
Abstract
Background
The oral and pharyngeal jaw of cichlid fishes are a classic example of evolutionary modularity as their functional decoupling boosted trophic diversification and contributed to the success of cichlid adaptive radiations. Most studies until now have focused on the functional, morphological, or genetic aspects of cichlid jaw modularity. Here we extend this concept to include transcriptional modularity by sequencing whole transcriptomes of the two jaws and comparing their gene coexpression networks.
Results
We show that transcriptional decoupling of gene expression underlies the functional decoupling of cichlid oral and pharyngeal jaw apparatus and the two units are evolving independently in recently diverged cichlid species from Lake Tanganyika. Oral and pharyngeal jaw coexpression networks reflect the common origin of the jaw regulatory program as there is high preservation of gene coexpression modules between the two sets of jaws. However, there is substantial rewiring of genetic architecture within those modules. We define a global jaw coexpression network and highlight jaw-specific and species-specific modules within it. Furthermore, we annotate a comprehensive in silico gene regulatory network linking the Wnt and AHR signalling pathways to jaw morphogenesis and response to environmental cues, respectively. Components of these pathways are significantly differentially expressed between the oral and pharyngeal jaw apparatus.
Conclusion
This study describes the concerted expression of many genes in cichlid oral and pharyngeal jaw apparatus at the onset of the independent life of cichlid fishes. Our findings suggest that – on the basis of an ancestral gill arch network—transcriptional rewiring may have driven the modular evolution of the oral and pharyngeal jaws, highlighting the evolutionary significance of gene network reuse. The gene coexpression and in silico regulatory networks presented here are intended as resource for future studies on the genetics of vertebrate jaw morphogenesis and trophic adaptation.
Similar content being viewed by others


Background
Evolutionary modularity, or the degree to which traits form higher-order units that evolve independently, has emerged as a central concept in evolutionary biology, especially in the last two decades [1,2,3,4,5,6,7,8]. It is now viewed as a major mechanism governing evolvability (i.e. potential for adaptive change). Units that exhibit strong within-module integration but relatively weak between-module integration are considered modular and there can be various degrees of modularity governing complex phenotypes [6]. Heterogeneity in integration enables adaptive flexibility, with low degrees of integration being associated with high phenotypic change [9]. Modularity is considered a major driver of phenotypic evolution [10, 11] and operates at many biological levels such as morphology, function, development, and genetic architecture (reviewed in [6]). Thus, modularity can also be observed at the molecular level, for example in gene regulatory networks [12]. Transcriptional modularity can bring about complex developmental changes and give biological systems greater ability to respond to changes with minimal interference with genomic complexity [13,14,15].
The vertebrate craniofacial anatomy is amongst the most complex musculoskeletal systems with highly modular compartmentalisation, representing a key innovation of the Craniota. Cichlid fishes are particularly diverse in terms of their craniofacial anatomy and are renowned for having two sets of jaws (oral and pharyngeal) that are a hallmark example of modularity [16]. The functional decoupling of these jaws is seen as a key innovation that permitted the oral jaw apparatus (OJA) to be solely dedicated to prey capture and the pharyngeal jaw apparatus (PJA) to prey processing, allowing for independent evolution. Their adaptive radiation in several East African lakes was hypothesised to be connected to their great efficiency to adapt to novel food sources [17]. Another factor facilitating adaptive radiation is phenotypic plasticity of the jaw apparatus that enabled rapid response to environmental changes [18]. Remarkably, similar craniofacial morphologies evolved in parallel across the radiations of the three Great East African Lakes (Lake Tanganyika, Malawi and Victoria) in response to similar selection regimes. The modularity of cichlid jaws has been an active area of research [19,20,21,22] as it can shed light not only on how cichlid jaws adapted to different diet regimes, but also to understand the role of modularity in facilitating diversification, even repeatedly [23]. The theory of morphological integration postulates that functionally modular traits will be inherited independently, and thus be genetically modular too [1]. So far, studies have provided evidence of cichlids jaws being not only functionally and mechanistically modular [19, 24] but also modular at the genetic level [16, 21, 25]. There is also mounting evidence of these two major units displaying high within-module integration [16, 26]. However, modularity at the transcriptional level in global gene expression has not been yet been investigated in detail.
Gene regulatory networks (GRNs) govern development in multicellular organisms. Modules (i.e. sub-circuits) within GRNs allow for parts of the network to evolve, without interfering with the rest of the network. Within each module, gene associations represent regulatory links that are mainly determined by presence of common transcription factor binding sites in their regulatory sequences [27]. Alterations of these links in the ancestral source structure, especially via cis-regulatory changes, can bring about diverse developmental and evolutionary change in derived structures [27]. The reuse and re-wiring of GRNs plays a major role in morphological evolution [28] and there are impressive examples in insects and crustaceans of GRN rewiring with varying levels of sub-circuit conservation (reviewed in [27]). In cichlids, it was previously shown that an ancient gene network was redeployed to give rise to oral and pharyngeal teeth [29]. However, it is not known if this also was the case for the two jaws.
During development and morphogenesis of the jaw musculoskeletal apparatus, a high number of highly interconnected molecular cascades (e.g. Wnt, BMP, Hedgehog, Notch, retinoic acid, growth factors and calcium dependent pathways) mediate their signals to regulate overlapping GRNs (reviewed in [30]). In cichlids, as one of the most diverse groups of vertebrates in jaw morphology, elucidating the related GRNs is an important step to understand the molecular basis of their functional modularity, phenotypic plasticity, and the evolution of parallel eco-morphologies. To date, the GRNs underlying cichlid jaw development are largely unknown, with only one attempt thus far using a set of candidate genes to described lower pharyngeal jaw plasticity at the GRN level [31].
By using data from whole transcriptome sequencing, it is possible to build gene coexpression networks (GCNs) and study the degrees of transcriptional connectivity at a broad scale [32]. GCNs are biologically interesting as they reveal clusters of genes that are expressed in symphony and thus may be under the control of the same transcriptional regulatory program [33]. Functionally annotating GCNs can shed light onto GRN architecture. Nodes in GCNs are represented by genes and highly interconnected (coexpressed) nodes are called modules. Genes that are coexpressed are considered to be co-regulated and be constituents of the same pathway or have the same upstream regulator(s), and thus be functionally related [34]. The direction of regulation cannot be deduced from GCNs however. This approach has been applied extensively in model organisms and recently successfully been applied to study key drivers of speciation and ecological phenotypes [35,36,37,38,39] and seems promising to identify the GCNs driving the functional modularity of cichlid oral and pharyngeal jaws.
