
Abstract
Variation in fin shape is one of the most prominent features of morphological diversity among fish. Regulation of fin growth has mainly been studied in zebrafish, and it is not clear whether the molecular mechanisms underlying shape variation are equally diverse or rather conserved across species. In the present study, expression levels of 37 candidate genes were tested for association with fin shape in the cichlid fish Lamprologus tigripictilis. The tested genes included members of a fin shape-associated gene regulatory network identified in a previous study and novel candidates selected within this study. Using both intact and regenerating fin tissue, we tested for expression differences between the elongated and the short regions of the spade-shaped caudal fin and identified 20 genes and transcription factors (including angptl5, cd63, csrp1a, cx43, esco2, gbf1, and rbpj), whose expression patterns were consistent with a role in fin growth. Collated with available gene expression data of two other cichlid species, our study not only highlights several genes that were correlated with fin growth in all three species (e.g., angptl5, cd63, cx43, and mmp9), but also reveals species-specific gene expression and correlation patterns, which indicate considerable divergence in the regulatory mechanisms of fin growth across cichlids.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The fins of fish contribute to various functions such as locomotion and communication (Wainwright et al., 2002; Lönnstedt et al., 2014), and variation in fin shape is one of the most prominent features of morphological diversity among fish species (e.g., Gilbert et al., 2022). In ray-finned fishes, the shape of the fins is determined predominantly by variation in the length of the individual fin rays. In many species, one or more of the fins are adorned by elongated filaments, which are formed by accelerated or prolonged growth of these fin regions during larval or later development. Less conspicuous shapes, such as spade- of fork-shaped caudal fins, are likewise the product of ray length variation within fins. Importantly, fin shape can regenerate completely after damage or experimental amputation throughout the lifetime of the fish, suggesting that it is under rather strict genetic control and that positional memory orchestrates the molecular factors necessary for fin regeneration (Rabinowitz et al., 2017). The fin rays of teleost fish are segmented, and both ontogenetic and regenerative growth involves the addition of bony segments at their distal ends. Ray length is therefore determined by both the length and the number of individual segments. At the molecular level, this involves components of various developmental pathways, such as WNT, FGF, Hedgehog, and retinoic acid (RA) signaling, as well as epigenetic, skeletogenic, and structural remodeling factors (Iovine, 2007; Yoshinari et al., 2009; Wehner & Weidinger, 2015; Sehring & Weidinger, 2020; Singh et al., 2021). Most of the ground-laying work on the genetic control of fin growth and shape has concentrated on zebrafish (Pfefferli & Jaźwińska, 2015), and it is not clear to which degree the molecular and anatomical basis of fin shape formation varies among fish species.
We have previously conducted a study of the anatomy and the molecular mechanisms underlying the ornamental fin shape of the cichlid fish Neolamprologus brichardi (Trewavas & Poll, 1952), an endemic of Lake Tanganyika in East Africa (Ahi et al., 2017; Ahi & Sefc, 2018). In this species, the distal tips of the dorsal and anal fins as well as the dorsal and ventral tips of the fork-shaped caudal fin are conspicuously elongated. Expression levels of candidate genes were compared between elongated and non-elongated (short) regions within the same fins, using intact as well as regenerating fin tissue sampled from adult fish. The observed gene expression patterns and correlations led to the proposition of a gene regulatory network (GRN) involved in the formation of the fin phenotype, hitherto referred to as N.b.-GRN. Members of the N.b.-GRN include genes reported to be involved in fin ray segmentation, angiogenesis, or neurogenesis such as cx43, mmp9, angptl5, angptl7, dpysl5a, csrp1a, and cd63 (Iovine et al., 2005; Monaghan et al., 2006; Nakatani et al., 2007; Sims et al., 2009; Ma et al., 2012; Ton & Iovine, 2012, 2013; Kang et al., 2015; Hagedorn et al., 2016), and several potential upstream regulators for this gene network including egr2, foxc1, foxd3, foxp1, irf8, and myc (Ahi & Sefc, 2018). Among the latter, myc, irf8, and foxd3 were already indicated in fin regeneration studies of other teleost fish (Christen et al., 2010; Li et al., 2012; Kang et al., 2015; Hasegawa et al., 2017; Huang & Chen, 2017). We predicted foxd3 as the key upstream regulator of the gene network in N. brichardi, since it consistently displayed significant expression correlation with all members of the network genes (Ahi & Sefc, 2018).
We next investigated whether members of the N.b.-GRN were also implicated in similar elongations of the dorsal and anal fins in another East African cichlid species, Steatocranus casuarius (Poll, 1939), a member of Steatocranini tribe which is a sister lineage to the Lake Tanganyika cichlids (Ahi et al., 2019a). Only a subset of the tested genes showed the expected expression level differences between short and elongated fin regions, indicating that the molecular mechanisms controlling fin elongation differed between the two cichlid species. The divergence in the molecular mechanisms of fin elongations between the two species was accompanied by anatomical differences. In N. brichardi, fin ray segments in the elongated fin regions were shorter or the same length as the fin ray segments in the short fin regions, suggesting that the elongation must be due to a larger number of segments in the elongated rays (Ahi et al., 2017). In S. casuarius, in contrast, segments of the elongated rays were longer than the fin ray segments in the short fin region (Ahi et al., 2019a).
With a divergence time of about 14 million years between their respective tribes (Irisarri et al., 2018), N. brichardi (tribe Lamprologini) and S. casuarius (tribe Steatocranini) represent two rather distantly related cichlids with convergent shapes of the dorsal and anal fins (Ahi et al., 2019a). In the present study, we ask whether the genetic control of fin growth is more conserved between two less divergent cichlid species. To this aim, we examine the caudal fin of Lamprologus tigripictilis (Schelly & Stiassny, 2004), which is reverse to the fork-shape of the caudal fin in N. brichardi—specifically, a spade-shaped caudal fin, in which the medial region is elongated compared to the short dorsal and ventral regions of the fin. L. tigripictilis is also a member of the Lamprologini and considerably more closely related to N. brichardi than S. casuarius, as the radiation of the Lamprologini started at only about 6 million years ago (Irisarri et al., 2018). If mechanisms of fin growth are shared between the two lamprologine cichlids, we expect that many of the N.b.-GRN member genes will show corresponding gene expression differences between the elongated and short caudal fin regions in L. tigripictilis. We first examined the caudal fin expression patterns of 16 members of the N.b.-GRN by comparing gene expression levels in short and elongated regions using qPCR. Since only few of the N.b.-GRN showed the expected expression patterns, we then expanded the set of candidate genes based on co-expression data and transcription factor prediction and tested their expression in the fin tissue samples of L. tigripictilis. We also measured the length of the fin ray segments in the short and elongated fin rays in order to assess the anatomical basis of fin elongation in L. tigripictilis.
Methods
Fin sampling for RNA isolation
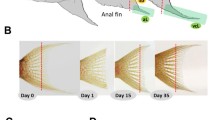
We used six captive bred adult males of L. tigripictilis (total length 7–10 cm). Prior to taking the fin biopsies, fish were anesthetized using 0.04 g of MS-222 per liter of water. Then, the caudal fin was cut in front of the first ray bifurcation (branching) under a stereomicroscope (red dashed lines in Fig. 1A, B). Next, three separate tissue samples were dissected from the severed fin (Fig. 1A): one comprised the most dorsal, branched fin ray and represented the dorsal short fin region (dS), one comprised the most ventral, branched fin ray and represented the ventral short fin region (vS), and one comprised the medial fin ray and represented the medial elongated fin region (mL). Each sample comprised the two branches of the selected fin ray. The tissue samples were stored frozen in RNA later (Qiagen) until RNA isolation. Biopsies were taken from the original fin tissue (stage 0) to study gene activity patterns associated with the maintenance of the phenotype, and then twice during regeneration, including a biopsy at day 20 after the first biopsy when the round shape of the fin started to appear (stage 1), and another biopsy at day 40 after the stage 1 biopsy, when the fin had become distinctly rounded (stage 2). The biopsy regime is illustrated in Fig. 1B. Anesthesia and fin biopsies were performed under permit number BMWFW-66.007/0028-WF/V/3b/2017 issued by the Federal Ministry of Science, Research and Economy of Austria (BMWFW). All methods were performed in accordance with the relevant guidelines and regulations of BMWFW.

