
Abstract
To improve clinical outcomes for patients with traumatic brain injury (TBI), it is necessary to explore the mechanism of traumatic brain injury (TBI)–induced neuroinflammation. Connective tissue growth factors (CTGF) have been reported to be involved in the process of inflammatory response or tissue repair, whereas whether and how CTGF participates in the astrocyte-mediated inflammation after TBI remains unclear. In the present study, the TBI-induced activation of astrocytes and augmentation of inflammatory response were simulated by stimulating rat astrocytes with TGF-β1 or CTGF in cultured conditions. TGF-β1 and CTGF both upregulated the expression of GFAP in astrocytes and facilitated the production of inflammatory cytokines and chemokines. Activation of astrocytes by CTGF is in an autocrine manner. According to the results of Boyden chamber assay, CTGF enhanced the recruitment of peripheral blood mononuclear cells (PBMCs) by reactive astrocytes. Besides, CTGF-mediated activation of astrocytes and augmentation of inflammatory response can be terminated by the inhibitor of ASK1 or p38 and JNK. Thus, our data suggested that CTGF could activate astrocytes in an autocrine manner and promote astrocyte-mediated inflammatory response by triggering the ASK1-p38/JNK-NF-κB/AP-1 pathways in astrocytes. Collectively, our study provided evidence that astrocyte-secreted CTGF serves as an amplifier of neuroinflammatory and could be a potential target for alleviating TBI-induced inflammation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Traumatic brain injury (TBI) has raised a huge public health concern, affecting individuals around the world [26]. After the initial injury is managed and resolved, 70~80% of TBI patients will suffer from the secondary pathology and develop long-lasting effects (e.g., changes in personality and cognition, anxiety, and depressive-like behaviors) [40]. Moreover, TBI also seems contribute to various chronic degenerative processes (e.g., chronic traumatic encephalopathy, Alzheimer disease, and Parkinson disease) [14]. One of the major driving forces of this secondary pathology appears to be the inflammatory reaction after TBI [13]. By measuring inflammatory biomarkers in serum or cerebrospinal fluid (CSF) of TBI patients, clinicians can predict prognosis and make decisions at the early stage of pathogenesis [15]. Intermediate products and signals of inflammatory response are critical targets for treating TBI [19]. Accordingly, the insights into the inflammatory mechanisms that drive the pathologic and consequent cognitive outcomes help to facilitate clinical outcomes for patients with TBI.
TBI-induced neuroinflammation refers to a cascade reaction encompassing complex interactions of multiple cell types and signal molecules, and astrocytes help orchestrate this response by acting as inflammatory amplifiers [23]. After brain injury, upregulation of glial fibrillary acidic protein (GFAP) induced by mechanical stress would mechanically activate sensitive cation channels and the release of danger signals, including ATP [1], High-mobility group box 1 (HMGB1) [39] and heat shock proteins 60 (HSP60) [41], from damaged cells. Astrocytes could recognize these autocrine or paracrine signals through their cell surface-expressed receptors, transmit these signals to intracellular effectors (NF-κB, p38, JNK, etc.) [12], and facilitate the production of inflammatory cytokines (TNF-α [49], IL-6, IL-1β [11], etc.) and chemokines (MCP-1 [45], RANTES [31], CXCL1 [51], etc.). Subsequently, inflammatory signals would recruit considerable peripheral immune cells across the blood-brain barrier (BBB), leading to pathological features (e.g., edema and diffuse axonal injury). Furthermore, reactive astrocytes could also produce neuroprotective growth factors (e.g., BDNF [21] and IGF-1 [32]), promoting endothelial-mediated neurovascular reconstruction and functional recovery of BBB. There are still questions about how astrocytes get involved in both protective and deleterious neuroinflammations.
Connective tissue growth factors (CTGF, also termed as CCN2), a member of the CCN family, were first discovered in 1991 as a secreted protein in the conditioned media of cultured human umbilical vascular endothelial cells [4]. CTGF have been reported to play various roles (e.g., angiogenesis and mesenchymal growth) in physiological processes [38]. Recently, glial-secreted CTGF has been reported critical for spinal cord repair in a zebrafish model [36]. However, CTGF was over-expressed restrictedly in a small proportion of glial cells, suggesting the delicate orchestration of CTGF function. Indeed, CTGF is abundantly expressed in various tissues, such as skin, blood vessels, liver, and kidney, and its over-expression is often associated with fibrosis, scarring diseases [47], and even tumorigenesis [10]. It has been revealed that CTGF can act as an inflammatory modulator, and it is involved in chronic inflammatory diseases [25]. Liu et al. showed that CTGF activated human synovial fibroblast cells through ASK1, p38/JNK, and NF-κB/AP-1 pathways and upregulated the IL-6 expression in these cells [28]. Since TBI can induce neuroinflammation, activate astrocytes, and cause CTGF accumulation [29], we attempted to explore the connections between CTGF, astrocytes, and inflammatory response in the TBI settings.
In this study, we got preliminary findings that CTGF can activate cultured rat astrocytes and facilitate the production of inflammatory cytokines and chemokines in a dose-dependent manner. CTGF treatment enhanced the recruitment of peripheral blood mononuclear cells (PBMCs) by astrocytes, indicating that CTGF could augment astrocyte-mediated inflammatory response. Like TGF-β1, CTGF activated astrocytes through ASK1-p38/JNK-AP-1/NF-κB signaling pathways. In conclusion, we implicated a role of CTGF in augmenting astrocyte-mediated inflammatory response and provided evidence for considering CTGF as a potential target for alleviating TBI-induced neuroinflammation.
