtome into 20-μm thick slides. The following indicated primary antibodies were used: Iba-1 (1:500, Wako, 016-26721, Japan) and GFAP (1:100, Cell Signaling Technology, no. 3670, MA, USA). The secondary antibodies used in this study included the following: goat anti-rabbit (1:3000, Alexa 488-conjugated, ThermoFisher, A32723, Waltham, MA) and goat anti-mouse (1:3000, Alexa 594-conjugated, A32740, ThermoFisher, Waltham, MA). DAPI (Abcam, Cambridge, no. 104139, UK) staining was used to determine the cell nuclei.
Histopathological Analysis
Ten percent buffered neutral formalin-embedded colon sections (5 μm) were stained with hematoxylin and eosin (H&E) and independently analyzed by pathologists in a blinded way.
Statistical Analysis
Data were expressed as the mean ± SE. SPSS 22.0 (SPSS Inc., Chicago, IL) was used to conduct all the statistical analyses. Mice were assigned to different treatment groups in a randomized manner. Multiple comparisons were carried out to determine the overall differences of pain behaviors at each time point. Repeated measures analysis of variance (ANOVA) was performed to assess the changes of pain behaviors over time. One-way ANOVA was used to determine differences in the results of colon length and immunofluorescence among groups.
In both cases, when significant main effects were observed, Bonferroni post hoc tests were conducted to determine the source(s) of these differences. P < 0.05 was considered statistically significant.
RESULTS
Downregulated PPARγ, Activated P65, and ERK Accompanied Release of Inflammatory Cytokines in Colonic Tissues of DSS-Induced Colitis Mice
To explore regulatory effect of PPARγ on the colon with intestinal inflammation, C57BL/6 mice were treated by 2.5% DSS for 7 days. DSS-treated mice showed more pronounced weight loss than that of control mice (Fig. 1a). Acutely DSS-treated mice also showed a significant change in colon length (Fig. 1b, c). Colonic levels of the inflammatory cytokines TNF-α and IL-6 as markers of disease activity were measured by using western blot, which were significantly upregulated in the acute phase of colitis (day 7). Then, the expressions of TNF-α and IL-6 continued to increase in early recovery phase (day 14) and decreased later on day 21. A previous study showed that MMP9 was highly expressed in UC patients compared with control tissues [28]. Thus, we detected the expression of MMP9 in colonic tissues. MMP9 was notably upregulated after DSS treatment on day 7 and day 14. Western blot analysis also showed that DSS treatment notably reduced PPARγ expression in day 7 (Fig. 1d). Consistent with the levels of TNF-α and IL-6, further analysis showed that P65 and ERK were strongly activated in DSS-treated mice on day 7 and day 14 (Fig. 1e). Studies indicated that PPARγ has been identified to activate the NF-κB pathway and MAPK pathway, thus decreasing the levels of pro-inflammatory cytokines [29, 30]. These results suggested that PPARγ in the colon participates in the progression of acute intestinal inflammation.

PPARγ expression is impaired during colitis with activation of ERK and P65. a Percentage of weight change during 2.5% DSS treatment and recovery. b Image of colon in 2.5% DSS-treated mice and control mice on day 7. c Quantification of colon length. d Increased IL-6, TNF-α, and MMP9 expression in inflamed colonic tissue of 2.5% DSS-treated mice on day 7 and day 14. Decreasing PPARγ expression on day 7. e Increased ERK and P65 activation in inflamed colonic tissue after DSS treatment. Immunoblot analysis is performed in tissue lysates with indicated antibodies. Each lane represents one mouse. *p < .05, **p < 0.01, ***p < 0.001 compared with CON and DSS group mice. Error bars represent ± SE.
Tight Junction Protein Impairment Alone with Activation of P65 and ERK
Histological changes on day 7 of acute DSS colitis were analyzed by H&E staining of paraffin-embedded colonic cross-sections. The results showed strong transmural inflammation with loss of crypt structure, severe epithelial erosions, and more neutrophilic infiltrates in DSS-treated mice. H&E analysis indicated a breakdown of epithelial barrier function after DSS treatment (Fig. 2a). It is reported that impairment of epithelial tight junction proteins is responsible for decreased barrier integrity leading to colitis [31]. ZO-1 and claudin-5 are important epithelial tight junction proteins [32]. Western blot analysis showed that the expressions of ZO-1 and claudin-5 were significantly downregulated in DSS-induced colitis mice compared with the control group on day 7, day 14, and day 21 (Fig. 2b), suggesting the dysfunction of epithelial barrier.