The caudal fin of L. tigripictilis. A Caudal fins were amputated along the dashed red line, and tissue samples representing the short dorsal (dS), the short ventral (vS), and the elongated medial (mL) regions of the dorsal fin were taken as indicated. B Biopsies were taken from the original fin tissue (day 0, representing stage 0) and from regenerating tissue at day 20 (representing stage 1) and day 40 (representing stage 2) after the preceding amputation. C Length of fin ray segments in the three regions of the caudal fin. Different box colors indicate significant differences in segment length
Fin ray segment length measurements
To measure the length of fin ray segments in elongated medial and the short dorsal and ventral regions of the caudal fin, biopsies were taken from six adult males in the same way as fin sampling for RNA isolation at stage 0 described above and stained with alizarin red. We modified the acid-free double staining protocol described by Walker & Kimmel (2007) and used 10% KOH in the clearing phase, increased the duration of the staining phase to 4 days and the duration of the clearing phase to 15 days. Using a Keyence VHX-5000 Digital Microscope, we measured the length of the 10 most distal, complete segments on one branch per each of two dorsal (short), two ventral (short), and four medial (elongated) fin rays. That is, measurements were taken from eight different rays, and not from branches pertaining to the same ray. The selected fin rays were the two most dorsal, branched rays, the two most ventral, branched rays (we did not use the most dorsal and the most ventral fin ray, as these were only rudimentary developed in some individuals) and the four medial fin rays. The branch representing each fin ray was either selected randomly or by avoiding irregularities in the segmentation pattern, which sometimes occurred in one of the branches. We used a linear mixed model (R package lmerTest) to compare segment length between the three fin regions, including segment length as response variable and fin region as predictor variable. To account for possible correlations within rays and individual fins, we included ‘fin ray’ nested in biological replicate (fish) as random factors (Supplementary Data 2, Table S1).
Candidate target and reference gene selection
The selection and analysis of candidate target genes and transcription factors (TF) was performed in three steps (see Ahi et al., 2015). First, we tested 16 genes of the N.b.-GRN described in the introduction (Ahi & Sefc, 2018). These genes were angptl5, angptl7, anxa2a, c1qtnl5a, cd63, csrp1a, cx43, dpysl5a, gnao1a, kif5a, mmp9, pfkpa, sema3d, txn, wnt10a, and wnt5b. Out of the first set of candidate genes, we picked the ones with the strongest expression differences between the elongated and the short fin regions (angptl5, cd63, csrp1a; see “Results” section) to search for genes co-expressed with all the three genes (top overlapping co-expressed genes) in the zebrafish co-expression database, COXPRESdb (http://coxpresdb.jp/) version 6.0 (Obayashi & Kinoshita, 2011). To attain a high degree of reliability, we filtered the genes co-expressed with each of the three genes by setting the Supportability score to a minimum of 1 (as described by Obayashi & Kinoshita, 2011) (Supplementary data 1). This step identified eight additional candidate genes (see results). Finally, we selected eight of the above 24 candidate genes, namely those with strongly increased expression in the elongated fin region (see results), for TF prediction. In order to predict the potential upstream regulators for these genes, we performed motif enrichment on 4 kb upstream sequences (promoter and 5’-UTR) of these genes as previously described by (Lecaudey et al., 2019, 2021) using the annotated genome of the Nile tilapia, Oreochromis niloticus (Flicek et al., 2013), and two algorithms: MEME (Bailey et al., 2009) and XXmotif (Luehr et al., 2012). The motifs that were present in the promoters of at least half of these genes were compared to position weight matrices (PWMs) from the TRANSFAC database (Matys et al., 2003) using STAMP (Mahony & Benos, 2007) to identify matching transcription factor (TF) binding sites (Supplementary data 1).
To identify stable reference genes, we selected 8 candidate reference genes with abundant expression in a range of tissues, which have already been investigated as reference genes in fins or other tissues containing skeletal structures or/and epidermis in fish (Table 1). Candidate reference genes were ranked according to expression stability by three different algorithms, BestKeeper (Pfaffl et al., 2004), NormFinder (Andersen et al., 2004), and geNorm (Vandesompele et al., 2002). The standard deviation (SD) based on Cq values of the fin regions was calculated by BestKeeper to determine the expression variation for each reference gene. In addition to ranking, BestKeeper also determines the stability of reference genes through a correlation calculation or BestKeeper index (r). GeNorm calculates mean pairwise variation between each gene and other candidates (the expression stability or M value) in a stepwise manner and NormFinder identifies the most stable genes (lowest expression stability values) based on the analysis of inter- and intra-group variation in expression levels variations (Ahi et al., 2018, 2019b). All three algorithms ranked rps18 and actb1 as the two most stable candidate reference genes (Table 1). Based on these results, we used the geometric mean of the expression of actb1 and rps18 for normalization of relative gene expression of candidate target genes.
Primer design
In order to design qPCR primers, we aligned the orthologues of each gene from different African cichlid tribes including one species of Tilapiini (Oreochromis niloticus; Linnaeus, 1758), one species of Lamprologini (N. brichardi), and one species of Haplochromini (Astatotilapia burtoni; Günther, 1894) (Brawand et al., 2014; Santos et al., 2016; Singh et al., 2017). The 1-to-1 orthologues were confirmed by blasting zebrafish mRNA REfSeq IDs against N. brichardi transcriptome in NCBI and cross-checking the top hits returned by BLAST in the Ensembl database for zebrafish and O. niloticus orthologues (http://www.ensembl.org). Next, we used the aligned sequences to identify conserved regions across the species (using CLC Genomic Workbench, CLC Bio, Denmark) and at the exon/exon boundaries (using annotated genome of O. niloticus in the Ensembl database. Primers with short amplicon sizes (< 250 bp) were designed using Primer Express 3.0 (Applied Biosystems, CA, USA) and OligoAnalyzer 3.1 (Integrated DNA Technology) (Supplementary data 1), as previously described (Ahi et al., 2020).
RNA isolation and real-time qPCR
RNA was isolated from individual tissue samples using the Trizol protocol described in (Ahi et al., 2017). DNA was removed enzymatically and RNA concentration was measured by spectrophotometry using a nanophotometer (IMPLEN GmbH, Munich, Germany). RNA quality (integrity number > 7) was ascertained in a R6K ScreenTape System on an Agilent 2200 TapeStation (Agilent Technologies). cDNA was prepared from 1000 ng of RNA using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems), according to the manufacturer's protocol. Negative controls, i.e., reactions without addition of reverse transcriptase (-RT samples), were prepared to confirm the absence of genomic DNA. cDNA was diluted 1:3 times in nuclease-free water for further use in quantitative real-time PCR.
The qPCR was conducted using Maxima SYBR Green/ROX qPCR Master Mix (2×) by following the manufacturer’s instruction (Thermo Fisher Scientific, St Leon-Rot, Germany) in 96-well PCR plates on an ABI 7500 real-time PCR System (Applied Biosystems). The experimental set-up per run followed the preferred sample maximization method (Hellemans et al., 2007). The primer efficiency analyses in LinRegPCR v11.0 (http://LinRegPCR.nl) (Ramakers et al., 2003) were conducted as described in our previous study (Ahi et al., 2017).