RESULTS
CTGF Alone Can Activate Rat Astrocytes in an Autocrine Manner
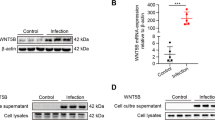
In order to investigate whether CTGF can induce the activation of rat astrocytes, we stimulated rat astrocyte RA with TGF-β1 or CTGF in vitro. Consistent with the results described previously [5], TGF-β1 significantly upregulated the transcription of CTGF, indicating that CTGF is a downstream effector of TGF-β1 (Fig. 1a). It has been reported that the expression of TGF-β1 was significantly elevated in CNS after TBI [37], and its over-expression was responsible for the upregulation of GFAP and the activation of astrocytes [54]. Therefore, the mRNA expression of GFAP in RA was measured at 24 h after stimulation with various concentrations of CTGF or 10 ng/ml TGF-β1. We found that CTGF increase the level of GFAP expression in a dose-dependent manner (Fig. 1b).

CTGF activated rat astrocytes through an autocrine-dependent manner in vitro. RA cells were cultured in the presence of 10 ng/ml recombinant TGF-β1, 5 μg/ml Brefeldin A, 10 μg/ml CTGF neutralizing antibody, or recombinant CTGF with indicated concentrations for 24 h, respectively, and the mRNA expressions of CTGF (a) and GFAP (b) were examined by quantitative real-time polymerase chain reaction (qRT-PCR). Results are expressed as the mean ± S.E. *p < 0.05, **p < 0.01, and ***p < 0.001. N.S. not significant.
Interestingly, CTGF alone can increase, even slightly, the auto-production of CTGF by cultured astrocytes (Fig. 1a), probably due to some positive feedback circuit. Upon TGF-β1 induction, CTGF secreted by astrocytes [9] rapidly affected surrounding cells through an autocrine or paracrine manner, resulting in the regulation of cell adhesion and intracellular signaling [48]. To further validate whether auto-production of CTGF activates astrocytes, the mRNA expression of GFAP and CTGF was measured after blocking the secretion of CTGF by Brefeldin A or neutralizing CTGF by the addition of specific antibody in the medium. We found that the activation of CTGF and GFAP by TGF-β1 was remarkably abrogated by Brefeldin A (Fig. 1a, b), and the activation of GFAP by TGF-β1 and CTGF was prevented by CTGF neutralization (Fig. 1b). Collectively, we showed here that CTGF could activate the expression of GFAP in rat astrocytes through an autocrine manner.
CTGF Alone Can Promote Astrocyte-Mediated Immune Responses
Upon activation, astrocytes release multiple types of cytokines and chemokines, leading to the initiation of an inflammatory cascade [19]. In order to study the effect of CTGF on the astrocyte-mediated immune response, RA was treated with 10 ng/ml TGF-β1 and 10 ng/ml CTGF. Supernatants were collected 24 h later and assayed for the production of cytokines TNF-α, IL-6, IL-1β, and chemokines CCL2 (MCP-1), CCL5 (RANTES), and CXCL1. Both TGF-β1 and CTGF treatments enhanced the production of TNF-α, IL-6, and IL-1β (Fig. 2a), as well as the production of MCP-1, RANTES, and CXCL1 (Fig. 2b). Unexpectedly, the production of neuroprotective growth factors IGF-1 and BDNF remained unchanged in either TGF-β1 or CTGF groups (Sup Fig. 1), implicating that both TGF-β1 and CTGF stimulations had no effect on astrocyte-mediated brain injury repair.

CTGF facilitated the production of cytokines and chemokines by astrocytes. RA cells were cultured in the presence of 10 ng/ml recombinant CTGF or TGF-β1 for 24 h, and supernatants were collected to measure the production of cytokines—TNF-α, IL-6, and IL-1β (a), and chemokines—MCP-1, RANTES, and CXCL1 (b). Results are expressed as the mean ± S.E. *: p < 0.05 as compared with solvent control group. *p < 0.05, **p < 0.01, and ***p < 0.001 as compared with solvent control group.
The major feature of immune response is the recruitment of immune cells to the damage sites. To confirm whether astrocytes are able to recruit immune cells, we used Boyden chamber techniques to study the migration of peripheral blood mononuclear cells (PBMCs) cocultured with RA (Fig. 3a). After stimulation with TGF-β1 or CTGF, activated RA can enhance the migration of PBMCs (Fig. 3b). Taken together, we found that CTGF facilitated the production of cytokines and chemokines and enhanced the recruitment of PBMCs, leading to locally augmented immune response.

CTGF enhanced the recruitment of PBMCs. a Schematic diagram of the Boyden chamber assay. b Effects of CTGF treatment on the chemotactic migration of PBMCs cocultured with RA. Number of invaded PBMCs during co-culture with RA was indicated on vertical axis. Results are expressed as the mean ± S.E. *p < 0.05 and **p < 0.01.
CTGF-Induced Inflammatory Responses Through ASK1-p38/JNK-NF-κB/AP-1 Pathways
In response to extracellular inflammatory signals, apoptosis signal-regulating kinase 1 (ASK1) plays an important role in activating innate immunity [35]. The activation of ASK1 is regulated by phosphorylation at Ther-845, as well as the dephosphorylation at different residues, such as Serine-967 [27]. Here, we found that both TGF-β1 and CTGF treatments increased the phosphorylation at T845 and reduced the phosphorylation at S967, resulting in ASK1 activation (Fig. 4a–b). To investigate the role of ASK1 in the neuroinflammatory response, we used the ASK1-specific inhibitor, GS-4997, to treat astrocytes, and examined the change of astrocyte-mediated immune response in condition of CTGF stimulation. The activation of NF-κB and AP-1 pathways is responsible for the production of cytokines and the amplification of immune response [6, 8]. p65 and c-Jun are key transcriptional factors in the NF-κB and AP-1 pathways, respectively. As indicated by Western blotting, the phosphorylation of p65 at S536 and phosphorylation of c-Jun at T239 were both increased while the expression of the protein p65 and c-Jun did not change after TGF-β1 and CTGF stimulation (Fig. 4a). Notably, the activation of ASK1, p65, and c-Jun induced by CTGF was abrogated by ASK1 inhibition (Fig. 4b), indicating that p65 and c-Jun were both downstream effectors of ASK1. IL-6 is an indicator of NF-κB/AP-1 activation. Quantitative real-time polymerase chain reaction (qRT-PCR) analysis revealed that enhanced expression of IL-6 induced by CTGF was completely impeded by ASK1 inhibition, indicating that ASK1 activation is responsible for astrocyte-mediated NF-κB/AP-1 activation (Fig. 4d).