Gut epithelial barrier dysfunction accompanied with decreased the expression of ZO-1 and claudin-5 in colitis. a H&E staining of a section obtained from the distal colons of CON and DSS-treated mice on day 7. b Decreased expression of ZO-1 and claudin-5 after DSS treatment. Each lane represents one mouse.
Acute Colonic Inflammation Leads to Persistent Pain, Increased Inflammatory Cytokines, and Phosphorylation of NR2B in the Spinal Cord
Chronic abdominal pain frequently happens in patients with inflammatory bowel disease [33]. Persistent abdominal pain also occurs in mice with experimental acute intestinal inflammation [34]. However, whether acute intestinal inflammation would result in hyperalgesia remains unclear. Thus, PWMT was performed to evaluate mechanical allodynia and hyperalgesia. After DSS treatment, the withdrawal threshold of both hind paws to mechanical stimuli was lower in experimental mice compared with control groups. Withdrawal thresholds in DSS-induced colitis mice significantly decreased between day 6 and day 21 (Fig. 3a, b), indicating the persistent discomfort even though body weight gradually recovered from day 10. These data indicated that DSS-induced acute colitis would lead to inflammation-related hyperalgesia.

Activation of astrocyte and microglia as well as NR2B signaling is required for colitis-reduced mechanical threshold. a Paw withdrawal mechanical threshold in response to von Frey filaments in CON and DSS-treated mice from day 1 to day 21. b Activation of spinal microglia and astrocyte. Immunofluorescent staining and corresponding quantification of staining intensity of Iba-1 (a microglia marker) and GFAP (an astrocyte marker) in the spinal cords performing in CON and DSS (day 7) group mice. c Increased expression of IL-6, TNF-α, and p-NR2B after DSS treatment. Spinal cord lysates of CON and DSS (day 7, day 14, and day 21) group mice. Each lane represents one mouse. All data are presented as the mean ± SE. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with CON and DSS group mice.
DSS treatment resulted in evident activation of microglia and astrocytes with increased levels of inflammatory cytokines TNF-α and IL-6 in the lumbar spinal cord (Fig. 3b, c). In addition, phosphorylation and activation of NR2B in the spine are vital in the maintenance of central sensitization and mechanical allodynia [35, 36]. Results of western blot showed that DSS treatment notably increased phosphorylation of NR2B in the spinal cord from day 7 to day 21, which was consistent with the changes of PWMT (Fig. 3c).
Pioglitazone Alleviates DSS-Induced Immune Response Through Downregulation of P65 and ERK
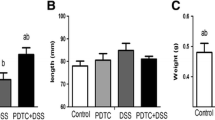
PPARγ, a member of the nuclear hormone receptor family, can be activated by anti-diabetic thiazolidinedione drugs, such as pioglitazone [37]. The treatment with pioglitazone or vehicle was introduced for 7 days during DSS administration. Mice with DSS-induced colitis receiving vehicle presented more pronounced weight loss, shorter length of colons, and more severe histologic alterations and macroscopic damages compared with those of controls. In the preventive group, pioglitazone successfully protected against weight loss and colonic shortening (Fig. 4b, c). Obviously, pioglitazone rescued DSS-induced reduction of PPARγ in colon tissues (Fig. 4d). A previous research has shown that PPARγ could regulate the NF-κB pathway and MAPK signaling activation, thus decreasing the levels of pro-inflammatory cytokines [38]. Western blot analysis showed that DSS-induced activation of P65 and ERK was significantly inhibited in pioglitazone preventive group, along with decreased expressions of IL-6, TNF-α, and MMP9 (Fig. 4d, e).

Pioglitazone promoted the recovery from intestinal inflammation. a Percentage of weight change in indicated groups. Daily IP injection of PIO during DSS administration. b Image of colons collected from DSS + Veh and DSS + PIO group mice (day 7). c Quantification of colon lengths in the different groups. d Colonic lysates of CON + Veh, DSS + Veh, and DSS + PIO (day 7, day 14, and day 21) group mice were analyzed by immunoblotting with IL-6, TNF-α, MMP9, and PPARγ antibodies, respectively. Each lane represents one mouse. d Colonic lysates of CON + Veh, DSS + Veh, and DSS + PIO (day 7, day 14, and day 21) group mice were analyzed by immunoblotting with p-ERK, ERK, p-P65, and P65 antibodies, respectively. Each lane represents one mouse. All data are presented as the mean ± SE. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with CON + Veh and DSS + PIO group mice. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with CON + Veh and DSS + Veh group mice. +p < 0.05, ++p < 0.01, +++p < 0.001 compared with DSS + Veh and DSS + PIO group mice.