Here we set out to delineate the gene coexpression networks of cichlid oral versus pharyngeal jaws and yield insight into the regulatory mechanisms underlying their evolutionary modularity. To address this, we sequenced whole transcriptomes of OJA and lower PJA (LPJA) from three cichlid species (Gnathochromis pfefferi, Ctenochromis horei, and Limnotilapia dardennii) with different prey spectres, prey capture strategies and distinct ecomorphologies. These species comprise a monophyletic and recently diverged clade (~ 3.56 MYA) [P. Singh, unpublished observations] from the tribe Tropheini from Lake Tanganyika, that are named after their impressive trophic diversity. G. pfefferi has a longer snout that is specialised for picking invertebrates such as shrimp/insects from the soft lake bottom [40]. C. horei is the most specialised fry-eater in the Tropheini with a terminal mouth and L. dardennii is an omnivore [57]. We used weighted gene coexpression network analysis (WGCNA) to identify major coexpression modules that may play a role in shaping their evolutionary and functional disparities with regards to skeletogenesis and environmental cues (i.e. plasticity). The monophyly and relatively recent coancestry of these species is advantageous as re-wiring of ancestral gene coexpression networks potentially driving oral and pharyngeal jaw adaptations will be easier to distinguish from background noise. Assuming some degree of conservation, we transferred the knowledge of functionally validated annotations from literature on model organisms to in silico annotated GRNs in cichlid jaws. We specifically focused on an ecologically informative developmental stage (stage 26, [41]) where post-embryonic development of both jaws is complete and the larvae are ready to feed independently and interact with the environment. We have previously shown that at this stage the jaws of species appear to be morphologically distinct [42]. Our new results provide insight into the transcriptional interactions that shaped oral and pharyngeal modularity in Lake Tanganyika cichlids.
Results
Gene expression patterns of oral and pharyngeal jaws
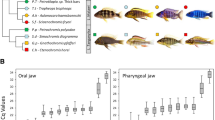
Approximately 11 million paired-end reads per sample (125 bp in length) were obtained from Illumina Hi-Sequencing Technology for four biological replicates per jaw per Lake Tanganyika species and one biological replicate per jaw for the Lake Malawi species (Table 1). After filtering, approximately 63% of the reads per sample mapped to the reference genome as proper pairs (Additional file 2: File S1). To identify the relationship between the transcriptomes of the jaws and species in this study (Fig. 1), we extracted the expression of 16,669 genes from each sample and conducted hierarchical clustering analysis based on Euclidean distance. A clear pattern emerged in the clustering dendrogram (Fig. 2). Gene expression of the samples showed jaw-specific clustering in both the PCA (Fig. 2a) and dendrogram (Fig. 2b), irrespective of trophic niche or lake of origin, indicating that the OJA and LPJA have highly conserved gene expression signatures. The gene expression of the LPJA suggests higher variance on both PC1 and PC2 compared to the OJA, even within the same species. Within the jaw-specific clusters in the dendrogram, the samples were clustered according to species. However, the species clustering of OJA samples was different from LPJA samples and neither reflected the phylogenetic relationship of the species shown in (Fig. 1 phylogeny based on Irisarri*, Singh* et al. 2018). In the OJA samples, C. horei and L. dardennii were closer to each other than the G. pfefferi but in the LPJA samples, G. pfefferi and L. dardennii were closer to each other than the C. horei. Notably, the Lake Malawi species, Petrotilapia sp. ‘yellow chin Chewere’, was embedded within the Lake Tanganyika samples, but held different positions within the OJA compared to LPJA. Petrotilapia sp. ‘yellow chin Chewere’ is a sister lineage to the Lake Tanganyika Tropheini (Fig. 1) and since we only one had one biological replicate per jaw for it, it was not included in the subsequent co-expression network and differential gene expression analyses.

adapted from Irisarri*, Singh* et al. 2018
Study design. The lake, trophic niche, phylogenetic relationships and number of cichlid species sequenced in this study. Fish photographs: W. Gessl (University of Graz). Phylogeny

a Principal component analysis (PCA) and b hierarchical clustering dendrogram of normalised expression counts of 16,669 O. niloticus annotated genes in 26 RNA-seq samples from the oral jaw apparatus (OJA) and lower pharyngeal jaw apparatus (LPJA). Ctenochromis horei (Ch), Gnathochromis pfefferi (Gp), Limnotilapia dardennii (Ld) and Petrotilapia sp. ‘yellow chin Chewere’ (Py)
Differential gene expression between oral and pharyngeal jaws
There were 9475 genes (FDR adjusted p-value < 0.05) differentially expressed genes between OJA and PJA (Additional file 2: File S1). Of these 4659 were upregulated in the OJA and 4816 were upregulated in the PJA (Additional file 2: File S1). The gene enrichment analysis for the differentially expressed genes resulted in 353 significant GOs for biological processes (FDR cut-off < 0.01, Additional file 3: File S2a). In particular, GOs related to muscle and skeletal system development and morphogenesis, as well as biosynthesis of organic compounds and response to stimulus were among the most abundantly enriched biological processes (Additional file 3: File S2a). To narrow down potential gene networks determining oral versus pharyngeal jaw apparatus, we only focused on GOs related to muscle and skeletal system development and morphogenesis. In addition, we also focused on GOs related to response to organic compounds (e.g., exogenous chemicals and nutritional compounds) which could potentially provide molecular links between mechanisms involved in jaw formation and activity of environmentally responsive signals exactly when feeding starts. We retrieved the genes in GOs related to each of these biological processes and grouped them together for next analysis steps (Additional file 3: File S2a).
Of the 225 genes enriched for skeletal GO terms, of note were ones from the Wnt, BMP, hox pathways and chondrocyte genes (col6a2, col6a1, colq, col9a2, col1a2, col11a1b, col8a1b, col28a2a) and dlx4b, dlx1a, sox10 genes (Additional file 3: File S2a). We also found predicted transcription factors of Wnt pathway to be enriched in the differentially expressed genes advocating the pivotal role of Wnt in OJA versus LPJA skeletal specification. Myomesin (myo1a, myo2a, myo18aa, myorg) and myosin (mybpha, mybphb, mybpc3) genes, the myogenic regulatory factor myf6; tbx20, tbx1; and troponin genes (tnnc1a, tnnc1b, tnnc2, tnni2a, tnnt2c, tnni3k) were notable genes out of the 105 genes enriched in the GOs related to muscle formation processes. The transcription factor (TF) predictions also found an enrichment for the TFs of these genes/pathways in the differentially expressed genes (Additional file 3:File S2a). The greatest number of genes (434) were associated with biogenesis of organic compounds and response to stimulus GO biological processes (Additional file 3: File S2a). Several components of the Ahr pathway, but not the pathway itself, were enriched. The TF prediction for these genes found 326 TF genes, among which 235 TFs displayed differential expression between the OJA and LPJA (Additional file 2: File S1, Additional file 3:File S2a). Many of these differentially expressed TFs also appeared to be involved in musculoskeletal formation.