CTGF activated the NF-κB and AP-1 through ASK1-p38/JNK pathways. a RA cells were incubated with 10 ng/ml CTGF or TGF-β1 for 24 h, and total protein extracts were collected. The expression of key molecules in ASK, p38/JNK, and NF-κB/AP-1 pathways and their phosphorylation states were examined by Western blotting. b RA cells were treated with either 10 μM ASK1 inhibitor GS-4997, or solvent control as indicated, for 30 min prior to stimulation with 20 ng/ml CTGF for 24 h. The expression of ASK, p65, and c-Jun and their phosphorylation states were examined by Western blotting. c RA cells were treated with either 5 μM SB20358 or 25 μM SP600125, or DMSO as control, for 30 min prior to stimulation with CTGF for 24 h, and total protein extracts were collected. The expression of ASK, p65, c-Jun and their phosphorylation states were examined by Western blotting. d RA cells were treated with chemical inhibitors or solvent control for 30 min prior to stimulation with 10 ng/ml CTGF for 24 h. The mRNA expression of IL-6 was analyzed using qRT-PCR. *p < 0.05 and **p < 0.01 as compared with CTGF group.
ASK1 belongs to the MAPKKK family and activates the p38 and JNK pathways via MKK3/6 and MKK4/7, respectively [20]. We found activation of JNK (phosphorylated at T183) and p38 (phosphorylated at T180/Y182) after TGF-β1 or CTGF stimulation (Fig. 4a). To further elucidate the role of p38 and JNK in ASK1-mediated immune activation, we blocked the p38 activity using selective inhibitor SB203580 and the JNK activity using SP600125, respectively. We found that addition of either SB203580 or SP600125 alone remarkably inhibited the phosphorylation of p65 and c-Jun induced by CTGF (Fig. 4c). The inhibition was further strengthened when SB203580 and SP600125 were simultaneously added to the medium (Fig. 4c). Notably, neither of these inhibitors can block the phosphorylation of ASK1 (Fig. 4c), indicating that ASK1 acts upstream of p38 and JNK. Likewise, inhibition of p38 by SB203580 or JNK by SP600125 significantly abrogated CTGF-induced IL-6 expression, while simultaneous inhibition of p38 and JNK further strengthened this abrogation (Fig. 4d). Therefore, these results suggested that CTGF can active NF-κB/AP-1 effectors through both ASK1-p38 and ASK1-JNK pathways.
To confirm whether CTGF activates astrocyte-mediated inflammatory response through ASK1-p38/JNK-NF-κB/AP-1 pathways, RA were treated with GS-4997, SB203580, or SP600125 for 30 min and cultured in the presence of CTGF for an extended period of 24 h. Supernatants were collected and assayed for the production of cytokines and chemokines, while migration of cocultured PBMCs was tested in Boyden chamber system. The increase of the production of cytokines—TNF-α (Fig. 5a), IL-6, IL-1β (Sup Fig. 2a), and chemokines—MCP-1 (Fig. 5b), RANTES, and CXCL1 (Sup Fig. 2b) by CTGF was partially abrogated after inhibition of p38 or JNK alone, but completely abrogated after simultaneously inhibition of p38 and JNK. As expected, the inhibition of ASK1 alone almost impeded the production of TNF-α (Sup Fig. 2c). Moreover, enhancement of the migration of cocultured PBMCs was partially impaired in p38 or JNK inhibition group, while completely impaired in p38 and JNK co-inhibition group (Fig. 5c) and in ASK1 inhibition group (Sup Fig. 2d). Taken together, we suggested that CTGF can promote the astrocyte-mediated inflammatory responses through ASK1-p38/JNK-NF-κB/AP-1 pathways (Fig. 6).

Augmentation of inflammation by CTGF is abrogated by p38 or JNK inhibitors. RA cells were treated with either 5 μM SB20358 or 25 μM SP600125, or DMSO as control, for 30 min prior to stimulation with CTGF. Supernatants were collected to measure the production of TNF-α (a) and MCP-1 (b), 24 h later. c RA cells were seeded in the lower wells of the Boyden chamber, then treated with inhibitors or DMSO for 30 min, followed by the treatment with 10 ng/ml CTGF. Thereafter, the upper wells seeded with PBMCs were placed on top of the lower wells and cells were cocultured for 96 h, then the invaded PBMCs under the membrane were counted. Results are expressed as the mean ± S.E. *p < 0.05, **p < 0.01, and ***p < 0.001 as compared with solvent control group. N.S. not significant.

Schematic presentation of the signaling pathways involved in CTGF-induced inflammatory augmentation of astrocytes.
DISCUSSION
Mechanisms regarding how astrocytes get involved in neuroinflammation after brain injury are complex and remain unclear. CTGF emerges as a new target and is intensely studied because of its strong association with chronic inflammatory disorder, such as atherosclerosis, arthritis, inflammatory kidney, and neuroinflammatory diseases [25]. However, the direct contribution of CTGF to astrocyte-mediated TBI-induced neuroinflammation is still unclear. In this study, we performed detailed research of molecular mechanisms and got preliminary data concerning the role of CTGF as an inflammatory amplifier in cultured conditions. We found that auto-production of CTGF can initiate and augment astrocyte-mediated inflammatory response through facilitating the production of inflammatory cytokines and chemokines and enhancing the recruitment of PBMCs. Moreover, our data suggested that CTGF-mediated activation of astrocytes is dependent on ASK1-p38/JNK-AP-1/NF-kB pathways, therefore providing references for manipulating CTGF signaling.