Analysis of qPCR data
The geometric mean of the Cq values (Vandesompele et al., 2002) of the two reference genes Cqreference was used to normalize Cq values of target genes in each sample (ΔCqtarget = Cqtarget – Cqreference). We randomly selected one biological replicate of the dorsal region of the caudal fin (stage 0) as calibrator sample, and in order to calculate ΔΔCq values, we subtracted the ΔCq values of all samples from the calibrator ΔCq value (ΔCqtarget – ΔCqcalibrator). Relative expression quantities (RQ values) were calculated as 2−ΔΔCq (Pfaffl, 2001).
RQ values were log-transformed for statistical analyses. For each target gene, differences in expression levels between dorsal (short) and medial (elongated), ventral (short) and medial (elongated), as well as dorsal (short) and ventral (short) regions of the caudal fin were tested in linear mixed models with log(RQ) as dependent variable, fin region as fixed factor and developmental stage nested within biological replicate (fish) as random factors (Supplementary data 2, Table S2). To account for multiple testing (N = 111 comparisons; 37 candidate genes times 3 fin region contrasts), P values for the effect of length were corrected using the Benjamini–Hochberg procedure (Benjamini & Hochberg, 1995). We fit analogous linear mixed models including only stage 1 and stage 2 data (i.e., the regenerating tissue but not the intact tissue). The log-transformed RQ values were also used for calculations of pairwise Pearson correlation coefficients (r) among the candidate genes. Finally, paired t-tests were used to conduct stage-specific comparisons between fin regions, again using log-transformed RQ values. Bonferroni–Hochberg corrections of p-values were conducted for multiple testing within each of the fin region contrasts at each stage (N = number of genes = 37), and corrected p-values were used to mark significant differences in the barplot figures illustrating gene expression levels (Figs. 2, 3, 4; see also Supplementary Data 2, Table S3).

Expression levels of candidate genes selected based on already identified GRN in N. brichardi. Means and standard deviations of RQ in three biological replicates are shown for the elongated and short regions of the caudal in original (stage 0) and regenerating tissue. See Fig. 1A for fin region codes; numbers 0 to 2 identify regeneration stages. Circles above bars indicate significantly elevated expression (P < 0.05 in paired t-tests; Supplementary data 2, Table S3) in comparisons between elongated and short fin region samples (i.e., compared to the bar matching the shade of the circle)

Expression levels of candidate genes selected based on co-expression with csrp1a, angptl5, and cd63. Means and standard deviations of RQ in three biological replicates are shown for the elongated and short regions of the caudal in original (stage 0) and regenerating tissue. See Fig. 1A for fin region codes; numbers 0 to 2 identify regeneration stages. Circles above bars indicate significantly elevated expression (P < 0.05 in paired t-tests; Supplementary data 2, Table S3) in comparisons between elongated and short fin region samples (i.e., compared to the bar matching the shade of the circle)

Expression levels of predicted upstream regulators. Means and standard deviations of RQ in three biological replicates are shown for the elongated and short regions of the caudal in original (stage 0) and regenerating tissue. See Fig. 1A for fin region codes; numbers 0 to 2 identify regeneration stages. Circles above bars indicate significantly elevated expression (P < 0.05 in paired t-tests; Supplementary data 2, Table S3) in comparisons between elongated and short fin region samples (i.e., compared to the bar matching the shade of the circle)
Results
Anatomical characterization of caudal fin rays of L. tigripictilis and N. brichardi
The lengths of the fin ray segments (Fig. 1; Supplementary data 2, Table S1) did not differ significantly between the elongated, medial fin region (mean ± sd = 309.4 ± 25.5 µm) and the short, ventral fin region (mean ± sd = 315.1 ± 25.2 µm; est. = 5.9, t = 1.6, P = 0.12). In contrast, the individual fin ray segments were significantly longer in the short, dorsal fin region (mean ± sd = 339.6 ± 28.6 µm), both compared to the medial fin region (est. = 30.5, t = 8.2, P = 5.6 × 10–10) and compared to the ventral fin region (est. = 24.6, t = 5.7, P = 1.2 × 10–6; complete model output is reported in Supplementary data 2, Table S1). Hence, comparisons between the medial and the ventral region of the caudal fin of L. tigripictilis represent a contrast between long and short fin rays that differ in the number of segments per ray. In contrast, comparisons involving the dorsal fin region involve differences in both segment length (longer in dS) and number (fewer in dS), with a larger difference in the number of segments between dS and mL than between dS and vS.
To enable a comparison with N. brichardi (i.e., the cichlid species, in which the GRN was originally reconstructed), we re-analyzed the fin ray segment length data of the caudal fin of N. brichardi (data from Ahi et al., 2017). In the original analysis (Ahi et al., 2017), data from the dorsal and the ventral edges of the caudal fin had been pooled for comparison with the medial region and no difference in segment length was detected. As for L. tigripictilis, we now distinguished between the dorsal elongated and the ventral elongated rays of the caudal fin of N. brichardi and compared each to the medial short rays. This analysis revealed that the segments of the (long) dorsal rays of the caudal fin of N. brichardi were longer than those of the (long) ventral rays (mean ± SL = 289.9 ± 54.3 µm, in the dorsal region; 238.7 ± 33.0 µm, in the ventral region; N = 5 segments of 2 rays from each of 3 fish per fin region; LM, est. = 53.5, t = 2.3, P = 0.05), but in contrast to L. tigripictilis, the segments in the (short) medial rays were equally long as those in the (long) dorsal rays (mean ± SL = 291.8 ± 29.0 µm, in the medial region, N = 5 segments of 2 rays from each of 4 fish; LM, est. = 2.1, t = 0.2, P = 0.87).
Expression analysis of candidate genes
Each of the three pairwise comparisons between fin regions represents a different phenotypic contrast (see above), and we therefore conducted three pairwise comparisons of candidate gene expression levels among the fin regions (as opposed to combining the data into a single ‘short’ versus ‘long’ comparison). In the following text, ‘expression in the medial elongated region of the caudal fin’ is abbreviated as ‘mL expression,’ ‘expression in the dorsal region’ is abbreviated as ‘dS expression,’ ‘expression in the ventral region’ is abbreviated as ‘vS expression,’ and the expression levels are reported as ‘increased’ or ‘decreased’ in comparison with the other regions. We obtained similar results in analyses that included all three stages (intact fin and regeneration stages 1 and 2) and in analyses restricted to the two regeneration stages and reported the analyses of the full data unless noted otherwise. Table 2 summarizes the significant expression level differences detected among the tested candidate genes, and the complete statistical analyses are reported in the supplementary material (Supplementary data 2, Table S2).
In the first step of our gene expression analysis, we examined the expression levels of 16 members of the N.b.-GRN. Among these, we detected increased mL expression compared to both vS- and dS expression for angptl5, cd63, csrp1a, cx43, and sema3d, and increased mL expression compared to vS expression for c1qtnf5 and dpysl5a (mainly in stage 1 of fin regeneration) (Table 2, Fig. 2; Supplementary data 2, Table S2). Additionally, dS expression of mmp9 was lower than mL expression. We also detected significant expression level differences for pfkpa (higher mL- than dS expression during regeneration).
The second step of our analysis was based on the three genes, angptl5, cd63, and csrp1a, which had the strongest expression differences between the elongated medial and the short fin regions in the above analysis. Using the zebrafish co-expression database, we identified eight additional candidate genes that are co-expressed with each of these genes and compared their expression levels between the caudal fin regions of L. tigripictilis (arpc1, cdc42ep3, colec12, ctsk, fstl1b, mmp14b, olfml3b, and rin3; Supplementary data 1). Among these, increased mL expression compared to both vS and dS expressions was detected for colec12, ctsk, fstl1b, mmp14b, and rin3, whereas increased mL expression compared to only one of the short regions (vS or dS) was detected for arpc1 and cdc42ep3, and no expression difference was detected for olfml3b (Table 2, Fig. 3; Supplementary Data 2, Table S2).