Conditional gene coexpression networks
To explore the preservation of GCN modules between cichlid OJA and LPJA transcriptomes, we first defined the GCN in one condition (i.e. OJA) and then verified the preservation of its modules in the second condition (i.e. LPJA), and vice versa. We analysed data of the OJA and LPJA samples separately and assessed module preservation between them using a composite Zsummary statistic. We identified seven OJA modules: yellow, brown2, lightyellow, darkslateblue, pink, orangered4 and lavenderblus3 (Fig. 3). Six of these seven modules showed strong preservation (i.e. many genes are shared between the modules of the two jaws) in the LPJA-GCN and one module showed moderate preservation (Fig. 3a). However, visualisation of the OJA modules in the LPJA-GCN showed that there was extensive re-wiring of genes of the OJA modules in the LPJ GCN (i.e. hierarchical clustering suggests the genes to be differently clustered between the two jaws) (Fig. 3b). A similar pattern was independently observed for the eight modules identified in LPJA-GCN and their preservation in the OJA-GCN (Additional file 1: Figure S4). Four LPJA modules were strongly preserved in OJA-GCN, two were moderately preserved, and two modules were not preserved. Overall, more OJA modules were preserved in the LPJA network than LPJA modules in the OJA network.

Conditional coexpression analysis: Preservation of modules in the GCNs underlying oral (OJA) and lower pharyngeal jaw apparatus (LPJA). a The colours represent identified OJA coexpression modules. Preservation of genes found in OJA modules in the LPJA coexpression network was calculated by a Zsummary statistic (y-axis) based on a permutation test that takes into account the connectivity and density of genes in a module. Zsummary < 2 represents lack of preservation (dotted blue line). Zsummary between 2 and 10 implies moderate preservation. Zsummary > 10 supports strong preservation of module. b Visual representation of module preservation. The upper panels of the dendrogram represents average linkage clustering tree based on topological overlap distance in gene expression profile. Each ‘leaf’ of the dendrogram corresponds to one gene. The lower panels of the dendrograms represent colours that correspond to OJA modules Top: OJA modules in the OJA GCN. Bottom: OJA modules in the LPJA GCN
Global jaw coexpression network
To construct a global GCN of cichlid jaw apparatus we conducted the analysis using both OJA and LPJA expression data as input for WGCNA. The signed hybrid network identified 19 gene coexpression modules, and genes that did not fit into any module were placed in the “grey” module (Fig. 4a, Additional file 1: Figure S5). The 19 modules ranged in size from 35 to 1992 genes). We correlated module eigengenes with jaw apparatus type (oral or lower pharyngeal) and species to identify jaw-specific and species-specific modules (Fig. 4a). The strength of correlation was measured by the gene significance and connectivity of genes in each module (Additional file 1: Figures S6, S7). Three modules were highly correlated (|r|> = 0.8) to jaw type were: Blue (r = -0.99, p-value = 9e-21), Cyan (r = 0.85, p-value = 5e-08) and Plum2 (r = 0.95, p-value = 8e-14). The two highest correlated C. horei specific modules were black and purple (|r|> = 0.8, p-value < 1e-6). The two highest correlated L. dardennii specific modules were floralwhite and darkgreen (|r|> = 0.64, p-value < 1e-3). The two highest correlated G. pfefferi specific modules were black and purple (|r|> = 0.8, p-value < 1e-5). Thus, the black and purple module was shared by the two carnivores G. pfefferi and C. horei. The black module was positively correlated with G. pfefferi and negatively correlated with C. horei, while the purple module was positively correlated with C. horei and negatively correlated with G. pfefferi. These two modules were not correlated with L. dardennii or jaw type.

Global coexpression analysis: Coexpression sub-modules with jaw or species specific expression patterns. a The colours represent the coexpression modules identified using data from bots jaws. Correlation (r) score and p-value significance of module to jaw type or species. Red denoted strong postive correlation and blue denotes strong negative correlation. Sub-modules of coexpressed genes within b Blue module c Plum2 and d Cyan modules. The red and blue connecting lines between the genes in each sub-modules indicate significant positive and negative expression correlations, respectively (P < 0.05). The predicted TF(s) at upstream of each each sub-module are depicted in square shape gene symbol with their related functions. The red, blue and white shadings for genes respectively indicate increased expression in oral and pharyngeal jaws, and no expression difference between the jaws
GO annotation of jaw-specific modules
Three coexpression gene modules displaying significant expression association in a jaw-specific manner were selected for further investigations of their potential regulatory networks (Fig. 4a). The Cyan and Plum2 modules showed positive correlation and Blue module had negative correlation. The largest module, Blue, consisted of 2024 genes and thus, the analysis of GO enrichment yielded a long list of significant biological processes (GOs with FDR < 0.001 listed in Additional file 4: File S2b). Among the enriched GOs, biological processes involving skeletogenesis, Wnt signalling pathway and biogenesis of organic compounds were selected for the next analysis step. While almost all of the genes enriched in GOs related to skeletogenesis/Wnt pathway had higher expression in OJA than LPJA, none of their predicted upstream TFs showed jaw-specific differential expression (Fig. 4b, Additional file 4: File S2b). Moreover, an exception within the enriched genes was observed for a paralogue of prdm1 gene, prdm1b, which appeared to have higher expression in LPJA (opposite to the other genes), whereas its paralogue, prdm1a, displayed higher OJA expression (Fig. 4b). This might suggest the possibility of sub-functionalisation of prdm1 paralogues in a jaw-specific and modular manner during cichlid development. We previously found prdm1a to exhibit modular gene expression in jaws of these carnivorous Tropheini cichlids as well as other haplochromines cichlids using qPCR [55]
Furthermore, in the Blue module, a long list TFs was predicted upstream of the genes with enriched GOs related to biogenesis of organic compounds. We found foxq1 and sox9, which were among the top 10 predicted TFs, to be differentially expressed between the jaws. Interestingly, the two paralogues of foxq1 gene (foxq1a and foxq1b) showed contrasting jaw-specific expression, i.e. foxq1a had higher expression in LPJA and foxq1b displayed higher expression in OJA, suggesting their modular sub-functionalisation in developing cichlid jaws (Fig. 4b). Most members of the two sub-modules regulated by foxq1 and sox9 were highly expressed in OJA with few exceptions such as tead3a, nr2f6a and prdm1b. Interestingly, the prdm1 paralogues were found to be enriched in all the investigated sub-circuits of Blue module indicating their potential role in linking the effects of organic compounds biogenesis to skeletogenesis in jaws.
The Plum2 module contained 1877 genes. Many of the top 20 enriched GOs for Plum2 module were related to biological processes involving nucleoside metabolism (GOs with FDR < 0.001 listed in Additional file 4: File 2b), therefore we selected these GOs for TF prediction steps. We identified only 2 TFs, sf1 and err1/esrra as upstream regulators of genes enriched in these GOs, and err1 displayed high expression in LPJA (Fig. 4c). Similarly, most genes in the err1 related sub-module appeared to have high expression in LPJA (Fig. 4c). We performed the same analysis steps for the Cyan module comprising 1277 genes and these resulted in GOs related to muscle development and myofibril assembly, and mef2 as top ranked predicted TF (Fig. 4d). Furthermore, we found mef2a and most of its downstream genes to be highly expressed in LPJA indicating more active muscle cell differentiation in the surrounding tissues of developing pharyngeal jaw compared to oral jaw (Fig. 4d).