Astrocytes were kind of star-shaped glial cells that widely distributed in the brain and spinal cords. Under physiological conditions, astrocytes are critical in supporting neuronal function, glial transmission, and signaling via Ca2+ release and uptake, as well as maintaining BBB integrity [23]. In the setting of TBI, an increase in astrocyte reactivity in response to mechanical injury and BBB breakdown is termed astrogliosis [50]. This response involves changes in morphology, increased expression of GFAP, heightened proliferation, and secretion of inflammatory mediators and growth factors. Reactive astrocytes can cause further BBB disruption through augmenting inflammatory response [16], or support repair and regeneration after CNS damage through producing neuroprotective growth factors [33]. Many of these factors act in an autocrine and paracrine fashion to facilitate astrocytic reactivity of the cells surrounding them. Therefore, the effect of astrocytes, either protective or deleterious, were determined by their surrounding microenvironment, illustrated by the variation of localizations, concentrations, and compositions of factors [23]. Still, the functions of these factors involved in this process remained to be determined. TGF-β1 can be synthesized by nearly all cells of the CNS and is upregulated in many CNS disorders including clinical and experimental TBI [37]. It has been reported that TGF-β1 promotes astrogliosis and the transcription of CTGF [17]. Thus, CTGF is an important downstream target of TGF-β1. Moreover, CTGF is also regulated by auto-products [43] and pro-inflammatory mediators, such as TNF-α [22] and IL-1β [34]. All the studies described above implied the critical roles of CTGF in shaping the microenvironment after TBI. In the present study, we found that TGF-β1-induced activation of rat astrocytes was CTGF-dependent. CTGF can upregulate the expression of GFAP and activate astrocytes in an autocrine-dependent manner. Therefore, our results suggested that CTGF could play important roles in amplifying TBI-induced neuroinflammation. However, we do not observe neuroprotective response since there is no significant alteration on the expression of BDNF and IGF-1. Further studies should be performed to examine the alterations of transcriptome to exclude the involvement of astrocytes in brain injury repair. In addition, the comprehensive effect of CTGF on the neuroinflammation and injury repair after TBI should be further examined in animal models.
Inflammation is a cascade response encompassing multiple types of immune cells. Neutrophils are an abundant population of circulating leukocytes that are usually among the first responders to tissue injuries in the periphery and CNS. Neutrophils are rapidly recruited to the CNS after TBI guided by chemokines, such as CXCL1 and CXCL2, and help prepare the damaged environment for repair [44]. CXCR2, the receptor of CXCL1, is dominantly expressed on the surface of neutrophils. Genetic deletion of CXCR2 prevented neutrophils infiltration and attenuated nerve injury shortly after TBI, while it had no effect on the long-term functional recovery of BBB [46]. On the other hand, neutrophils enhance the recruitment of monocytes [24], which are capable of crossing the BBB into the injured brain as a result of MCP-1 and differentiating into macrophages or dendritic cells. MCP-1, another pro-inflammatory chemokines, is significantly increased within 24 h in the CSF of patients with TBI [45]. Examination of MCP-1−/− mice after TBI revealed no changes in lesion size within the first week of injury, but had improved functional recovery after a longer period of 2 to 4 weeks, suggesting a pathogenic role for macrophages during the chronic phase of TBI [45]. T cells play important roles in both innate and adaptive immunities. Although considerable amounts of T cells were recruited to the sites of injury after TBI, their roles are still unclear. RANTES, which is majorly chemotactic for T cells, is elevated in the cortex and the plasma of TBI patients and its concentration may correlate with poor outcome in TBI patients [31]. However, functional studies using RAG1−/− mice, which lack T and B cells, revealed no difference in any pathologic or neurologic parameters [53]. In this study, we found that the productions of CXCL1, MCP-1, and RANTES were all significantly elevated within 24 h after TGF-β1 or CTGF stimulation. Our results implicated that astrocytes activated by CTGF can recruit multiple types of immune cells, including neutrophils, monocytes, and T cells. It might be interesting to further investigate the role of CTGF on specific populations of these immune cells, as well as on the reconstitution of CNS microenvironment, using the animal model of TBI.
In mammalians, ASK1-p38, ASK1-JNK, NF-κB, and AP-1 pathways all contribute to the regulation of innate immunity [2]. ASK1 plays various roles in oxidative stress, endoplasmic reticulum stress, and apoptosis [52]. It has been reported that knockdown of ASK1 prevented the CTGF-mediated activation of p38 and JNK pathways and inhibited the production of IL-6 in human synovial fibroblasts, a major mesenchymal cells contributed to osteoarthritis [28]. Consistent with these results, we found that CTGF-mediated activation of astrocytes is ASK1-dependent. However, deletion of ASK1 blocked only the LPS-induced p38 activation, while had no effect on the activation of JNK and NF-κB signaling [18]. In this study, we found that CTGF-induced activation of ASK1 transduced the signals to both p38 and JNK pathways. Therefore, the difference in signaling transduction might be due to the difference in cell surface receptors and extracellular signals and thus contribute to the divergent functions in astrocytes. Further studies should be performed to examine which receptors are responsible for CTGF-induced activation of astrocytes. In the TBI models, NF-κB and AP-1 pathways regulate cell death and neuroinflammation [42]. NF-κB is a transcriptional factor containing p65 and p50 subunits, while AP-1 is another transcriptional factor containing Jun (c-Jun, JunB, and JunD) and Fos (c-Fos, Fra-1, Fra-2, and FosB) family members. In response to extracellular signals, NF-κB and AP-1 can both be activated by intracellular kinase, such as p38 and JNK, and thus obtain DNA-binding capacity [3, 7]. Inflammatory mediators, such as TNF-α, IL-1β, IL-6, and MCP-1, were potential targets of NF-κB and AP-1. In this study, we found that NF-κB and AP-1 were both activated by CTGF, suggesting that they both play roles in CTGF-mediated activation of astrocytes.