Expression analysis of candidate upstream regulators
Predicted upstream regulators for eight genes with the most strongly increased mL expression in the above analyses (angptl5, cd63, ctsk, cx43, csrp1a, fstl1b, mmp14b, and rin3) included the 12 transcription factors alx4a, ap4, egr1, egr2, foxd3, foxp1, gbf1, heb, patz1, rbpj, srf, and znf219 (Supplementary Data 1). Additionally, we tested the expression of esco2 (which was not among the predicted TFs) because it regulates cx43 and sema3d in a gene regulatory network involved in the formation, growth, and regeneration of fin ray segments and joints (Iovine et al., 2005; Govindan & Iovine, 2014, 2015; Banerji et al., 2016; Govindan et al., 2016). Among the 13 tested TFs, decreased mL expression (compared to both vS and dS expressions) was detected for gbf1, esco2, and rbpj when data of all three developmental stages were pooled, but not when only stage 1 and 2 (regeneration) were analyzed. Finally, znf219 and alx4a showed higher dS than vS expression, but only in analyses across all three developmental stages (Table 2, Fig. 4; Supplementary data 2, Table S2).
Gene expression correlations
We tested for expression correlations among those of the candidate genes and upstream regulators, which had shown significant expression differences between elongated and short fin regions. A number of significant pairwise correlations as well as clusters of correlated genes were detected (Fig. 5). For instance, a cluster of positively correlated gene expression levels comprised the genes cx43, fstl1b, mmp14b, c1qtnl5a, rin3, angptl5, cd63, and ctsk. These genes showed higher expression in the mL region in comparison with one or both of the short fin regions (dS and/or vS). The expression levels of these genes were negatively correlated with that of the transcription factor rbpj and positively with that of the TF esco2. We also detected strong positive expression correlations among the transcription factors foxp1, gbf1, and rbpj. Expression of each of these TFs was negatively correlated with the expression of the TF esco2, and positively correlated with the expression of pfkpa, colec12, cdc42ep3, and dpysl5a.

Correlation analysis reveals significant positive or negative co-expression of the candidate genes. Pearson correlation coefficient (r) was used to assess the pairwise expression similarity between the candidate genes during craniofacial development. Blue represents positive and red represents negative expression correlation. *P < 0.05; **P < 0.01; ***P < 0.001
Discussion
Convergence across species in a morphological trait may, but need not, coincide with shared mechanisms at an anatomical and genetic level (Chan et al., 2010; Elmer & Meyer, 2011; Colombo et al., 2013; Conith et al., 2019). The present study addressed the genetic control of the differential growth of fin regions, which underlies the formation of the fin shape, in a cichlid fish species. If mechanisms of fin shape formation were strongly conserved, we would expect congruent gene expression patterns in elongated compared to short fin regions, both across types of fins and across species. The expression patterns of some of the N.b.-GRN genes that had previously been found to be associated with fin shape in the cichlid fish N. brichardi were indeed correlated with fin shape in the present focal species, L. tigripictilis. Many of them, however, were not, and some of the expression patterns and inferred regulatory interactions differed from N. brichardi, which suggests a certain level of divergence in the pathways controlling fin shape between related cichlid species. We therefore searched for further candidate genes and upstream regulators that might be involved in fin shape formation in L. tigripictilis and detected several promising candidates. In the following, we first discuss fin shape-associated expression patterns of the N.b.-GRN genes in the focal species, L. tigripictilis, in relation to available data from two other cichlid species. We then attend to the new candidate genes brought forth in this study and review published data that support their role in fin growth.
Expression patterns of the N.b.-GRN genes in L. tigripictilis fins
Slightly more than half of the N.b.-GRN members showed significant expression level differences between the fin regions of L. trigripictilis, but not all of these expression differences were in the direction predicted based on the patterns observed in N. brichardi (Ahi et al., 2017, Ahi & Sefc, 2018; summarized in Table 2). Fin shape-associated expression levels of the N.b.-GRN member genes have previously also been examined in the cichlid S. casuarius (Ahi et al., 2019a), and the set of genes with significant expression differences shared between N. brichardi and L. tigripictilis in the present study overlapped only partially with the genes that had shared expression differences between N. brichardi and S. casuarius (Ahi et al., 2019a; summarized in Table 2). An interesting congruence across all three species was detected for mmp9, which encodes a matrix remodeling enzyme with a role in fin regeneration (Yoshinari et al., 2009; LeBert et al., 2015). Expression of mmp9 was consistently lowest in those fin regions with the longest ray segments (i.e., the dorsal short region in L. tigripictilis, the short fin regions in N. brichardi and the elongated fin regions in S. casuarius; Table 2), suggesting that mmp9 expression might be associated with reduced ray segment length in the three cichlid species. In zebrafish, mmp9 is negatively regulated by cx43 (Ton & Iovine, 2013), which encodes a subunit of the gap junction protein complex and whose expression is positively correlated with segment length in the fin rays of zebrafish (Iovine et al., 2005; Sims et al., 2009). cx43 is a member of the N.b.-GRN and showed elevated expression in the fin regions with shorter segments in the L. tigripictilis and N. brichardi, suggesting that the interaction between cx43 and mmp9 and the role of cx43 in segment growth may differ between the zebrafish and the lamprologine cichlids. Regardless of segment length, however, cx43 was consistently more strongly expressed in the elongated fin regions of each of the three cichlid species (Table 2), which corroborates the gene as a strong candidate for a regulator of fin growth.
In zebrafish, cx43 is also a positive regulator of another of the tested N.b.-GRN genes, sema3d, which mediates the cx43-dependent functions in cell proliferation, joint formation, and phenotypic changes of zebrafish fins (Ton & Iovine, 2012). In humans, mutations in cx43 and sema3d are associated with defects in finger growth [brachydactyly, Kjaer et al., 2004; Jamsheer et al., 2014); ectrodactyly (Sivasankaran et al., 2015)]. The elevated mL expression of both sema3d and cx43 in L. tigripictilis is therefore concordant with known roles of these genes in skeletal growth. However, the functional relationship between the two genes appears to be different in N. brichardi and S. casuarius, where sema3d and cx43 showed opposite expression differences between L and S fin regions (Table 2).
Increased expression in elongated fin regions across all three studied cichlid species was detected for cd63, which induces spinal cord regeneration in the axolotl (Monaghan et al., 2006), and for angptl5, which encodes an angiopoietic protein. Increased expression of angptl5 was also observed during exaggerated elongation of the caudal fin in swordtail fish (Kang et al., 2015). In humans, the expression of angptl5 in endothelial cells is highly induced through interaction with osteoblasts during osteogenesis and bone remodeling (Simunovic et al., 2013). These data indicate that angptl5 expression during skeletal outgrowth is a marker for induced angiogenesis in the skeletal tissue across vertebrate taxa.
In summary, the present data collated with previous results (Ahi & Sefc, 2018; Ahi et al., 2019a) highlight a set of genes, including angptl5, cd63, cx43, and mmp9, whose expression levels were consistently correlated with fin elongation across three cichlid species. However, numerous differences between species with respect to which gene expression levels co-varied with fin shape and how gene expression levels were correlated with one another suggest that networks of genes involved in fin shape formation are not fully congruent between species. Our data also indicate variation in regulatory interactions, as only one of the upstream regulators of the N.b.-GRN, foxp1, displayed significant expression differences among fin regions in L. tigripictilis. Since the investigated fin types and their shapes differed between the three cichlid species, some of the inter-specific incongruences in gene expression patterns may reflect positional effects, for instance, when different genes control the growth of medial fin regions compared to elongations of the dorsal and ventral edges. Some of the incongruent gene expression patterns are likely associated with differences in the anatomical basis of fin ray elongation, specifically the lengths of fin ray segments in short and elongated fin regions. In the two lamprologine cichlid species, segment length variation was decoupled from fin ray length, whereas in S. casuarius, longer rays consisted of longer segments than shorter rays (Table 22). Consequently, expression patterns of some of the tested genes are expected to differ between species contingent on whether the gene controls the length (as, for instance, indicated for mmp9) or the number of ray segments, and functional studies are needed to elucidate their possible functions in fin growth.