Enhanced Tight Junction Expression and Recovered Intestinal Barrier Function Induced by Pioglitazone
Pathological injury in mouse colorectum was independently examined by experienced pathologists. H&E staining (magnification 100×) of colorectum tissues from 3 mice in each group was conducted on day 7 (Fig. 5a). Within intestinal samples from mice in each group, expressions of claudin-5 and ZO-1 were significantly downregulated in the intestines of DSS-treated mice, whereas the expression levels of them were dramatically elevated in the intestines of the pioglitazone preventive group (Fig. 5b).

Pioglitazone-alleviated tissue injury was histologically evident by increasing the expression of ZO-1 and claudin-5. a H&E staining of a section obtained from the distal colons of CON + Veh, DSS + Veh, and DSS + PIO mice on day 7. b Colonic lysates of CON + Veh, DSS + Veh, and DSS + PIO (day 7, day 14, and day 21) group mice were analyzed by immunoblotting with ZO-1 and claudin5 antibodies, respectively. Each lane represents one mouse.
Pioglitazone Reduces Mechanical Hypersensitivity in Colitis Mice
Pioglitazone-treated mice showed decreased mechanically hypersensitivity and gradual recovery of inflammation during 2 weeks post-DSS discontinuation (Fig. 6a). Less activated microglia and astrocytes in spinal dorsal cord could also be observed in pioglitazone-treated mice compared with DSS-treated mice, which were consistent with protein level changes of Iba1 and GFAP in the spinal dorsal cord (Fig. 6b, c). Western blot analysis of spinal dorsal cord also showed that pioglitazone treatment remarkably decreased the levels of TNF-α and IL-6 (Fig. 6c). Further analysis showed that phosphorylation of NR2B was notably suppressed after pioglitazone intervention, which was in accordance with the findings of pain-related behaviors (Fig. 6d).

The antihyperalgesic effect of pioglitazone is mediated by inhibiting colitis-induced spinal inflammation. a Paw withdrawal mechanical threshold in response to von Frey filaments in indicated groups (CON + Veh, DSS + Veh, and DSS + PIO) from day 1 to day 21. b Immunofluorescent staining and corresponding quantification of staining intensity of Iba-1 (a microglia marker) and GFAP (an astrocyte marker) in the spinal cords in indicated groups (CON + Veh, DSS + Veh, and DSS + PIO) on day 7. c Spinal cord lysates of CON + Veh, DSS + Veh, and DSS + PIO (day 7, day 14, and day 21) group mice were analyzed by immunoblotting with IL-6, TNF-α, GFAP, and Iba1 antibodies. d Immunoblotting was performed to analyze the expressions of p-NR2B and NR2B in spinal cord of CON + Veh, DSS + Veh, and DSS + PIO (day 7, day 14, and day 21) group mice. All data are presented as the mean ± SE. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with CON + Veh and DSS + PIO group mice. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with CON + Veh and DSS + Veh group mice. +p < 0.05, ++p < 0.01, +++p < 0.001 compared with DSS + Veh and DSS + PIO group mice.
DISCUSSION
This study demonstrated that systematic administration of pioglitazone, an agonist of PPARγ, could alleviate DSS-induced colitis, attenuate colitis-associated mechanical hyperalgesia, and improve integrity of the intestinal mucosal barrier by directly upregulating tight junction proteins.
Increasing effects have been made on clarifying the possible role of gut-brain-axis in the complex regulation of pain [39, 40]. However, it is still necessary to explore more crosstalks between the intestinal tract and the central nervous system or enteric nervous system in pain regulation. The etiology of IBD-associated chronic pain remains enigmatic. Visceral hypersensitivity and systematic mechanical hyperalgesia have been well explored in the researches of IBD-associated pain [41]. Hypersensitivity is driven by peripheral and central mechanisms [42], involving the participation of the intestinal wall, spinal cord, and brain centers [43]. Nonetheless, contemporary histopathology studies in IBD patients, as well as the established murine models of intestinal inflammation, implicate a causative combination of progressive destruction of the intestinal mucosal barrier and altered mucosal immune responses [44].