GO annotation of species-specific modules
The most important species-specific modules in our analysis were the Black and Purple modules containing 2117 and 1358 genes respectively (Fig. 4a) that showed the opposite correlation patterns in the piscivore C. horei and the invertivore G. pfefferi. The black module, which was positively correlated with G. pfefferi has genes annotated for several important pathways (Additional file 1: Figure S8), the most interesting of which was hypertrophy (2.5 enrichment ratio), suggesting an increase in muscle mass in G. pfefferi. The other interesting pathways were the androgen receptor pathway (1.39 enrichment ratio) and the ERK/MAPK cascade (1.25 enrichment ratio). The purple module, which was negatively correlated with G. pfefferi, but positively correlated with C. horei was enriched for the polyol pathway (6.0 enrichment ratio), selenium metabolism (1.9 enrichment ratio), estrogen signalling (enrichment ratio 1.6) and most notably, the delta-notch signalling pathway (1.5 enrichment ratio, Additional file 1: Figure S8).
Discussion
There is keen interest to identify the genes controlling the craniofacial anatomy of vertebrates in evolutionary biology, particularly those underlying divergent and convergent trophic adaptations. This is especially true for cichlid fishes where the decoupling of the oral and pharyngeal jaw are considered a key innovation driving trophic diversification and adaptive radiation [17]. Jaw morphogenesis, like many ecologically and evolutionarily relevant phenotypes, is a quantitative trait governed by myriad genes. As genes do not function in isolation, understanding gene to gene relationships and their hierarchy is critical to better understand the regulatory pathways underlying adaptive traits. This can be accomplished by linking evolutionary theory with systems biology [56, 57]. GCN analysis is a systems biology approach that transcends single gene level analysis and provides more information about gene–gene relationships underlying complex traits [32]. Here we demonstrate its application in identifying the network of genes underlying a key cichlid ecological adaptation: the functional decoupling of oral and pharyngeal jaw apparatuses. We show that transcriptional modularity of the OJA and LPJA recapitulates their form and functional modularity, exhibiting independently evolving gene expression patterns in the Tropheini cichlids from Lake Tanganyika. Our results suggest that rewiring of GCNs may have facilitated the functional and evolutionary modularity of cichlid jaws. Furthermore, we identify several jaw- or species-specific coexpressed gene modules and incorporate functional annotations to define in silico GRNs governing cichlid jaw morphogenesis.
Cichlid OJA and LPJA are transcriptionally decoupled and evolving independently
Modularity is a central concept in evolutionary biology as it can enhance ‘evolvability’. The hierarchical clustering of transcriptomes sequenced in this study revealed that gene expression signatures of the OJA and LPJA are conserved across different trophic niches as well as across Lake Tanganyika and Lake Malawi. This suggests that the two jaws have distinct gene regulatory environments, and that this dissociation underlies the key innovation that is their functional decoupling. Based on the divergence time of the cichlid species in this study, this conservation spans at an evolutionary distance of ~ 5.0 MYA (divergence time of Lake Malawi cichlids and the Lake Tanganyika Tropheini) [P. Singh, unpublished data; 58] but this regulatory homology most likely arose as deep as the modification of the gill arches in the craniota. Our findings are consistent with previous studies in mammals showing that tissue-specific clustering of gene expression dominates within major clades, especially in tissues that demarcate a clade [11, 59]. Additionally, in mammals it was also observed that gene expression clustering is consistent with the phylogeny until about ~ 5 – 7 MYA [11]. However, neither the species clustering in the OJA, nor in the LPJA, followed the known phylogeny of these species [58]. We speculate that this lack of consolidation of the gene sequence with gene expression is the result of the recent divergence of these species and jaw adaptations. Analysing transcriptomes from a range of recent and older diverged cichlid species would show if such a consolidation exists along the phylogeny.
The species clustering of LPJA gene expression differed from the OJA suggesting that gene expression, and thus gene regulation of these two units is evolving independently, as would be the case under the model of modularity. This further reinforces the idea that OJA and LPJA are transcriptionally decoupled. As the study species exhibit two highly specialised carnivorous and one omnivorous (i.e. generalist) diet regimes, the differential clustering of the jaws may reflect the unique post-divergence trajectory of each species towards trophic specialisation. In the OJA gene expression clustering, C. horei and L. dardennii were closer to each other than to G. pfefferi, which supports the distinct mouth form and prey catching strategy of the invertebrate picker G. pfefferi [60]. Furthermore, in both the OJA and LPJA samples, the Lake Malawi algae grazer P. sp. ‘yellow chin Chewere’ clustered closely with the Lake Tanganyika omnivore, L. dardennii, and not as sister group to all three Lake Tanganyika endemic Tropheini species, like in the species phylogeny. This is interesting not only because both species largely feed on algae [40] but because Lake Malawi cichlids comprise a related but independent adaptive radiation that is thought be seeded by the same ancestral lineage that seeded the Tropheini in Lake Tanganyika. Instead, the clustering hints at perhaps repeated evolution of gene expression in convergent trophic morphologies.
Conserved but rewired coexpression networks underlie oral and pharyngeal jaw origin
The jaws of gnathostomes (jawed vertebrates) are ancient structures that evolved from the pharyngeal arches [61, 62]. The number of arches is variable across vertebrates [63] and cichlid fishes belong to the teleost group that have seven pharyngeal arches [64]. The OJ is derived from the first pharyngeal arch, the hyomandibular complex is derived from the second pharyngeal arch, and the most posterior seventh arch forms the PJ [64, 65]. Given that the origin of elements in both the OJ and PJ lies in serially homologous pharyngeal arches, we hypothesised that ancient regulatory networks were modified to yield two novel morphologies, as already demonstrated for the dentition of the two jaws [29, 66,67,68]. Reorganisation of existing GRNs may have permitted rapid phenotypic evolution in the Tropheini. In particular, modification of cis-regulatory modules that govern gene expression within a GRN are known to have significant effect on development and phenotype [27, 69,70,71]. For example, it was shown that rewiring of GRNs played a role in the body segment modification in insects [72] and the acquisition of an embryonic exoskeleton in sea urchins [73]. Our findings show that the same suite of coexpressed gene modules is deployed in both the OJA and LPJA, suggesting that the gene composition of these modules may have been derived from ancestral pharyngeal arch GRN in jawless vertebrates. Evolutionary rewiring of these ancient GRNs may have first given rise to the OJA from the first pharyngeal arch and subsequently to the pharyngeal jaw from the seventh pharyngeal arch. Furthermore, we show that rewiring manifests not only in the architecture of gene connectivity, but also in the form of differential gene expression and sub-functionalisation of gene duplicates (details discussed below). As rewiring of an ancient gene network has already been suggested for the evolution of teeth on the oral and pharyngeal jaws [29], given that teeth and jaws co-evolved, it is not surprising that similar mechanisms may have driven the evolution of the two jaw structures.