Novel fin shape candidate genes detected in L. tigripictilis
Given that our data linked only few of the original candidate genes from the N.b.-GRN to fin shape formation in L. tigripicilis, we extended our candidate gene set based on existing co-expression data in zebrafish. Seven of the new candidate genes showed gene expression differences between the fin regions of L. tigripictilis (Table 2). Three of the new genes, fstl1, colec12, and mmp14, have already been implicated in skeletal morphogenesis in mammals or zebrafish. Follistatin-like 1, fstl1, encodes a potent antagonist of the BMP pathway during skeletogenesis in vertebrates (Sylva et al., 2011) and has been shown to affect digit formation in mammals (Lorda-Diez et al., 2013; Sylva et al., 2013). Colec12 (previously known as collectin placenta protein 1 gene, clp1) encodes collectin-12, which is involved in vasculogenesis and has been shown to be positively associated with body elongation in zebrafish during development; i.e., knockdown of colec12 caused shortened body length in zebrafish (Fukuda et al., 2011). The metalloprotease encoding mmp14 gene is involved in human skeletogenesis with effects on finger and toe morphology (Wilkinson et al., 2012; De Vos et al., 2018, 2019). In zebrafish, mutations in mmp14b can lead to skeletal anomalies including shortening of body and fin, prominent frontal bone and skeletal curvatures (De Vos et al., 2018). The elevated expression levels of mmp14 and colec12 in elongated compared to short fin regions in L. tigripictilis are consistent with the mutant zebrafish phenotypes.
We also extended our candidate gene set by predicting transcription factors for genes with strongly increased mL expression in L. tigripictilis. TFs predicted from genes with strongly increased mL expression are expected to display expression correlations with these genes and therefore to display either increased or decreased mL expression. The expected pattern was observed for gbf1 and rbpj, both of which showed reduced mL expression compared to both dS and vS fin regions. The expression levels of gbf1 and rbpj were also significantly correlated with the majority of the genes involved in the prediction procedure (Fig. 5). Golgi brefeldin A-resistant factor 1 gene, gbf1, encodes a protein that functions as a guanine nucleotide exchange factor and plays important roles in regulating organelle structure and cargo-selective vesicle trafficking (Manolea et al., 2008). During zebrafish development, gbf1 is involved in vascular system formation, pigmentation, and morphogenesis of the caudal fin (Chen et al., 2017). Knockdown of GBF1 in mammalian cells leads to a range of structural anomalies which eventually inhibit trafficking of transmembrane proteins and cell death (Citterio et al., 2008).
The second TF, rbpj, encodes a transcription factor with dual regulatory activities, which can regulate skeletogenic process both as an activator and as a repressor, depending on its interaction with Notch signal proteins (Castel et al., 2013). For instance, the Notch intracellular domain (NICD) and rbpj form a complex that can act as transcription repressor and negatively regulate chondrocyte differentiation (Chen et al., 2013). On the other hand, the NICD-rbpj complex acts as transcriptional activator inducing osteoblast proliferation (Tao et al., 2010). Moreover, rbpj has been shown to inhibit osteoclastogenesis and bone resorption (Zhao et al., 2012; Miller et al., 2016). It should be noted that Notch signal activity and rbpj transcription are both required for maintaining blastema cells in a plastic, undifferentiated, and proliferative state, which is essential for fin regeneration in zebrafish (Münch et al., 2013). In human, mutation in Rbpj is shown to be associated with etiology of Adams-Oliver syndrome (AOS) which is identified with multiple-malformation disorders, and particularly, with terminal limb defects (Hassed et al., 2012; Nakayama et al., 2014). The terminal limb defects in AOS are characterized by shortening of the end of the fingers (brachydactyly) and curvature of the digits (clinodactyly) (Nakayama et al., 2014). In mammals, rbpj can act as upstream regulator of mmp14 expression (Gao et al., 2016; Nus et al., 2016). The two genes show opposite expression patterns (mmp14 with elevated and rbpj with reduced mL expression, respectively) and significantly negative expression correlations (r = − 0.72, P > 0.001) in L. tigripictilis, suggesting that a regulatory interaction also exists in this cichlid species.
Finally, expression correlations suggested that the TF esco2 is connected with several of the tested TFs and genes, notably angptl5 and cd63, which our data strongly implicated in fin elongation. In contrast to evidence from zebrafish (Banerji et al., 2016), where esco2 is a positive regulator of cx43, there was no evidence for a regulatory link between esco2 and cx43 in L. tigripictilis. This is consistent with previous data from N. brichardi (Ahi and Sefc 2018), but opposite to S. casuarius, where expression levels of esco2 were positively correlated with those of cx43. These findings suggest a role for esco2 in the formation of fin shape in cichlid fish, but divergence with respect to the underlying molecular mechanisms.
Conclusions
The present study reports genes and transcription factors, whose expression levels were statistically linked to fin shape in the cichlid species L. tigripitilis. Among the tested candidate genes were members of a GRN that had previously been proposed to be involved in the filamentous elongation of fin edges in another cichlid species, N. brichardi. Only few of the N.b.-GRN genes showed the expected expression patterns in elongated and short regions of the L. tigripictilis caudal fin. Moreover, the predicted upstream regulators of the genes, whose expression was statistically associated with elongated fin regions, were also different in L. tigripictilis (gbf1 and rbpj) from those found in N. brichardi. Considering gene functions and interactions known in zebrafish as well as the gene expression data from previous work on cichlid fishes, the present data demonstrate some degree of conservation, but also substantial differences in the regulation of fin growth among cichlid fishes and also in comparison with zebrafish.
Data availability
All data generated during this study are included in the supplementary material accompanying this published article.
References
Ahi, E. P. & K. M. Sefc, 2018. Towards a gene regulatory network shaping the fins of the Princess cichlid. Scientific Reports 8: 9602.
Ahi, E. P., S. S. Steinhäuser, A. Pálsson, S. R. Franzdóttir, S. S. Snorrason, V. H. Maier & Z. O. Jónsson, 2015. Differential expression of the aryl hydrocarbon receptor pathway associates with craniofacial polymorphism in sympatric Arctic charr. EvoDevo 6: 27.
Ahi, E. P., F. Richter & K. M. Sefc, 2017. A gene expression study of ornamental fin shape in Neolamprologus brichardi, an African cichlid species. Scientific Reports 7: 17398.
Ahi, E. P., P. Singh, L. A. Lecaudey, W. Gessl & C. Sturmbauer, 2018. Maternal mRNA input of growth and stress-response-related genes in cichlids in relation to egg size and trophic specialization. EvoDevo 9: 23.
Ahi, E. P., F. Richter, L. A. Lecaudey & K. M. Sefc, 2019a. Gene expression profiling suggests differences in molecular mechanisms of fin elongation between cichlid species. Scientific Reports 9: 9052.
Ahi, E. P., P. Singh, A. Duenser, W. Gessl & C. Sturmbauer, 2019b. Divergence in larval jaw gene expression reflects differential trophic adaptation in haplochromine cichlids prior to foraging. BMC Evolutionary Biology 19: 150.
Ahi, E. P., L. A. Lecaudey, A. Ziegelbecker, O. Steiner, W. Goessler & K. M. Sefc, 2020. Expression levels of the tetratricopeptide repeat protein gene ttc39b covary with carotenoid-based skin colour in cichlid fish. Biology Letters 16: 20200629.