Previous studies have indicated a potential linkage between genetic mutations or deletions in tight junction-associated proteins and development of IBD [45]. Further studies also have proven that stabilizing junctional complex in intestinal barrier could attenuate intestinal inflammation [46]. In this paper, tight junction proteins claudin-5 and ZO-1 were notably downregulated in mice after DSS treatment, and further H&E identically supported our findings, indicating the impairment of intestinal mucosal barrier in DSS-induced colitis mice. Interestingly, decreased protein level of PPARγ was corresponding to the severity of intestinal inflammation. Based on the differences in mechanical thresholds observed between controls and experimental mice, potential correlation between intestinal inflammation and changes in mechanical hypersensitivity was investigated. Because the PPARγ system is a relevant target for the treatment of inflammatory diseases and pain, molecular mechanisms underlying the healing of intestinal mucosal injury and analgesic effect of pioglitazone in DSS-induced colitis mice were clarified here.
In our analysis, the PPARγ antagonist pioglitazone was administered once a day in mice from day 1 to day 7 alone or in combination with DSS. Our data suggested that the anti-inflammatory effects of pioglitazone were closely linked to its regulatory ability on tight junction protein expressions. The prevention of intestinal mucosal barrier injury observed in pioglitazone-treated mice with colitis was also associated with increased expressions of tight junction proteins and reduced protein levels of TNF-α, IL-6, and MMP9, which are soluble mediators controlled by the ERK-NF-κB [38]. However, the reduction of these inflammatory mediators in colon tissue could be simply a consequence of a decreased infiltration of neutrophils, which were beneficial from recovered intestinal mucosal barrier after pioglitazone treatment. Our data also demonstrated a marked inhibition of the p65 NF-κB subunit and ERK in the colons of pioglitazone-treated mice with colitis. It is possible that impaired intestinal mucosal barrier allowed the passage of inflammatory mediators in DSS-treated mice, contributing to the systemically immune-mediated activation. In the current study, relative levels of TNF-α and IL-6 in mouse spinal cord after DSS treatment were significantly enhanced. It is well known that multiple inflammatory mediators could activate microglia and astrocytes [47], which maintain central sensitization and mechanical hyperalgesia [48]. In addition, activation of NMDA receptor in the spinal cord dorsal horn is one of key events, driving central sensitization and pain hypersensitivity [49]. Our results showed a significant colitis-induced activation of NMDA receptor-NR2B subunit, which is of importance role in regulating spinal synaptic plasticity in persistent pain conditions. In our study, systemically pioglitazone treatment inhibited the activation of microglia and astrocytes, and DSS-induced phosphorylation of NR2B subunit in the spinal cord, thus contributing to pain relief.
LIMITATIONS
In the present study, there are several limitations. Firstly, our results are not the first to describe the anti-inflammatory effect of pioglitazone on colitis. Secondly, although it has been reported that intrathecal or systematical of pioglitazone attenuates hyperalgesia in a neuropathic pain model [20, 50]. However, we only observe the effects of systematical administration of pioglitazone on colitis and related pain hypersensitivity. Thirdly, in fact, there are much more possible mechanisms of colitis-associated chronic pain. Further evidences indicated that colitis-associated pain can result from sympathetic nerves to the spinal cord via the dorsal roots [51]. Except for intestinal mucosa injury, the commensal microbiota-gut-brain axis has been found to be both ecologically and functionally perturbed in colitis and related pain [52]. The pain processing of colitis patients with abdominal pain is tightly associated with gut-derived neurochemical metabolites [53]. A further study on the specific mechanism of brain-gut-network in colitis-associated pain has been conducted in our laboratory.
CONCLUSIONS
In summary, we demonstrated that systemic administration of pioglitazone markedly alleviated pain hypersensitivity by decreasing inflammatory mediators coming into the spinal cord from the injured intestinal mucosa in DSS-induced acute colitis. Pioglitazone can ameliorate colitis-associated intestinal barrier function by upregulating tight junction proteins, which may result in the reduction of spinal levels of inflammatory mediators. We proposed that the PPARγ tight junction protein signaling might be a potential therapeutic target for the treatment of colitis-associated chronic pain.