In conclusion, our study indicated a role of CTGF in astrocyte activation and inflammatory augmentation and provided preliminary evidences that CTGF could be valid therapeutic targets for alleviating neuroinflammation and improving the outcomes of TBI.
MATERIALS AND METHODS
Cell Lines and Reagents
Rat primary cortical astrocyte cell line RA was purchased from Sigma-Aldrich (Shanghai, China). Recombinant rat CTGF was obtained from R&D Systems with purity > 85% and endotoxin level < 0.1 EU/ml. Recombinant rat TGF-β1 (purity > 90% and endotoxin level < 1 EU/ml) was obtained from Sino Biological, Inc. (Beijing, China). CTGF neutralizing antibody was purchased from R&D Systems. Brefeldin A was purchased from Sigma-Aldrich (Shanghai, China). Selective inhibitors of ASK1 (GS-4997) were purchased from Selleck Inc. (Shanghai, China), and p38 (SB20358) and JNK (SP600125) were purchased from InvivoGen, Inc. (Beijing, China).
Cell Culture and Stimulation
RA was routinely maintained in DMEM/F12 medium with 10% FBS, penicillin, and streptomycin and cultured at 37 °C in 5% CO2. Cells were seeded in 24-well plates with the density of 1 × 106 per well. Prior to stimulation, cells were treated with either 10 μM GS-4997, 5 μM SB20358, 25 μM SP600125, or DMSO as control, for 30 min. Then, supernatants were replaced with refresh DMEM medium containing 10 ng/ml recombinant TGF-β1, 5 μg/ml Brefeldin A, 10 μg/ml CTGF neutralizing antibody, or recombinant CTGF with various concentrations, respectively. Supernatants were collected and assayed for the production of cytokines and chemokines, 24 h later. After stimulation, cells were dissolved in Trizol (Invitrogen, Shanghai, China) for RNA extraction and quantitative real-time polymerase chain reaction (qRT-PCR) analysis. Alternatively, cells were washed with PBS and dissolved in lysis buffer and stored at − 80 °C for Western blotting analysis.
Peripheral blood mononuclear cells (PBMCs) were purified using Ficoll centrifugation (Ficoll-Paque Plus, GE Healthcare) from the blood of adult Sprague-Dawley (SD) rats (obtained from model animal research center, Nanjing University, China). Cells were maintained in RPMI 1640, supplemented with 10% heat-inactivated FBS, penicillin, and streptomycin, and cultured at 37 °C in 5% CO2.
Quantitative Real-time Polymerase Chain Reaction
Total RNA was extracted from RA using a Trizol kit (Invitrogen, Shanghai, China). The reverse transcription reactions were performed using 2 μg of total RNA that was reverse transcribed into cDNA using oligo (dT) and random mixed primers (Tiangen, China). The quantitative real-time PCR (qPCR) analysis was carried out using Taqman® one-step PCR Master Mix (Applied Biosystems, Shanghai, China) as described previously [28]. Sequences for all target gene primers and probes were listed in Table S1 (β-actin was used as internal control). The threshold was set above the non-template control background and within the linear phase of the target gene amplification to calculate the cycle number at which the transcript was detected (denoted CT). Quantification was performed by normalizing Ct values with β-actin Ct and analyzed with the 2−ΔΔCT method [30].
Western Blotting
Astrocytes were washed once with ice-cold PBS and lysed in RIPA buffer containing a protease inhibitor mixture (Roche Applied Science, China). Protein concentrations were determined using Bradford reagent (Bio-Rad, China). In total, 20 μg of protein extract was resolved by 10–12% SDS-PAGE and proteins were transferred to a polyvinylidene difluoride (PVDF) membrane according to standard protocols. Primary antibodies against p65, p-p65 (S536), c-Jun, p-c-Jun (T239), ASK1, p-ASK 1 (T845), p-ASK1 (S967), p38, p-p38 (T180/Y182), JNK, p-JNK (T183), and β-actin were all purchased from Affinity Bioscience, Inc. (Shanghai, China) and diluted according to the recommended concentration in product manuals. All antibodies were diluted in tris-buffered saline (TBS) containing 0.5% Tween-20 and were incubated overnight at 4 °C. On the following day, the membranes were incubated with horseradish peroxidase-conjugated goat anti-rat IgG (1:5000; Bio-Rad) for 2 h at room temperature. The blots were developed with the use of an ECL detection kit (Invitrogen, Shanghai, China).
Measurement of Cytokines and Chemokines in Supernatants
Rat IL-1β, IL-6, and TNF-α and MCP-1, RANTES, and CXCL1 in culture supernatants in response to various stimuli were detected by ELISA kits (BD Biosciences) according to the manufacturers’ standard protocols.
The Boyden Chamber (Chemotaxis) Assay
The Boyden chamber (chemotaxis) assay were performed in a 48-well-modified Boyden chamber (Neuroprobe, Shanghai, China) on 10-μm-thick uncoated Nucleopore membrane (Neuroprobe, Shanghai, China) with a pore diameter of 8 μm. Migration was measured by assessment of the monocyte migration rate through the membrane, where the PBMCs and the astrocytes were grown without direct cell-to-cell contact. About 1 × 105 PBMCs were seeded into the upper wells (inserts) of the chamber, while 1 × 106 astrocytes were seeded into the lower wells. The astrocytes were treated with either inhibitors or DMSO for 30 min, followed by the treatment with 10 ng/ml CTGF. Thereafter, the upper wells were placed on top of the lower wells and cells were cocultured for 96 h, then the invaded PBMCs under the membrane were counted. Controls were performed by omission of the astrocytes. For better clearness, invasion is shown as % of control (= 100%).