Andersen, C. L., J. L. Jensen & T. F. Ørntoft, 2004. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Research 64: 5245–5250.
Bailey, T. L., M. Boden, F. A. Buske, M. Frith, C. E. Grant, L. Clementi, J. Ren, W. W. Li & W. S. Noble, 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Research 37: W202–W208.
Banerji, R., D. M. Eble, M. K. Iovine & R. V. Skibbens, 2016. Esco2 regulates cx43 expression during skeletal regeneration in the zebrafish fin. Developmental Dynamics 245: 7–21.
Benjamini, Y. & Y. Hochberg, 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society 57: 289–300.
Brawand, D., C. E. Wagner, Y. I. Li, M. Malinsky, I. Keller, S. Fan, O. Simakov, A. Y. Ng, Z. W. Lim, E. Bezault, J. Turner-Maier, J. Johnson, R. Alcazar, H. J. Noh, P. Russell, B. Aken, J. Alföldi, C. Amemiya, N. Azzouzi, J.-F. Baroiller, F. Barloy-Hubler, A. Berlin, R. Bloomquist, K. L. Carleton, M. A. Conte, H. D’Cotta, O. Eshel, L. Gaffney, F. Galibert, H. F. Gante, S. Gnerre, L. Greuter, R. Guyon, N. S. Haddad, W. Haerty, R. M. Harris, H. A. Hofmann, T. Hourlier, G. Hulata, D. B. Jaffe, M. Lara, A. P. Lee, I. MacCallum, S. Mwaiko, M. Nikaido, H. Nishihara, C. Ozouf-Costaz, D. J. Penman, D. Przybylski, M. Rakotomanga, S. C. P. Renn, F. J. Ribeiro, M. Ron, W. Salzburger, L. Sanchez-Pulido, M. E. Santos, S. Searle, T. Sharpe, R. Swofford, F. J. Tan, L. Williams, S. Young, S. Yin, N. Okada, T. D. Kocher, E. A. Miska, E. S. Lander, B. Venkatesh, R. D. Fernald, A. Meyer, C. P. Ponting, J. T. Streelman, K. Lindblad-Toh, O. Seehausen & F. Di Palma, 2014. The genomic substrate for adaptive radiation in African cichlid fish. Nature 513: 375–381.
Castel, D., P. Mourikis, S. J. J. Bartels, A. B. Brinkman, S. Tajbakhsh & H. G. Stunnenberg, 2013. Dynamic binding of RBPJ is determined by notch signaling status. Genes and Development 27: 1059–1071.
Chan, Y. F., M. E. Marks, F. C. Jones, G. Villarreal, M. D. Shapiro, S. D. Brady, A. M. Southwick, D. M. Absher, J. Grimwood, J. Schmutz, R. M. Myers, D. Petrov, B. Jónsson, D. Schluter, M. A. Bell & D. M. Kingsley, 2010. Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a pitxl enhancer. Science 327: 302–305.
Chen, S., J. Tao, Y. Bae, M.-M. Jiang, T. Bertin, Y. Chen, T. Yang & B. Lee, 2013. Notch gain of function inhibits chondrocyte differentiation via Rbpj-dependent suppression of Sox9. Journal of Bone and Mineral Research 28: 649–659.
Chen, J., X. Wu, L. Yao, L. Yan, L. Zhang, J. Qiu, X. Liu, S. Jia & A. Meng, 2017. Impairment of cargo transportation caused by gbf1 mutation disrupts vascular integrity and causes hemorrhage in zebrafish embryos. Journal of Biological Chemistry 292: 2315–2327.
Christen, B., V. Robles, M. Raya, I. Paramonov & J. C. Izpisúa Belmonte, 2010. Regeneration and reprogramming compared. BMC Biology 8: 5.
Citterio, C., A. Vichi, G. Pacheco-Rodriguez, A. M. Aponte, J. Moss & M. Vaughan, 2008. Unfolded protein response and cell death after depletion of brefeldin A-inhibited guanine nucleotide-exchange protein GBF. Proceedings of the National Academy of Sciences 105: 2877–2882.
Colombo, M., E. T. Diepeveen, M. Muschick, M. E. Santos, A. Indermaur, N. Boileau, M. Barluenga & W. Salzburger, 2013. The ecological and genetic basis of convergent thick-lipped phenotypes in cichlid fishes. Molecular Ecology 22: 670–684.
Conith, M. R., A. J. Conith & R. C. Albertson, 2019. Evolution of a soft-tissue foraging adaptation in African cichlids: roles for novelty, convergence, and constraint. Evolution Society for the Study of Evolution 73: 2072–2084.
De Vos, I. J. H. M., E. Y. Tao, S. L. M. Ong, J. L. Goggi, T. Scerri, G. R. Wilson, C. G. M. Low, A. S. W. Wong, D. Grussu, A. P. A. Stegmann, M. Van Geel, R. Janssen, D. J. Amor, M. Bahlo, N. R. Dunn, T. J. Carney, P. J. Lockhart, B. J. Coull & M. A. M. Van Steensel, 2018. Functional analysis of a hypomorphic allele shows that MMP14 catalytic activity is the prime determinant of the Winchester syndrome phenotype. Human Molecular Genetics 27: 2775–2788.
de Vos, I. J. H. M., A. S. W. Wong, T. J. M. Welting, B. J. Coull & M. A. M. van Steensel, 2019. Multicentric osteolytic syndromes represent a phenotypic spectrum defined by defective collagen remodeling. American Journal of Medical Genetics, Part A 179(8): 1652–1664.
Elmer, K. R. & A. Meyer, 2011. Adaptation in the age of ecological genomics: insights from parallelism and convergence. Trends in Ecology and Evolution. 26(6): 298–306.
Flicek, P., I. Ahmed, M. R. Amode, D. Barrell, K. Beal, S. Brent, D. Carvalho-Silva, P. Clapham, G. Coates, S. Fairley, S. Fitzgerald, L. Gil, C. García-Girón, L. Gordon, T. Hourlier, S. Hunt, T. Juettemann, A. K. Kähäri, S. Keenan, M. Komorowska, E. Kulesha, I. Longden, T. Maurel, W. M. McLaren, M. Muffato, R. Nag, B. Overduin, M. Pignatelli, B. Pritchard, E. Pritchard, H. S. Riat, G. R. S. Ritchie, M. Ruffier, M. Schuster, D. Sheppard, D. Sobral, K. Taylor, A. Thormann, S. Trevanion, S. White, S. P. Wilder, B. L. Aken, E. Birney, F. Cunningham, I. Dunham, J. Harrow, J. Herrero, T. J. P. Hubbard, N. Johnson, R. Kinsella, A. Parker, G. Spudich, A. Yates, A. Zadissa & S. M. J. Searle, 2013. Ensembl 2013. Nucleic Acids Research 41: D48–D55.
Fukuda, M., K. Ohtani, S. J. Jang, T. Yoshizaki, K. I. Mori, W. Motomura, I. Yoshida, Y. Suzuki, Y. Kohgo & N. Wakamiya, 2011. Molecular cloning and functional analysis of scavenger receptor zebrafish CL-P1. Biochimica Et Biophysica Acta 1810: 1150–1159.
Gao, X., D. Han & W. Fan, 2016. Down-regulation of RBP-J mediated by microRNA-133a suppresses dendritic cells and functions as a potential tumor suppressor in osteosarcoma. Experimental Cell Research 349: 264–272.
Gilbert, M. C., C. S. Lerose, A. J. Conith & R. C. Albertson, 2022. Breaking constraints: the development and evolution of extreme fin morphology in the Bramidae. Evolution & Development 24: 109–124.
Govindan, J. & M. K. Iovine, 2014. Hapln1a is required for Connexin43-dependent growth and patterning in the regenerating fin skeleton. PLoS ONE 9: e88574.