Abbreviations
- PPARγ:
-
proliferator-activated receptor-γ
- DSS:
-
dextran sodium sulfate
- PWMT:
-
paw withdrawal mechanical threshold
- IBD:
-
inflammatory bowel disease
- H&E:
-
hematoxylin and eosin
References
Neurath, M.F. 2019. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nature Immunology 20 (8): 970–979.
Larrosa Pardo, F., E. Bondesson, Schelin MEC, and A. Joud. 2019. A diagnosis of rheumatoid arthritis, endometriosis or IBD is associated with later onset of fibromyalgia and chronic widespread pain. European Journal of Pain 23: 1563.
Eberhardson, M., C.R.H. Hedin, M. Carlson, L. Tarnawski, Y.A. Levine, and P.S. Olofsson. 2019. Towards improved control of inflammatory bowel disease. Scandinavian Journal of Immunology 89 (3): e12745.
Heiss, C.N., and L.E. Olofsson. 2019. The role of the gut microbiota in development, function and disorders of the central nervous system and the enteric nervous system. Journal of Neuroendocrinology 31 (5): e12684.
Vermeulen, W., J.G. De Man, P.A. Pelckmans, and B.Y. De Winter. 2014. Neuroanatomy of lower gastrointestinal pain disorders. World Journal of Gastroenterology 20 (4): 1005–1020.
Na, Y.R., M. Stakenborg, S.H. Seok, and G. Matteoli. 2019. Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nature Reviews. Gastroenterology & Hepatology 16: 531.
Lopetuso, L.R., C. De Salvo, L. Pastorelli, N. Rana, H.N. Senkfor, V. Petito, L. Di Martino, F. Scaldaferri, A. Gasbarrini, F. Cominelli, D.W. Abbott, W.A. Goodman, and T.T. Pizarro. 2018. IL-33 promotes recovery from acute colitis by inducing miR-320 to stimulate epithelial restitution and repair. Proceedings of the National Academy of Sciences of the United States of America 115 (40): E9362–E9370.
Cseko, K., B. Beckers, D. Keszthelyi, and Z. Helyes. 2019. Role of TRPV1 and TRPA1 Ion channels in inflammatory bowel diseases: Potential therapeutic targets? Pharmaceuticals (Basel)12 (2): 48.
Morales-Soto, W., and B.D. Gulbransen. 2019. Enteric glia: A new player in abdominal pain. Cellular and Molecular Gastroenterology and Hepatology 7 (2): 433–445.
Wirtz, S., V. Popp, M. Kindermann, K. Gerlach, B. Weigmann, S. Fichtner-Feigl, and M.F. Neurath. 2017. Chemically induced mouse models of acute and chronic intestinal inflammation. Nature Protocols 12 (7): 1295–1309.
Pastorelli, L., C. De Salvo, J.R. Mercado, M. Vecchi, and T.T. Pizarro. 2013. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: Lessons learned from animal models and human genetics. Frontiers in Immunology 4: 280.
Shigetomi, K., and J. Ikenouchi. 2019. Cell adhesion structures in epithelial cells are formed in dynamic and cooperative ways. Bioessays 41 (7): e1800227.
Zhu, L., J. Han, L. Li, Y. Wang, Y. Li, and S. Zhang. 2019. Claudin family participates in the pathogenesis of inflammatory bowel diseases and colitis-associated colorectal cancer. Frontiers in Immunology 10: 1441.
Da Silva, S., A.V. Keita, S. Mohlin, S. Pahlman, V. Theodorou, I. Pahlman, J.P. Mattson, and J.D. Soderholm. 2018. A novel topical PPAR gamma agonist induces PPAR gamma activity in ulcerative colitis mucosa and prevents and reverses inflammation in induced colitis models. Inflammatory Bowel Diseases 24 (4): 792–805.
Zhang, C., X. Luo, J. Chen, B. Zhou, M. Yang, R. Liu, D. Liu, H.F. Gu, Z. Zhu, H. Zheng, L. Li, and G. Yang. 2019. Osteoprotegerin promotes liver steatosis by targeting the ERK-PPAR-γ-CD36 pathway. Diabetes 68 (10): 1902–1914. https://doi.org/10.2337/db18-1055.