Statistical Analysis
Results were expressed as the mean ± standard deviation (SD). Differences between groups were examined for statistical significance using two-tailed Student’s t test. P values of less than 0.05 are considered to be significant.
References
Ahmed, S.M., B.A. Rzigalinski, K.A. Willoughby, H.A. Sitterding, and E.F. Ellis. 2000. Stretch-induced injury alters mitochondrial membrane potential and cellular ATP in cultured astrocytes and neurons. Journal of Neurochemistry 74 (5): 1951–1960.
Akira, S., and K. Takeda. 2004. Toll-like receptor signalling. Nature Reviews. Immunology 4 (7): 499–511. https://doi.org/10.1038/nri1391.
Basak, S., and A. Hoffmann. 2008. Crosstalk via the NF-kappa B signaling system. Cytokine & Growth Factor Reviews 19 (3–4): 187–197. https://doi.org/10.1016/j.cytogfr.2008.04.005.
Bradham, D.M., A. Igarashi, R.L. Potter, and G.R. Grotendorst. 1991. Connective tissue growth factor: a cysteine-rich mitogen secreted by human vascular endothelial cells is related to the SRC-induced immediate early gene product CEF-10. The Journal of Cell Biology 114 (6): 1285–1294.
Brunner, A., J. Chinn, M. Neubauer, and A.F. Purchio. 1991. Identification of a gene family regulated by transforming growth factor-beta. DNA and Cell Biology 10 (4): 293–300. https://doi.org/10.1089/dna.1991.10.293.
Cahill, C.M., and J.T. Rogers. 2008. Interleukin (IL) 1beta induction of IL-6 is mediated by a novel phosphatidylinositol 3-kinase-dependent AKT/IkappaB kinase alpha pathway targeting activator protein-1. The Journal of Biological Chemistry 283 (38): 25900–25912. https://doi.org/10.1074/jbc.M707692200.
Cargnello, M., and P.P. Roux. 2011. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiology and Molecular Biology Reviews 75 (1): 50–83. https://doi.org/10.1128/MMBR.00031-10.
Chen, H., F. Wang, H. Mao, and X. Yan. 2014. Degraded lambda-carrageenan activates NF-kappaB and AP-1 pathways in macrophages and enhances LPS-induced TNF-alpha secretion through AP-1. Biochimica et Biophysica Acta 1840 (7): 2162–2170. https://doi.org/10.1016/j.bbagen.2014.03.011.
Chen, Y., P. Segarini, F. Raoufi, D. Bradham, and A. Leask. 2001. Connective tissue growth factor is secreted through the Golgi and is degraded in the endosome. Experimental Cell Research 271 (1): 109–117. https://doi.org/10.1006/excr.2001.5364.
Chu, C.Y., C.C. Chang, E. Prakash, and M.L. Kuo. 2008. Connective tissue growth factor (CTGF) and cancer progression. Journal of Biomedical Science 15 (6): 675–685. https://doi.org/10.1007/s11373-008-9264-9.
Clausen, F., A. Hanell, C. Israelsson, J. Hedin, T. Ebendal, A.K. Mir, H. Gram, and N. Marklund. 2011. Neutralization of interleukin-1beta reduces cerebral edema and tissue loss and improves late cognitive outcome following traumatic brain injury in mice. The European Journal of Neuroscience 34 (1): 110–123. https://doi.org/10.1111/j.1460-9568.2011.07723.x.
Daniele, S., E. Da Pozzo, E. Zappelli, and C. Martini. 2015. Trazodone treatment protects neuronal-like cells from inflammatory insult by inhibiting NF-kappaB, p38 and JNK. Cellular Signalling 27 (8): 1609–1629. https://doi.org/10.1016/j.cellsig.2015.04.006.
Das, M., S. Mohapatra, and S.S. Mohapatra. 2012. New perspectives on central and peripheral immune responses to acute traumatic brain injury. Journal of Neuroinflammation 9: 236. https://doi.org/10.1186/1742-2094-9-236.
Faden, A.I., and D.J. Loane. 2015. Chronic neurodegeneration after traumatic brain injury: Alzheimer disease, chronic traumatic encephalopathy, or persistent neuroinflammation? Neurotherapeutics 12 (1): 143–150. https://doi.org/10.1007/s13311-014-0319-5.
Gao, J., and Z. Zheng. 2015. Development of prognostic models for patients with traumatic brain injury: a systematic review. International Journal of Clinical and Experimental Medicine 8 (11): 19881–19885.
Gao, W., Z. Zhao, G. Yu, Z. Zhou, Y. Zhou, T. Hu, R. Jiang, and J. Zhang. 2015. VEGI attenuates the inflammatory injury and disruption of blood-brain barrier partly by suppressing the TLR4/NF-kappaB signaling pathway in experimental traumatic brain injury. Brain Research 1622: 230–239. https://doi.org/10.1016/j.brainres.2015.04.035.
Grotendorst, G.R., H. Okochi, and N. Hayashi. 1996. A novel transforming growth factor beta response element controls the expression of the connective tissue growth factor gene. Cell Growth & Differentiation 7 (4): 469–480.
Guo, X., C. Harada, K. Namekata, A. Matsuzawa, M. Camps, H. Ji, D. Swinnen, et al. 2010. Regulation of the severity of neuroinflammation and demyelination by TLR-ASK1-p38 pathway. EMBO Molecular Medicine 2 (12): 504–515. https://doi.org/10.1002/emmm.201000103.
Helmy, A., M.G. De Simoni, M.R. Guilfoyle, K.L. Carpenter, and P.J. Hutchinson. 2011. Cytokines and innate inflammation in the pathogenesis of human traumatic brain injury. Progress in Neurobiology 95 (3): 352–372. https://doi.org/10.1016/j.pneurobio.2011.09.003.