Govindan, J. & M. K. Iovine, 2015. Dynamic remodeling of the extra cellular matrix during zebrafish fin regeneration. Gene Expression Patterns 19: 21–29.
Govindan, J., K. M. Tun & M. K. Iovine, 2016. Cx43-Dependent skeletal phenotypes are mediated by interactions between the Hapln1a-ECM and Sema3d during fin regeneration. PLOS ONE 11: e0148202.
Hagedorn, M., G. Siegfried, K. B. Hooks & A.-M. Khatib, 2016. Integration of zebrafish fin regeneration genes with expression data of human tumors in silico uncovers potential novel melanoma markers. Oncotarget 7: 71567–71579.
Hasegawa, T., C. J. Hall, P. S. Crosier, G. Abe, K. Kawakami, A. Kudo & A. Kawakami, 2017. Transient inflammatory response mediated by interleukin-1β is required for proper regeneration in zebrafish fin fold. Life 6: e22716.
Hassed, S. J., G. B. Wiley, S. Wang, J. Y. Lee, S. Li, W. Xu, Z. J. Zhao, J. J. Mulvihill, J. Robertson, J. Warner & P. M. Gaffney, 2012. RBPJ mutations identified in two families affected by Adams-Oliver syndrome. American Journal of Human Genetics 91: 391–395.
Hellemans, J., G. Mortier, A. De Paepe, F. Speleman & J. Vandesompele, 2007. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biology 8: R19.
Huang, J. & L. Chen, 2017. IL-1β inhibits osteogenesis of human bone marrow-derived mesenchymal stem cells by activating FoxD3/microRNA-496 to repress wnt signaling. Genesis. https://doi.org/10.1002/dvg.23040.
Iovine, M. K., 2007. Conserved mechanisms regulate outgrowth in zebrafish fins. Nature Chemical Biology 3: 613–618.
Iovine, M. K., E. P. Higgins, A. Hindes, B. Coblitz & S. L. Johnson, 2005. Mutations in connexin43 (GJA1) perturb bone growth in zebrafish fins. Developmental Biology 278: 208–219.
Irisarri, I., P. Singh, S. Koblmüller, J. Torres-Dowdall, F. Henning, P. Franchini, C. Fischer, A. R. Lemmon, E. M. Lemmon, G. G. Thallinger, C. Sturmbauer & A. Meyer, 2018. Phylogenomics uncovers early hybridization and adaptive loci shaping the radiation of Lake Tanganyika cichlid fishes. Nature Communications 9: 1–12.
Jamsheer, A., A. Sowińska-Seidler, M. Socha, A. Stembalska, C. Kiraly-Borri & A. Latos-Bieleńska, 2014. Three novel GJA1 missense substitutions resulting in oculo-dento-digital dysplasia (ODDD)—further extension of the mutational spectrum. Gene 539: 157–161.
Kang, J. H., T. Manousaki, P. Franchini, S. Kneitz, M. Schartl & A. Meyer, 2015. Transcriptomics of two evolutionary novelties: how to make a sperm-transfer organ out of an anal fin and a sexually selected “sword” out of a caudal fin. Ecology and Evolution 5: 848–864.
Kjaer, K. W., L. Hansen, H. Eiberg, P. Leicht, J. M. Opitz & N. Tommerup, 2004. Novel Connexin 43 (GJA1) mutation causes oculo-dento-digital dysplasia with curly hair. American Journal of Medical Genetics 127A: 152–157.
LeBert, D. C., J. M. Squirrell, J. Rindy, E. Broadbridge, Y. Lui, A. Zakrzewska, K. W. Eliceiri, A. H. Meijer & A. Huttenlocher, 2015. Matrix metalloproteinase 9 modulates collagen matrices and wound repair. Development (cambridge) 142: 2136–2146.
Lecaudey, L. A., C. Sturmbauer, P. Singh & E. P. Ahi, 2019. Molecular mechanisms underlying nuchal hump formation in dolphin cichlid, Cyrtocara moorii. Scientific Reports 9: 20296.
Lecaudey, L. A., P. Singh, C. Sturmbauer, A. Duenser, W. Gessl & E. P. Ahi, 2021. Transcriptomics unravels molecular players shaping dorsal lip hypertrophy in the vacuum cleaner cichlid Gnathochromis permaxillaris. BMC Genomics 22: 506.
Li, L., B. Yan, Y.-Q. Shi, W.-Q. Zhang & Z.-L. Wen, 2012. Live imaging reveals differing roles of macrophages and neutrophils during zebrafish tail fin regeneration. The Journal of Biological Chemistry 287: 25353–25360.
Lönnstedt, O. M., M. C. O. Ferrari & D. P. Chivers, 2014. Lionfish predators use flared fin displays to initiate cooperative hunting. Biology Letters 10: 20140281.
Lorda-Diez, C. I., J. A. Montero, J. Rodriguez-Leon, J. A. Garcia-Porrero & J. M. Hurle, 2013. Expression and functional study of extracellular BMP antagonists during the morphogenesis of the digits and their associated connective tissues. PLoS ONE 8: e60423.
Luehr, S., H. Hartmann & J. Söding, 2012. The XXmotif web server for eXhaustive, weight matriX-based motif discovery in nucleotide sequences. Nucleic Acids Research 40: W104–W109.
Ma, L., Y.-M. Yu, Y. Guo, R. P. Hart & M. Schachner, 2012. Cysteine- and glycine-rich protein 1a is involved in spinal cord regeneration in adult zebrafish. The European Journal of Neuroscience 35: 353–365.
Mahony, S. & P. V. Benos, 2007. STAMP: a web tool for exploring DNA-binding motif similarities. Nucleic Acids Research 35: W253–W258.
Manolea, F., A. Claude, J. Chun, J. Rosas & P. Melançon, 2008. Distinct functions for Arf guanine nucleotide exchange factors at the Golgi complex: GBF1 and BIGs are required for assembly and maintenance of the Golgi stack and trans-Golgi network, respectively. Molecular Biology of the Cell 19: 523–535.
Matys, V., E. Fricke, R. Geffers, E. Gössling, M. Haubrock, R. Hehl, K. Hornischer, D. Karas, A. E. Kel, O. V. Kel-Margoulis, D.-U. Kloos, S. Land, B. Lewicki-Potapov, H. Michael, R. Münch, I. Reuter, S. Rotert, H. Saxel, M. Scheer, S. Thiele & E. Wingender, 2003. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Research 31: 374–378.
Miller, C. H., S. M. Smith, M. Elguindy, T. Zhang, J. Z. Xiang, X. Hu, L. B. Ivashkiv & B. Zhao, 2016. RBP-J-regulated miR-182 promotes TNF-α-induced osteoclastogenesis. The Journal of Immunology 196: 4977–4986.
Monaghan, J. R., J. A. Walker, R. B. Page, S. Putta, C. K. Beachy & S. R. Voss, 2006. Early gene expression during natural spinal cord regeneration in the salamander Ambystoma mexicanum. Journal of Neurochemistry 101: 27–40.
Münch, J., A. González-Rajal & J. L. de la Pompa, 2013. Notch regulates blastema proliferation and prevents differentiation during adult zebrafish fin regeneration. Development (cambridge, England) 140: 1402–1411.
Nakatani, Y., M. Nishidate, M. Fujita, A. Kawakami & A. Kudo, 2007. Migration of mesenchymal cell fated to blastema is necessary for fish fin regeneration. Development, Growth & Differentiation 50: 71–83.
Nakayama, T., H. Saitsu, W. Endo, A. Kikuchi, M. Uematsu, K. Haginoya, N. Hino-fukuyo, T. Kobayashi, M. Iwasaki, T. Tominaga, S. Kure & N. Matsumoto, 2014. RBPJ is disrupted in a case of proximal 4p deletion syndrome with epilepsy. Brain and Development Elsevier 36: 532–536.