Jaw-specific co-expression modules
Many members of the Wnt pathway in the jaw-specific Blue module were associated with muscle/skeletal morphogenesis. Cross-talks between Wnt pathway and a dozen other signalling pathways have been described in detail during teleost fish jaw development [30]. As we were not able to identify differentially expressed TFs at upstream of this network, it is likely that one of these interacting pathways influences the transcription of Wnt signal components. In addition, the higher OJA expression of genes like ap2 (tfap2) and wnt9b can be assertive evidence for modular specificity of the Blue module, since these genes are determinants of anterior viscerocranium and upper jaw, respectively [74, 75]. Another gene in the Blue module, hapln1a, has been shown to be repressed by activated AHR pathway in developing zebrafish jaw [76] and AHR (a major pathway mediating metabolism of cyclic compounds) is among the pathways showing direct cross-talk with Wnt pathway in different zebrafish tissues [77,78,79]. Additionally, GOs related to organic compounds biosynthesis in the Blue module were highly expressed in oral jaw, and interestingly, paralogues of predicted TFs, sox9b and foxq1b, are known to be direct downstream targets of activated AHR pathway during zebrafish jaw development [76, 80]. In each sub-module of the Blue module, there were genes with potentially important implications for the divergent morphogenesis of oral versus pharyngeal jaw. For instance, the higher expression six1b and pax3a/b in sox9-related sub-module could indicate the presence of fast-twitch muscle fibres and anterior viscerocranial specific muscle in the surrounding oral jaw tissues [81, 82]. However, the higher expression of prdm1a in oral jaw muscles might lead to opposite outcome of decreased fast-twitch muscle fibres [83]. In zebrafish, prdm1a is also required for posterior pharyngeal skeletal formation [84], on the contrary, we found increased oral jaw expression of prdm1a whereas its paralogue, prdm1b, was highly expressed in pharyngeal jaw. This could potentially be an indication for sub-functionalisation of the paralogues as well as potential changes in modular function of prdm1a along the anterior–posterior axis in viscerocranial skeleton of cichlids compared to zebrafish. Using qPCR, it was shown that cichlids from all major East African radiations express prdm1a modularly and differentially in oral and pharyngeal jaws [55]. It should be noted that prdm1a/b were enriched in all the three Blue sub-modules (Fig. 4b). A similar event was observed for foxq1 paralogues, i.e. foxq1a was higher in oral jaw but foxq1b showed increased expression in pharyngeal jaw, and again in zebrafish an opposite expression pattern is reported for foxq1b along the viscerocranial anterior–posterior axis [80].
The other examples of interesting genes were lhx6 and nr2f1 in sox9-related Blue sub-module with pivotal role in zebrafish dentition and upper jaw specification, respectively [85, 86] (Fig. 4b). Also, a highly-conserved gene with crucial role in coordination oral jaw development and elongation, satb2, was enriched in foxq1-related sub-module (Fig. 4b) [87, 88]. Satb2 delineates a developmental module within oral jaw by controlling the formation of distal elements of both the upper and lower jaws. The later evidence suggests satb2 as a prime candidate for modular evolvability of vertebrate oral jaw by generating variations in length of the distal elements [89]. The above findings provide first insights into potential transcriptional regulation of these important genes by sox9 and foxq1a/b during oral jaw development in fish.
Genes in the Cyan and Plum2 modules had opposite gene expression direction to genes in the Blue module (i.e. increased expression in LPJA apparatus) (Fig. 4c, d). The TFs, err1/esrra and mef2a, with increased LPJA expression were predicted at upstream of these modules. Esrra is a nuclear receptor regulating estrogen mediated signalling pathway in musculoskeletal system [90, 91]. Esrra is required for the formation of mainly posterior viscerocranial skeleton [91], however, its own expression is not induced by activated estrogen pathway during zebrafish viscerocranial development [92]. Markedly, esrra is shown to be a potential target of AHR pathway in zebrafish [93], and at the same time it can repress the activity of Wnt pathway during osteoblast differentiation [94]. Therefore, esrra seems to be yet another candidate for mediating cross-talk between AHR and Wnt pathways, which appeared to be distinctly activated in oral and pharyngeal jaw apparatus of the cichlids. The enriched GOs for esrra-related sub-module were involved in nucleic acid metabolism but little is known about the effects of estrogen signal on nucleic acid metabolism during development, even though such effects are already implicated in adult zebrafish and carp [95, 96]. The enriched GOs for mef2a-related sub-module were involved in skeletal muscle formation and mef2a is a well-known TF controlling skeletal muscle growth and differentiation [97]. Importantly, mef2a participates in adaptive mechanisms by which skeletal myofibers acquire specialised contractile properties (fast- or slow-twitch muscles) [98]. These mechanisms are tightly dependent on the activity of calcium/calmodulin pathway [99] which also plays a role in adaptive morphological changes in the jaw skeleton [100,101,102].
It should be noted that calcium/calmodulin pathway antagonizes the canonical Wnt pathway in different tissues during vertebrate development [103] whereas it can enhance AHR pathway activity [104, 105]. To this end, we found a GO related to cation/ion transport in the list of enriched GOs for the genes differentially expressed between oral and pharyngeal jaw apparatus. In addition to several genes encoding calcium transport channels (e.g. cacng6b, cacna1sb, pkd2 and atp2b1a) that are differentially expressed, other genes encoding calcium/calmodulin-dependent protein kinases including camkk1a/b, camk1da, camkk2, and camk2b1 also show differential expression between oral and pharyngeal jaws. This raises the possibility of differentially activated calcium/calmodulin pathway acting upstream of both AHR and Wnt pathways, which in turn leads to distinct transcriptional pattern of these two pathways between oral and pharyngeal jaws.
Species-specific coexpression modules
Interestingly, the Black and Purple coexpression modules had significant but opposite correlations with C. horei and G. pfefferi. These two sister species are both carnivores but with high specialised and distinct ecomorphologies [60] and diets [40]. G. pfefferi has a longer snout that is specialised for picking invertebrates from the soft lake bottom [40]. The Black module that was positively correlated with G. pfefferi was enriched for hypertrophy pathways that are known to result in increased muscle mass. Extreme hypertrophied lips have evolved repeatedly in several cichlid radiations to exploit novel food resources [106]. The black module contained dlx5a (Additional file 4: File S2b), a transcription factor involved in soft tissue craniofacial morphogenesis that was also differentially expressed in cichlids with nuchal humps [107]. The Black module was also enriched for the ERK/MAPK cascade that plays a key role in the development of the mesoderm, teeth and skeleton and have been shown to be trophic modulators in mice [108]. The MAPK pathway also interacts with the BMP and Wnt pathways, which are both involved in craniofacial development [30]. The Purple module was positively correlated with C. horei, a specialised fry-eater with a terminal mouth. The most prominent pathway in this module was the polyol pathway, which is associated with skeletal muscle atrophy [109] and also the Selenium metabolism pathway that has been linked to bone metabolism [110]. Interestingly, the estrogen pathway enriched in the purple module is associated with sex-specific trophic viscerocranial morphogenesis in cichlids [111]. The estrogen pathway interacts with the Ahr pathway (Fig. 5). Finally, the enriched Delta-Notch Pathway in this module is well-established for its role in craniofacial development [112]. Based on our findings, it is likely that the genes in the Blue and Purple modules, play modular roles in determining divergent carnivorous trophic morphologies in the Tropheini.