Ichijo, H., E. Nishida, K. Irie, P. ten Dijke, M. Saitoh, T. Moriguchi, M. Takagi, K. Matsumoto, K. Miyazono, and Y. Gotoh. 1997. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 275 (5296): 90–94.
Kaplan, G.B., J.J. Vasterling, and P.C. Vedak. 2010. Brain-derived neurotrophic factor in traumatic brain injury, post-traumatic stress disorder, and their comorbid conditions: role in pathogenesis and treatment. Behavioural Pharmacology 21 (5–6): 427–437. https://doi.org/10.1097/FBP.0b013e32833d8bc9.
Karger, A., B. Fitzner, P. Brock, G. Sparmann, J. Emmrich, S. Liebe, and R. Jaster. 2008. Molecular insights into connective tissue growth factor action in rat pancreatic stellate cells. Cellular Signalling 20 (10): 1865–1872. https://doi.org/10.1016/j.cellsig.2008.06.016.
Karve, I.P., J.M. Taylor, and P.J. Crack. 2016. The contribution of astrocytes and microglia to traumatic brain injury. British Journal of Pharmacology 173 (4): 692–702. https://doi.org/10.1111/bph.13125.
Kolaczkowska, E., and P. Kubes. 2013. Neutrophil recruitment and function in health and inflammation. Nature Reviews. Immunology 13 (3): 159–175. https://doi.org/10.1038/nri3399.
Kular, L., J. Pakradouni, P. Kitabgi, M. Laurent, and C. Martinerie. 2011. The CCN family: a new class of inflammation modulators? Biochimie 93 (3): 377–388. https://doi.org/10.1016/j.biochi.2010.11.010.
Ling, H., J. Hardy, and H. Zetterberg. 2015. Neurological consequences of traumatic brain injuries in sports. Molecular and Cellular Neurosciences 66 (Pt B): 114–122. https://doi.org/10.1016/j.mcn.2015.03.012.
Liu, Q., B.J. Wilkins, Y.J. Lee, H. Ichijo, and J.D. Molkentin. 2006. Direct interaction and reciprocal regulation between ASK1 and calcineurin-NFAT control cardiomyocyte death and growth. Molecular and Cellular Biology 26 (10): 3785–3797. https://doi.org/10.1128/MCB.26.10.3785-3797.2006.
Liu, S.C., C.J. Hsu, H.T. Chen, H.K. Tsou, S.M. Chuang, and C.H. Tang. 2012. CTGF increases IL-6 expression in human synovial fibroblasts through integrin-dependent signaling pathway. PLoS One 7 (12): e51097. https://doi.org/10.1371/journal.pone.0051097.
Liu, Y., Z. Liu, X. Li, B. Luo, J. Xiong, W. Gan, M. Jiang, Z. Zhang, H.J. Schluesener, and Z. Zhang. 2014. Accumulation of connective tissue growth factor+ cells during the early phase of rat traumatic brain injury. Diagnostic Pathology 9: 141. https://doi.org/10.1186/1746-1596-9-141.
Livak, K.J., and T.D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25 (4): 402–408. https://doi.org/10.1006/meth.2001.1262.
Lumpkins, K., G.V. Bochicchio, B. Zagol, K. Ulloa, J.M. Simard, S. Schaub, W. Meyer, and T. Scalea. 2008. Plasma levels of the beta chemokine regulated upon activation, normal T cell expressed, and secreted (RANTES) correlate with severe brain injury. The Journal of Trauma 64 (2): 358–361. https://doi.org/10.1097/TA.0b013e318160df9b.
Madathil, S.K., S.W. Carlson, J.M. Brelsfoard, P. Ye, A.J. D’Ercole, and K.E. Saatman. 2013. Astrocyte-specific overexpression of insulin-like growth factor-1 protects hippocampal neurons and reduces behavioral deficits following traumatic brain injury in mice. PLoS One 8 (6): e67204. https://doi.org/10.1371/journal.pone.0067204.
Martin-Lopez, E., J. Garcia-Marques, R. Nunez-Llaves, and L. Lopez-Mascaraque. 2013. Clonal astrocytic response to cortical injury. PLoS One 8 (9): e74039. https://doi.org/10.1371/journal.pone.0074039.
Masuko, K., M. Murata, K. Yudoh, H. Shimizu, M. Beppu, H. Nakamura, and T. Kato. 2010. Prostaglandin E2 regulates the expression of connective tissue growth factor (CTGF/CCN2) in human osteoarthritic chondrocytes via the EP4 receptor. BMC Research Notes 3: 5. https://doi.org/10.1186/1756-0500-3-5.
Matsuzawa, A., K. Saegusa, T. Noguchi, C. Sadamitsu, H. Nishitoh, S. Nagai, S. Koyasu, K. Matsumoto, K. Takeda, and H. Ichijo. 2005. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nature Immunology 6 (6): 587–592. https://doi.org/10.1038/ni1200.
Mokalled, M.H., C. Patra, A.L. Dickson, T. Endo, D.Y. Stainier, and K.D. Poss. 2016. Injury-induced ctgfa directs glial bridging and spinal cord regeneration in zebrafish. Science 354 (6312): 630–634. https://doi.org/10.1126/science.aaf2679.
Morganti-Kossmann, M.C., V.H. Hans, P.M. Lenzlinger, R. Dubs, E. Ludwig, O. Trentz, and T. Kossmann. 1999. TGF-beta is elevated in the CSF of patients with severe traumatic brain injuries and parallels blood-brain barrier function. Journal of Neurotrauma 16 (7): 617–628. https://doi.org/10.1089/neu.1999.16.617.