Ormseth, M.J., A.M. Oeser, A. Cunningham, A. Bian, A. Shintani, J. Solus, S.B. Tanner, and C.M. Stein. 2014. Reversing vascular dysfunction in rheumatoid arthritis: Improved augmentation index but not endothelial function with peroxisome proliferator-activated receptor gamma agonist therapy. Arthritis & Rhematology 66 (9): 2331–2338.
Hasegawa-Moriyama, M., T. Kurimoto, M. Nakama, K. Godai, M. Kojima, T. Kuwaki, and Y. Kanmura. 2013. Peroxisome proliferator-activated receptor-gamma agonist rosiglitazone attenuates inflammatory pain through the induction of heme oxygenase-1 in macrophages. Pain 154 (8): 1402–1412.
Bouguen, G., A. Langlois, M. Djouina, J. Branche, D. Koriche, E. Dewaeles, A. Mongy, J. Auwerx, J.F. Colombel, P. Desreumaux, L. Dubuquoy, and B. Bertin. 2015. Intestinal steroidogenesis controls PPAR gamma expression in the colon and is impaired during ulcerative colitis. Gut 64 (6): 901–910.
Pedersen, G., and J. Brynskov. 2010. Topical rosiglitazone treatment improves ulcerative colitis by restoring peroxisome proliferator-activated receptor-gamma activity. The American Journal of Gastroenterology 105 (7): 1595–1603.
Khasabova, I.A., S.G. Khasabov, J.K. Olson, M.L. Uhelski, A.H. Kim, A.M. Albino-Ramirez, C.L. Wagner, V.S. Seybold, and D.A. Simone. 2019. Pioglitazone, a PPAR gamma agonist, reduces cisplatin-evoked neuropathic pain by protecting against oxidative stress. Pain 160 (3): 688–701.
Saito, T., M. Hasegawa-Moriyama, T. Kurimoto, T. Yamada, E. Inada, and Y. Kanmura. 2015. Resolution of inflammation by resolvin D1 is essential for peroxisome proliferator-activated receptor-gamma-mediated analgesia during postincisional pain development in type 2 diabetes. Anesthesiology 123 (6): 1420–1434.
Bento, A.F., R. Marcon, R.C. Dutra, R.F. Claudino, M. Cola, D.F. Leite, and J.B. Calixto. 2011. Beta-caryophyllene inhibits dextran sulfate sodium-induced colitis in mice through CB2 receptor activation and PPAR gamma pathway. The American Journal of Pathology 178 (3): 1153–1166.
Griggs, R.B., R.R. Donahue, J. Morgenweck, P.M. Grace, A. Sutton, L.R. Watkins, and B.K. Taylor. 2015. Pioglitazone rapidly reduces neuropathic pain through astrocyte and nongenomic PPARgamma mechanisms. Pain 156 (3): 469–482.
Okayasu, I., S. Hatakeyama, M. Yamada, T. Ohkusa, Y. Inagaki, and R. Nakaya. 1990. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 98 (3): 694–702.
Hontecillas, R., W.T. Horne, M. Climent, A.J. Guri, C. Evans, Y. Zhang, B.W. Sobral, and J. Bassaganya-Riera. 2011. Immunoregulatory mechanisms of macrophage PPAR-gamma in mice with experimental inflammatory bowel disease. Mucosal Immunology 4 (3): 304–313.
Schaefer, K.L., S. Denevich, C. Ma, S.R. Cooley, A. Nakajima, K. Wada, J. Schlezinger, D. Sherr, and L.J. Saubermann. 2005. Intestinal antiinflammatory effects of thiazolidinedione peroxisome proliferator-activated receptor-gamma ligands on T helper type 1 chemokine regulation include non transcriptional control mechanisms. Inflammatory Bowel Diseases 11 (3): 244–252.
Chaplan, S.R., F.W. Bach, J.W. Pogrel, J.M. Chung, and T.L. Yaksh. 1994. Quantitative assessment of tactile allodynia in the rat paw. Journal of Neuroscience Methods 53 (1): 55–63.
Koukos, G., C. Polytarchou, J.L. Kaplan, A. Morley-Fletcher, B. Gras-Miralles, E. Kokkotou, M. Baril-Dore, C. Pothoulakis, H.S. Winter, and D. Iliopoulos. 2013. MicroRNA-124 regulates STAT3 expression and is down-regulated in colon tissues of pediatric patients with ulcerative colitis. Gastroenterology 145 (4): 842–852 e842.