In silico cichlid jaw regulatory networks: highlights of important interactions between in silico annotated gene regulatory networks derived from OJA vs LPJA differentially expressed genes. The in silico annotations and interactions are deduced based on available data for vertebrates in FunCoup 3.0 [51], STRING v10 [52], and a meticulous literature search. The solid lines with arrow and blocked ends indicate potential inductive or repressive regulatory interactions with evidential bases from studies of vertebrates. The dashed lines indicate potential regulatory interaction found in this study but without functional support. The predicted TF(s) upstream of each each sub-module are depicted in square shape gene symbol with their related functions marked. The red, blue and white shadings for genes respectively indicate increased expression in oral and pharyngeal jaws, and no expression difference between the jaws
In silico GRNs annotated in differentially expressed oral versus pharyngeal jaw genes
The identification of GRNs promises opportunities to unravel the details of complex molecular mechanisms driving phenotypic diversification. Recent efforts have been undertaken to identify GRNs underlying morphological variations of craniofacial skeletal structures in non-model teleost fishes [31, 36, 113, 114]. However, little is known about the details of GRNs determining oral versus pharyngeal jaw structures. In this study, we focused on genes that were differentially expressed between OJA and LPJA, and were also enriched for GOs related to musculoskeletal system formation as well as metabolic response to organic compounds. The latter could provide molecular links between mechanisms involved in jaw formation and sensing extrinsic/intrinsic chemical cues, as well as metabolic processes. Based on a thorough literature review and vertebrate databases, we propose an in silico GRN of genes identified in the enriched muscle, skeleton, and response to organic substances biological processes from the differentially expressed genes between OJA and PJA (Fig. 5).
WNT is a major skeletogenesis and bone remodelling pathway [115] and the AHR pathway mediates the effects of environmental chemicals and nutritional compounds during jaw musculoskeletal development through transcriptional regulation of a variety of relevant TFs in Arctic charr [116]. We found distinct activation of components of two major upstream pathways, Wnt and AHR, in oral and pharyngeal jaws, respectively (Fig. 5). Antagonistic cross-talk between the two pathways might be a reason for this discrepancy, for instance, through direct repression of axin2, a major TF target of Wnt signal, by activated AHR pathway [79], repression of Wnt by notch1 after its AHR-dependent induction [117, 118], and/or through AHR-dependent induction of esrra, another Wnt pathway repressor (see section below). Genes with modular function in the formation of upper and lower oral jaws, wnt9b and pbx1, were found to be highly expressed in oral jaw compartment [75, 119], whereas fgfr1a determinant of posterior pharyngeal skeleton displayed increased expression in pharyngeal jaw [120]. Finally, we found increased expression of Ap1 complex genes (fos and jun) in oral jaw which can also mediate the effects of mechanical stressors in jaw [114, 121]. Altogether, these observations represent the complex interaction of genes mainly regulated by AHR and Wnt pathways with the potential to respond to environmental changes that determine distinct musculoskeletal system in cichlid jaws. The in silico GRN presented here provides several candidates for functional and molecular evolutionary studies on cichlid trophic adaptation in the future. An in silico GRN related to skeletal muscle formation displayed increased expression in LPJA apparatus for most of its gene constituents (Fig. 5). This suggests that more active muscle formation occurs in this region, possibly due to higher musculature in the LPJA as this jaw is more active in mastication. In contrast, most downstream members of an identified in silico GRN related to skeletogenesis had increased expression in the OJA, suggesting more active skeletal formation in the oral region. A major mediator of the Wnt signalling pathway during osteogenesis, tcf3, and a marker of specific skeletal elements in oral jaw, tfap2/ap2, were found to have increased expression in oral jaw [122, 123].
The enriched GO terms with molecular response to stimulus by organic compounds had the highest number of genes and predicted upstream TFs. Of note were components of the Ahr pathway, which can also be activated by diverse endogenous chemicals [124] and thus it perhaps provides a link between exposure to a range of compounds (from metabolic products to environmental toxicants) and jaw skeletogenesis. Remarkably most of the predicted TFs of genes involved in stimulus by organic compounds had oral/pharyngeal jaw differential expression in our analysis and are known to play a variety of roles in skeletogenesis. Two of the TFs in this in silico GRN, mafaa and mafbb, are of paramount importance because of their responsiveness to presence of extrinsic/intrinsic chemicals and activating signaling cascades required for metabolisms of organic compounds in fish [125]. This has significant implications for understanding the regulatory networks governing adaptive phenotypic plasticity in cichlid jaws [126].
Limitations of our study
Our study design is correlational and based upon discrete transcriptomes taken at one particular life stage from non-inbred fish reared under standardised conditions. At this stage no information regarding the phenotype – aside of the choice of model species and tissue – were used in constructing the networks. Our analysis is intended to complement those connecting quantitative traits and their genetic basis by providing a wider transcriptomic perspective. The suspensorium of gnathostomes can be traced back in ancestry to the gill arches of the Chordata that show serial homology, which also applies to the apomorphic modifications of the Gnathostomata [63]. The oral jaws originated via modification of the third ceratobranchial, the pharyngeal jaws from modifications of the fifth ceratobranchial arch [17, 62, 65]. Thus, it is possible that sets of conserved ancestral gene networks are acting in all gill arches as well as structures that are derived from it. Another limitation is that we had to restrict the dissection of the pharyngeal jaws to the lower section, which actually represents the ventral portion of the fifth ceratobranchial in terms of structural homology [65]. It would be informative to add transcriptomic data from other less modified gill arches to coexpression network studies in the future to elucidate the set of plesiomorphic conserved genes.
Conclusion
According to R. A. Raff [127, 128], modularity enhances evolvability by allowing three evolution mechanisms to operate: dissociation (decoupling), co-option (or rewiring), and duplication and divergence. Our study finds that all three of these mechanisms are putatively in play at the transcriptional level in the gene coexpression networks of cichlid jaw apparatuses. We expect that the coexpression modules and in silico jaw regulatory network presented here will be an important resource for futures studies on jaw morphogenesis; especially for those seeking to understand the link between environmental cues and skeletogenesis. Our findings on the OJA are valuable as most cichlids whole-transcriptome studies have focused on pharyngeal jaws. Our findings on the LPJA are also valuable as most vertebrate jaw model systems do not possess pharyngeal jaws. Given the insights gained here, we propose the necessity to embrace a systems biology approach to better understand the complex gene interactions underlying ecologically relevant phenotypes and their evolution.