Moussad, E.E., and D.R. Brigstock. 2000. Connective tissue growth factor: what’s in a name? Molecular Genetics and Metabolism 71 (1–2): 276–292. https://doi.org/10.1006/mgme.2000.3059.
Pedrazzi, M., M. Patrone, M. Passalacqua, E. Ranzato, D. Colamassaro, B. Sparatore, S. Pontremoli, and E. Melloni. 2007. Selective proinflammatory activation of astrocytes by high-mobility group box 1 protein signaling. Journal of Immunology 179 (12): 8525–8532.
Ponsford, J., K. Draper, and M. Schonberger. 2008. Functional outcome 10 years after traumatic brain injury: its relationship with demographic, injury severity, and cognitive and emotional status. Journal of the International Neuropsychological Society 14 (2): 233–242. https://doi.org/10.1017/S1355617708080272.
Qu, J., X.Y. Tao, P. Teng, Y. Zhang, C.L. Guo, L. Hu, Y.N. Qian, C.Y. Jiang, and W.T. Liu. 2017. Blocking ATP-sensitive potassium channel alleviates morphine tolerance by inhibiting HSP70-TLR4-NLRP3-mediated neuroinflammation. Journal of Neuroinflammation 14 (1): 228. https://doi.org/10.1186/s12974-017-0997-0.
Raghupathi, R. 2004. Cell death mechanisms following traumatic brain injury. Brain Pathology 14 (2): 215–222.
Riser, B.L., M. Denichilo, P. Cortes, C. Baker, J.M. Grondin, J. Yee, and R.G. Narins. 2000. Regulation of connective tissue growth factor activity in cultured rat mesangial cells and its expression in experimental diabetic glomerulosclerosis. Journal of the American Society of Nephrology 11 (1): 25–38.
Roth, T.L., D. Nayak, T. Atanasijevic, A.P. Koretsky, L.L. Latour, and D.B. McGavern. 2014. Transcranial amelioration of inflammation and cell death after brain injury. Nature 505 (7482): 223–228. https://doi.org/10.1038/nature12808.
Semple, B.D., N. Bye, M. Rancan, J.M. Ziebell, and M.C. Morganti-Kossmann. 2010. Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2−/− mice. Journal of Cerebral Blood Flow and Metabolism 30 (4): 769–782. https://doi.org/10.1038/jcbfm.2009.262.
Semple, B.D., N. Bye, J.M. Ziebell, and M.C. Morganti-Kossmann. 2010. Deficiency of the chemokine receptor CXCR2 attenuates neutrophil infiltration and cortical damage following closed head injury. Neurobiology of Disease 40 (2): 394–403. https://doi.org/10.1016/j.nbd.2010.06.015.
Shi-Wen, X., A. Leask, and D. Abraham. 2008. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine & Growth Factor Reviews 19 (2): 133–144. https://doi.org/10.1016/j.cytogfr.2008.01.002.
Shi-wen, X., L.A. Stanton, L. Kennedy, D. Pala, Y. Chen, S.L. Howat, E.A. Renzoni, et al. 2006. CCN2 is necessary for adhesive responses to transforming growth factor-beta1 in embryonic fibroblasts. The Journal of Biological Chemistry 281 (16): 10715–10726. https://doi.org/10.1074/jbc.M511343200.
Shojo, H., Y. Kaneko, T. Mabuchi, K. Kibayashi, N. Adachi, and C.V. Borlongan. 2010. Genetic and histologic evidence implicates role of inflammation in traumatic brain injury-induced apoptosis in the rat cerebral cortex following moderate fluid percussion injury. Neuroscience 171 (4): 1273–1282. https://doi.org/10.1016/j.neuroscience.2010.10.018.
Sofroniew, M.V., and H.V. Vinters. 2010. Astrocytes: biology and pathology. Acta Neuropathologica 119 (1): 7–35. https://doi.org/10.1007/s00401-009-0619-8.
Szmydynger-Chodobska, J., N. Strazielle, B.J. Zink, J.F. Ghersi-Egea, and A. Chodobski. 2009. The role of the choroid plexus in neutrophil invasion after traumatic brain injury. Journal of Cerebral Blood Flow and Metabolism 29 (9): 1503–1516. https://doi.org/10.1038/jcbfm.2009.71.
Takeda, K., R. Shimozono, T. Noguchi, T. Umeda, Y. Morimoto, I. Naguro, K. Tobiume, M. Saitoh, A. Matsuzawa, and H. Ichijo. 2007. Apoptosis signal-regulating kinase (ASK) 2 functions as a mitogen-activated protein kinase kinase kinase in a heteromeric complex with ASK1. The Journal of Biological Chemistry 282 (10): 7522–7531. https://doi.org/10.1074/jbc.M607177200.
Weckbach, S., M. Neher, J.T. Losacco, A.L. Bolden, L. Kulik, M.A. Flierl, S.E. Bell, V.M. Holers, and P.F. Stahel. 2012. Challenging the role of adaptive immunity in neurotrauma: Rag1(−/−) mice lacking mature B and T cells do not show neuroprotection after closed head injury. Journal of Neurotrauma 29 (6): 1233–1242. https://doi.org/10.1089/neu.2011.2169.
Yu, Z., P. Yu, H. Chen, and H.M. Geller. 2014. Targeted inhibition of KCa3.1 attenuates TGF-beta-induced reactive astrogliosis through the Smad2/3 signaling pathway. Journal of Neurochemistry 130 (1): 41–49. https://doi.org/10.1111/jnc.12710.
Funding
This study was supported by the Hangzhou science and technology plan guidance project (Grant No. 20171226Y87).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lu, M., Yan, XF., Si, Y. et al. CTGF Triggers Rat Astrocyte Activation and Astrocyte-Mediated Inflammatory Response in Culture Conditions. Inflammation 42, 1693–1704 (2019). https://doi.org/10.1007/s10753-019-01029-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-019-01029-7