Zhu, W., H. Yan, S. Li, W. Nie, F. Fan, and J. Zhu. 2016. PPAR-gamma agonist pioglitazone regulates dendritic cells immunogenicity mediated by DC-SIGN via the MAPK and NF-kappaB pathways. International Immunopharmacology 41: 24–34.
Takagi, T., Y. Naito, N. Tomatsuri, O. Handa, H. Ichikawa, N. Yoshida, and T. Yoshikawa. 2002. Pioglitazone, a PPAR-gamma ligand, provides protection from dextran sulfate sodium-induced colitis in mice in association with inhibition of the NF-kappaB-cytokine cascade. Redox Report 7 (5): 283–289.
Friedrich, M., L. Gerbeth, M. Gerling, R. Rosenthal, K. Steiger, C. Weidinger, J. Keye, H. Wu, F. Schmidt, W. Weichert, B. Siegmund, and R. Glauben. 2019. HDAC inhibitors promote intestinal epithelial regeneration via autocrine TGFbeta1 signalling in inflammation. Mucosal Immunology 12 (3): 656–667.
Bhat, A.A., S. Uppada, I.W. Achkar, S. Hashem, S.K. Yadav, M. Shanmugakonar, H.A. Al-Naemi, M. Haris, and S. Uddin. 2018. Tight junction proteins and signaling pathways in Cancer and inflammation: A functional crosstalk. Frontiers in Physiology 9: 1942.
Srinath, A., E. Young, and E. Szigethy. 2014. Pain management in patients with inflammatory bowel disease: Translational approaches from bench to bedside. Inflammatory Bowel Diseases 20 (12): 2433–2449.
Wang, C.Z., H. Yao, C.F. Zhang, L. Chen, J.Y. Wan, W.H. Huang, J. Zeng, Q.H. Zhang, Z. Liu, J. Yuan, Y. Bi, C. Sava-Segal, W. Du, M. Xu, and C.S. Yuan. 2018. American ginseng microbial metabolites attenuate DSS-induced colitis and abdominal pain. International Immunopharmacology 64: 246–251.
Wang, X.Y., H.R. Zhou, S. Wang, C.Y. Liu, G.C. Qin, Q.Q. Fu, J.Y. Zhou, and L.X. Chen. 2018. NR2B-Tyr phosphorylation regulates synaptic plasticity in central sensitization in a chronic migraine rat model. The Journal of Headache and Pain 19 (1): 102.
Li, S., J. Zeng, X. Wan, Y. Yao, N. Zhao, Y. Yu, C. Yu, and Z. Xia. 2017. Enhancement of spinal dorsal horn neuron NMDA receptor phosphorylation as the mechanism of remifentanil induced hyperalgesia: Roles of PKC and CaMKII. Molecular Pain 13: 1744806917723789.
Rigano, D., C. Sirignano, and O. Taglialatela-Scafati. 2017. The potential of natural products for targeting PPARalpha. Acta Pharmaceutica Sinica B 7 (4): 427–438.
Tilg, H., A.R. Moschen, A. Kaser, A. Pines, and I. Dotan. 2008. Gut, inflammation and osteoporosis: Basic and clinical concepts. Gut 57 (5): 684–694.
Filpa, V., E. Moro, M. Protasoni, F. Crema, G. Frigo, and C. Giaroni. 2016. Role of glutamatergic neurotransmission in the enteric nervous system and brain-gut axis in health and disease. Neuropharmacology 111: 14–33.
Ji, R.R., A. Chamessian, and Y.Q. Zhang. 2016. Pain regulation by non-neuronal cells and inflammation. Science 354 (6312): 572–577.
Ciesielczyk, K., A. Furgala, L. Dobrek, K. Juszczak, and P. Thor. 2017. Altered sympathovagal balance and pain hypersensitivity in TNBS-induced colitis. Archives of Medical Science 13 (1): 246–255.
Woller, S.A., K.A. Eddinger, M. Corr, and T.L. Yaksh. 2017. An overview of pathways encoding nociception. Clin Exp Rheumatol 35 Suppl 107 (5): 40–46.
Ceuleers, H., H. Van Spaendonk, N. Hanning, J. Heirbaut, A.M. Lambeir, J. Joossens, K. Augustyns, J.G. De Man, I. De Meester, and B.Y. De Winter. 2016. Visceral hypersensitivity in inflammatory bowel diseases and irritable bowel syndrome: The role of proteases. World Journal of Gastroenterology 22 (47): 10275–10286.