Nus, M., B. Martinez-Poveda, D. MacGrogan, R. Chevre, G. D’Amato, M. Sbroggio, C. Rodriguez, J. Martinez-Gonzalez, V. Andrés, A. Hidalgo & J. L. De La Pompa, 2016. Endothelial Jag1-RBPJ signalling promotes inflammatory leucocyte recruitment and atherosclerosis. Cardiovascular Research 112: 568–580.
Obayashi, T. & K. Kinoshita, 2011. COXPRESdb: a database to compare gene coexpression in seven model animals. Nucleic Acids Research 39: D1016–D1022.
Pfaffl, M. W., 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research 29: e45.
Pfaffl, M. W., A. Tichopad, C. Prgomet & T. P. Neuvians, 2004. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnology Letters 26: 509–515.
Pfefferli, C. & A. Jaźwińska, 2015. The art of fin regeneration in zebrafish. Regeneration 2: 72–83.
Rabinowitz, J. S., A. M. Robitaille, Y. Wang, C. A. Ray, R. Thummel, H. Gu, D. Djukovic, D. Raftery, J. D. Berndt & R. T. Moon, 2017. Transcriptomic, proteomic, and metabolomic landscape of positional memory in the caudal fin of zebrafish. Proceedings of the National Academy of Sciences of the United States of America 114: E717–E726.
Ramakers, C., J. M. Ruijter, R. H. L. Deprez & A. F. M. Moorman, 2003. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neuroscience Letters 339: 62–66.
Santos, M. E., L. Baldo, L. Gu, N. Boileau, Z. Musilova & W. Salzburger, 2016. Comparative transcriptomics of anal fin pigmentation patterns in cichlid fishes. BMC Genomics 17: 712.
Sehring, I. M. & G. Weidinger, 2020. Recent advancements in understanding fin regeneration in zebrafish. Wires Developmental Biology 9: e367.
Sims, K., D. M. Eble & M. K. Iovine, 2009. Connexin43 regulates joint location in zebrafish fins. Developmental Biology 327: 410–418.
Simunovic, F., D. Steiner, D. Pfeifer, G. B. Stark, G. Finkenzeller & F. Lampert, 2013. Increased extracellular matrix and proangiogenic factor transcription in endothelial cells after cocultivation with primary human osteoblasts. Journal of Cellular Biochemistry 114: 1584–1594.
Singh, P., C. Börger, H. More & C. Sturmbauer, 2017. The role of alternative splicing and differential gene expression in cichlid adaptive radiation. Genome Biology and Evolution 9: 2764–2781.
Singh, P., E. P. Ahi & C. Sturmbauer, 2021. Gene coexpression networks reveal molecular interactions underlying cichlid jaw modularity. BMC Ecology and Evolution 21: 1–17.
Sivasankaran, A., A. Srikanth, P. S. Kulshreshtha, D. Anuradha, J. S. Kadandale & C. R. Samuel, 2015. Split hand/foot malformation associated with 7q21.3 microdeletion: a case report. Molecular Syndromology 6: 287–296.
Sylva, M., V. S. W. Li, A. A. A. Buffing, J. H. van Es, M. van den Born, S. van der Velden, Q. Gunst, J. H. Koolstra, A. F. M. Moorman, H. Clevers & M. J. B. van den Hoff, 2011. The BMP Antagonist follistatin-like 1 is required for skeletal and lung organogenesis. PLoS ONE 6: e22616.
Sylva, M., A. F. M. Moorman & M. J. B. van den Hoff, 2013. Follistatin-like 1 in vertebrate development. Birth Defects Research Part c: Embryo Today: Reviews 99: 61–69.
Tao, J., S. Chen, T. Yang, B. Dawson, E. Munivez, T. Bertin & B. Lee, 2010. Osteosclerosis owing to Notch gain of function is solely Rbpj-dependent. Journal of Bone and Mineral Research 25: 2175–2183.
Ton, Q. V. & K. M. Iovine, 2012. Semaphorin3d mediates Cx43-dependent phenotypes during fin regeneration. Developmental Biology 366: 195–203.
Ton, Q. V. & M. K. Iovine, 2013. Identification of an evx1-dependent joint-formation pathway during FIN regeneration. PLoS ONE Public Library of Science 8: e81240.
Vandesompele, J., K. De Preter, F. Pattyn, B. Poppe, N. Van Roy, A. De Paepe & F. Speleman, 2002. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biology 3: 4–6.
Wainwright, P. C., D. R. Bellwood & M. W. Westneat, 2002. Ecomorphology of locomotion in labrid fishes. Environmental Biology of Fishes Springer 65: 47–62.
Walker, M. & C. Kimmel, 2007. A two-color acid-free cartilage and bone stain for zebrafish larvae. Biotechnic & Histochemistry 82: 23–28.
Wehner, D. & G. Weidinger, 2015. Signaling networks organizing regenerative growth of the zebrafish fin. Trends in Genetics 31(6): 336–343.
Wilkinson, J. M., R. K. Davidson, T. E. Swingler, E. R. Jones, A. N. Corps, P. Johnston, G. P. Riley, A. J. Chojnowski & I. M. Clark, 2012. MMP-14 and MMP-2 are key metalloproteases in Dupuytren’s disease fibroblast-mediated contraction. Biochimica Et Biophysica Acta – Molecular Basis of Disease Elsevier 1822: 897–905.
Yoshinari, N., T. Ishida, A. Kudo & A. Kawakami, 2009. Gene expression and functional analysis of zebrafish larval fin fold regeneration. Developmental Biology 325: 71–81.
Zhao, B., S. N. Grimes, S. Li, X. Hu & L. B. Ivashkiv, 2012. TNF-induced osteoclastogenesis and inflammatory bone resorption are inhibited by transcription factor RBP-J. Journal of Experimental Medicine 209: 319–334.
Acknowledgements
The authors thank Wolfgang Gessl (www.pisces.at) for his responsible management of our fish. We also thank Holger Zimmermann and Stephan Koblmüller for sharing their precious knowledge on cichlid fishes of Lake Tanganyika, and Martin Grube and his lab for technical assistance and access to their real-time PCR System. The authors acknowledge the financial support by the University of Graz.
Funding
Open Access funding provided by University of Helsinki including Helsinki University Central Hospital. The study was funded by the University of Graz. Additionally, K.M.S. acknowledges funding by the Austrian Science Fund (FWF; Grant P28505-B25) and the Austrian Science Fund (FWF; Grant I3535). The funding bodies had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
EPA and KMS designed the study, analyzed the data, and wrote the manuscript. EPA conducted the fin dissections, RNA extraction, and qPCR laboratory experiments. EPA and KMS prepared figures and tables. FR conducted fin skeletal staining, photography, and measurement of fin ray segments as well as the schematic drawing of fish (Fig. 1A and B).
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
All experimental protocols were approved by the Federal Ministry of Science, Research and Economy of Austria (BMWFW) under permit BMWFW-66.007/0004-WF/V/3b/2016.
Consent to participate
All methods were carried out in accordance with relevant guidelines and regulations of the Austrian animal welfare law. All methods are reported in accordance with ARRIVE guidelines (https://arriveguidelines.org) for the reporting of animal experiments.
Additional information
Handling editor: Sidinei M. Thomaz
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Guest editors: S. Koblmüller, R. C. Albertson, M. J. Genner, K. M. Sefc & T. Takahashi / Advances in Cichlid Research V: Behavior, Ecology and Evolutionary Biology
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visithttp://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ahi, E.P., Richter, F. & Sefc, K.M. Gene expression patterns associated with caudal fin shape in the cichlid Lamprologus tigripictilis. Hydrobiologia 850, 2257–2273 (2023). https://doi.org/10.1007/s10750-022-05068-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-022-05068-4