Methods
Study design and sampling
For this study we used three cichlid species (Gnathochromis pfefferi, Ctenochromis horei and Limnotilapia dardennii) from the haplochromine tribe Tropheini in Lake Tanganyika and one haplochromine species from Lake Malawi (Petrotilapia sp. ‘yellow chin Chewere’) that were purchased from the aquarium trade or private breeders and reared in the certified aquarium facility at the University of Graz (Austria) (Fig. 1). The fish were raised in standardised aquaria and standardised temperature, light and water conditions, and received the same diet of Spirulina flakes, minimising the effects of plasticity. Once young adult stage was reached, indicated by when mating behaviour was first observed, all individuals were carefully monitored to identify spawning period. Immediately (± 3 h) after the end of spawning, we removed the eggs from the mouth of the females using moderate manual pressure on their cheeks. The eggs were placed in an oxygenated, low speed shaker. Once the eggs hatched and the larvae reached developmental stage 26, which marks the end of postembryonic development as the yolk-sac is absorbed into the body cavity [41], the larvae were euthanized in water containing 0.2 g MS-222/litre and then stored in RNAlater. By the end of the study the parents of larvae were sacrificed in water containing 0.8 g MS-222/litre. In nature, stage 26 is when the larvae leave the buccal cavity of the mother to begin foraging independently [41]. As we were not focusing explicitly on the developmental pathways, our choice to sample at stage 26 provides a snapshot of growth where the transcriptomes can also provide information on distinct skeletogenesis related processes that are responsive to environmental cues. For each individual larva, the OJA and LPJA were dissected as two separate units and treated as separate samples from here on (Additional file 1: Figure S1). This was an improved design from our previous study where the two jaws were not separated during dissection [42]. The new dissection design here enabled us to study the two units separately and address questions on modularity. The OJA included the upper and lower units of the oral jaw and surrounding soft tissue such as muscle and cartilage that is difficult to remove. The LPJA only included the lower part of the pharyngeal jaw and surrounding soft tissue (muscle, cartilage) and not the upper pharyngeal jaw due to difficulties in dissecting the upper unit. We account for this drawback by avoiding direct comparison of the dorso-ventral patterning of gene expression. Additionally, including bone and soft tissue in the dissection does not violate any biological assumptions about modularity as we know that there is cross-talk between the bone, cartilage, muscle pathways of the trophic apparatus [30]. As we only had one individual of the Lake Malawi P. sp. ‘yellow chin Chewere’ species, it was included as the phylogenetic outgroup for the hierarchical clustering analysis in Fig. 2, but excluded from subsequent coexpression and differential expression analyses due to a lack of biological replicates, and because the focus of our study were the Tropheini from Lake Tanganyika (see Fig. 1 for details on the study design and number of biological replicates). Please note that the sampling here was conducted from a different batch of fish and tissues as that in [42] and all new sequencing data was generated for this study.
RNA library preparation and sequencing
The preserved samples were homogenized by using Lysing Matrix A tubes (MP Bio) in a FastPrep®-24 (MP Bio) homogenizer, further extraction was done using RNeasy Mini Kit (Qiagen). Total RNA qualities and quantities were measured first with NanoPhotometer (Implen) and later with 2200 TapeStation (Agilent) either using High Sensitivity RNA ScreenTapes® or RNA SreenTapes®. Only individuals with a RIN value higher than 7 in one of their extracts were included in this study.
TruSeq® RNA Sample Prep Kit V2 (Illumina) was used to individually indexed paired-end libraries for each sample with unique adapters. At stage 26 in Tropheini species, the pharyngeal jaw is completely ossified and so are mandible, maxilla, and premaxilla of the oral jaw. The articular part of the oral jaw is not ossified, as it is a cartilage (unpublished data from our lab based on histological data from bone and cartilage staining). Thus, the dissection of the OJA (bone and surrounding tissues) of one individual larva produced enough RNA for TrueSeq v2 Kit. We also obtained enough RNA from the lower PJA (bone and surrounding tissues) of one. Therefore, pooling of individuals was not required. Library qualities were checked using D1000 ScreenTapes®, diluted, and 26 libraries were multiplexed to peak molarities and paired-end sequenced on two lanes of Illumina HiSeq® (2 × 125 bp) at the Biomedical Sequencing Facility in Vienna (Austria).
Differential gene expression analysis
Approximately 11 million paired-end 125 bp sequences were obtained for each sample (315,933,795 PE reads in total; (Additional file 2: File S1). The FastX toolkit (version 0.0.13; http://hannon-lab.cshl.edu/fastx_toolkit/) was used to trim the reads to a length of 92 bp and reads containing 20% or more bases with a Phred Quality score of < 30 were discarded. High quality, filtered reads were mapped to the Oreochromis niloticus Broad Institute reference genome [43] using TopHat2 (version 2.1) [44]. Gene expression counts were calculated using HT-seq (version 0.6.1) [45]. Prefiltering was conducted to remove genes with less than 10 total reads across all samples to avoid biases introduced by low coverage genes. Differential gene expression was conducted using DESeq2 [48]. All libraries were normalised simultaneously using default settings recommended by Love et al. [48]. Differential gene expression between OJAand PJA of all species was conducted using the following model: ~ species + jawtype. Wald’s test was used to calculate p.values for the log2-fold change difference. False discovery rate (FDR) [49] correction was used to account for multiple testing (p.adjust value cutoff of < 0.05).
Gene coexpression network (GCN) analysis
The filtered gene expression table used for differential gene expression analysis was transformed using VarianceStabilizingTransformation implemented in DESeq2 [46], as recommended by the best practices of Weighted Gene Coexpression Network Analysis (WGCNA version 1.68) R-package (version 3.2.1) [32]. As our interest was in the OJA and LPJA comparison, samples from all three species were used as biological replicates for the two jaw units. This provided sufficient sampling power for WGCNA. Hierarchical clustering of samples based on gene expression was conducted to identify sample relationships and outliers. Signed hybrid coexpression networks were constructed using the following steps: (1) Pearson correlation coefficients were used a measure of gene coexpression (2) The Coefficients were raised to a softpower to create an adjacency matrix that was determined with reference to a scale free topology (3) The adjacency matrix is used to calculate the topological overlap distance matrix (4) The topological overlap distance was then used to hierarchically cluster genes (method = average) (5) Groups of coexpressed gene (i.e. modules) were identified using the cutTreeDynamic function with a minimum module size of 30 genes (6) Each module was assigned a colour, and the first principal component (module eigengene) of each module represented the gene expression profile of that module (6) Based on module eigengene (ME) dissimilarity, highly similar modules at a ME distance threshold of 0.25 were merged to obtain the final set of coexpressed gene modules.
Two different sets of coexpression analyses were conducted: (1) Conditional coexpression analysis: OJA and LPJA coexpression networks were separately constructed to investigate the preservation of OJA modules in the PJA network and vice versa. A softpower of 18 was used to construct adjacency matrix (Additional file 1: Figure S2). Module preservation statistics were computed using WGCNA to see how the density and connectivity of modules defined in the reference dataset (i.e. OJA) were preserved in the query dataset (i.e. LPJA) [47]. A permutation test was used to repeatedly permute genes in the query network and Zscore was used as a measure of significance. Individual Z scores from all permutations (200) were summarised as a Zsummary statistic. (2) Global coexpression analysis: one coexpression network was constructed with both the OJA and LPJA data. A softpower of 6 was used to construct adjacency matrix (Additional file 1: Figure S3). The global jaw modules were correlated with jaw type (i.e. OJA or LPJA) and species in order to identify modules that were jaw-specific and species-specific. In order to do this a gene significance (GS) value based on the Pearson correlation between genes and traits was calculated. Species-specific correlations were coded as follows: C. horei vs others, L. dardennii vs others, G. pfefferi versus others, as recommended for non-binary traits by WGCNA best practices.