Coufal, S., N. Galanova, L. Bajer, Z. Gajdarova, D. Schierova, Z. Jiraskova Zakostelska, K. Kostovcikova, Z. Jackova, Z. Stehlikova, P. Drastich, H. Tlaskalova-Hogenova, and M. Kverka. 2019. Inflammatory bowel disease types differ in markers of inflammation, gut barrier and in specific anti-bacterial response. Cells 8 (7). https://doi.org/10.3390/cells8070719.
Stremmel, W., S. Staffer, M.J. Schneider, H. Gan-Schreier, A. Wannhoff, N. Stuhrmann, A. Gauss, H. Wolburg, A. Mahringer, A. Swidsinski, and T. Efferth. 2017. Genetic mouse models with intestinal-specific tight junction deletion resemble an ulcerative colitis phenotype. Journal of Crohn's & Colitis 11 (10): 1247–1257.
Chen, Y., H.S. Zhang, G.H. Fong, Q.L. Xi, G.H. Wu, C.G. Bai, Z.Q. Ling, L. Fan, Y.M. Xu, Y.Q. Qin, T.L. Yuan, H. Sun, and J. Fang. 2015. PHD3 stabilizes the tight junction protein occludin and protects intestinal epithelial barrier function. The Journal of Biological Chemistry 290 (33): 20580–20589.
Zusso, M., V. Lunardi, D. Franceschini, A. Pagetta, R. Lo, S. Stifani, A.C. Frigo, P. Giusti, and S. Moro. 2019. Ciprofloxacin and levofloxacin attenuate microglia inflammatory response via TLR4/NF-kB pathway. Journal of Neuroinflammation 16 (1): 148.
Ji, R.R., A. Nackley, Y. Huh, N. Terrando, and W. Maixner. 2018. Neuroinflammation and central sensitization in chronic and widespread pain. Anesthesiology 129 (2): 343–366.
Raja, S.N., E. Sivanesan, and Y. Guan. 2019. Central sensitization, N-methyl-D-aspartate receptors, and human experimental pain models: Bridging the gap between target discovery and drug development. Anesthesiology 131 (2): 233–235.
Mohseninejad, L., C. van Gils, C.A. Uyl-de Groot, E. Buskens, and T. Feenstra. 2015. Evaluation of patient registries supporting reimbursement decisions: The case of oxaliplatin for treatment of stage III colon cancer. Value in Health 18 (1): 84–90.
Camilleri, M., and G. Boeckxstaens. 2017. Dietary and pharmacological treatment of abdominal pain in IBS. Gut 66 (5): 966–974.
Powell, N., M.M. Walker, and N.J. Talley. 2017. The mucosal immune system: Master regulator of bidirectional gut-brain communications. Nature Reviews. Gastroenterology & Hepatology 14 (3): 143–159.
Lv, K., W. Song, R. Tang, Z. Pan, Y. Zhang, Y. Xu, B. Lv, Y. Fan, and M. Xu. 2018. Neurotransmitter alterations in the anterior cingulate cortex in Crohn’s disease patients with abdominal pain: A preliminary MR spectroscopy study. Neuroimage Clin 20: 793–799.
Availability of Data and Materials
All data and materials are available. All authors take full responsibility for the data and materials.
Funding
This study was supported by the National Natural Science Foundation of China (Nos. 81671087, 81471129, 81870871, 81500954), National Key R&D Program of China (SQ2018YFC200044), and a Grant from Jiangsu Commission of Health (Jiangsu Provincial Key Medical Discipline).
Author information
Authors and Affiliations
Contributions
Conception and design: Zhengliang Ma, Xiaoping Gu, and Yulin Huang. Acquisition of data: Yulin Huang, Chenchen Wang, Yanting Mao, Xinyu Tian, and Bailin Hou. Data analysis and interpretation: Yulin Huang and Chenchen Wang. Manuscript draft: Yulin Huang. Revision for intellectual content: Yu’e Sun. Final approval of the completed manuscript: Yulin Huang and Chenchen Wang.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, Y., Wang, C., Tian, X. et al. Pioglitazone Attenuates Experimental Colitis-Associated Hyperalgesia through Improving the Intestinal Barrier Dysfunction. Inflammation 43, 568–578 (2020). https://doi.org/10.1007/s10753-019-01138-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-019-01138-3