Functional annotation: gene ontology and pathway enrichment analysis
Genes in coexpression network modules and differentially expressed genes were functionally annotated using gene ontology (GO) enrichment analysis and upstream transcriptional regulators/factors (TFs) using WebGestalt, a knowledge-based network enrichment analysis tool for vertebrates [50]. Recent literature on regulatory interactions between the genes of interest, in addition to information available on two comprehensive databases for vertebrate interactome, FunCoup (version 3.0) [51] and STRING (version 10) [52], were utilised to deduce relevant networks of gene interactions. Moreover, the phenotype information of gene mutations was cross-checked with the zebrafish database (ZFIN.org) [53] in order to infer their potential role in jaw skeletogenesis. To do this, we converted gene IDs of the differentially expressed genes to zebrafish orthologues gene IDs with well annotated coding and regulatory sequences (e.g. annotated promoter, introns and 5′/3′-UTR sequences) using the BioMart package [54]. This integrative approach allowed us to in silico annotate enriched genes and pathways within the coexpression modules and differentially expressed genes.
Availability of data and materials
Raw sequence reads are deposited on NCBI SRA (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA626001) and code is available on GitHub (github.com/poojasingh09/cichlid_jaw_modularity_Singh_et_al).
Abbreviations
- OJA:
-
Oral jaw apparatus
- LPJA:
-
Lower pharyngeal apparatus
- GRNs:
-
Gene regulatory networks
- GCN:
-
Gene coexpression networks
- TFs:
-
Transcription factors
- GS:
-
Gene significance
- ME:
-
Module eigengene
- GO:
-
Gene Ontology
References
Olson E, Miller R. Morphological integration. Chicago: University of Chicago Press; 1958.
Mayr E. The growth of biological thought: diversity, evolution, and inheritance. Cambridge: Harvard University Press; 1982.
Wagner GP. Homologues, natural kinds and the evolution of modularity. Am J Zool. 1996;36:36–43.
Pigliucci M, Preston K. Phenotypic integration: studying the ecology and evolution of complex phenotypes. New York: Oxford University Press; 2004.
Wagner GP, Pavlicev M, Cheverud JM. The road to modularity. Nat Rev Genet. 2007;8:921–31.
Klingenberg CP. Morphological integration and developmental modularity. Annu Rev Ecol Evol Syst. 2008;39:115–32.
Klingenberg CP. Studying morphological integration and modularity at multiple levels: concepts and analysis. Philos Trans R Soc B Biol Sci. 2014;369:20130249–20130249. https://doi.org/10.1098/rstb.2013.0249.
Melo D, Porto A, Cheverud JM, Marroig G. Modularity: Genes, Development, and Evolution. Annu Rev Ecol Evol Syst. 2016;47:463–86. https://doi.org/10.1146/annurev-ecolsys-121415-032409.
Klingenberg CP. Evolution and development of shape: integrating quantitative approaches. Nat Rev Genet. 2010;11:623–35. https://doi.org/10.1038/nrg2829.
King M, Wilson AC. Evolution at two levels in humans and chimpanzees. Science. 1975;188:107–16.
Brawand D, Soumillon M, Necsulea A, Julien P, Csárdi G, Harrigan P, et al. The evolution of gene expression levels in mammalian organs. Nature. 2011;478:343–8.
Somogyi R, Fuhrman S, Anderson G, Madill C, Greller L, Chang B. Systematic exploration and mining of gene expression data provides evidence for higher-order, modular regulation. In: Schlosser G, Wagner G, editors. Modularity in development and evolution. Chicago, IL: University of Chicago Press; 2004. p. 203–21.
Raff RA, Sly BJ. Modularity and dissociation in the evolution of gene expression territories in development. Evol Dev. 2000;2:102–13.
Lorenz DM, Jeng A, Deem MW. The emergence of modularity in biological systems. Phys Life Rev. 2011;8:129–60. https://doi.org/10.1016/j.plrev.2011.02.003.
Jiménez A, Cotterell J, Munteanu A, Sharpe J. A spectrum of modularity in multi-functional gene circuits. Mol Syst Biol. 2017;13:925. https://doi.org/10.15252/msb.20167347.
Albertson RC, Streelman JT, Kocher TD, Yelick PC. Integration and evolution of the cichlid mandible: the molecular basis of alternate feeding strategies. PNAS. 2005;102:16287–92. https://doi.org/10.1073/pnas.0506649102.
Liem K. Modulatory multiplicity in the functional repertoire of the feeding mechanism in cichlid fishes. J Morphol. 1978;158:323–60.
Meyer A. Phenotypic plasticity and heterochrony in Cichlasoma managuense (Pisces, Cichlidae) and their implication for speciation in cichlid fishes. Evolution (N Y). 1987;41:1357–69.
Hulsey CD, de García León FJ, Rodiles-Hernández R. Micro- and macroevolutionary decoupling of cichlid jaws: a test of Liem’s key innovation hypothesis. Evolution. 2006;60:2096–109.
Cooper WJ, Wernle J, Mann K, Albertson RC. Functional and genetic integration in the skulls of Lake Malawi cichlids. Evol Biol. 2011;38:316–34.
Parsons KJ, Ma E, Albertson RC. Constraint and opportunity: the genetic basis and evolution of modularity in the cichlid madible. Am Nat. 2012;179:64–78.
Le Pabic P, Cooper WJ, Schilling TF. Developmental basis of phenotypic integration in two Lake Malawi cichlids. EvoDevo. 2016;7:1–26.
Parsons KJ, Cooper WJ, Albertson RC. Modularity of the oral jaws is linked to repeated changes in the craniofacial shape of African cichlids. Int J Evol Biol. 2011. https://doi.org/10.4061/2011/641501.
Liem KF. Evolutionary strategies and morphological innovations: cichlid pharyngeal jaws. Syst Zool. 1973;22:425–41. https://doi.org/10.2307/2412950.
Hu Y, Parsons KJ, Albertson RC. Evolvability of the cichlid jaw: new tools provide insights into the genetic basis of phenotypic integration. Evol Biol. 2014;41:145–53.
Albertson RC, Streelman JT, Kocher TD. Directional selection has shaped the oral jaws of Lake Malawi cichlid fishes. PNAS. 2003;100:5252–7. https://doi.org/10.1073/pnas.0930235100.
Peter IS, Davidson EH. Evolution of gene regulatory networks controlling body plan development. Cell. 2011;144:970–85. https://doi.org/10.1016/j.cell.2011.02.017.
Erwin DH, Davidson EH. The evolution of hierarchical gene regulatory networks. Nat Rev Genet. 2009;10:141–8.
Fraser GJ, Hulsey CD, Bloomquist RF, Uyesugi K, Manley NR, Streelman JT. An ancient gene network is co-opted for teeth on old and new jaws. PLoS Biol. 2009;7:e31. https://doi.org/10.1371/journal.pbio.1000031.
Ahi EP. Signalling pathways in trophic skeletal development and morphogenesis: insights from studies on teleost fish. Dev Biol. 2016;420:11–31. https://doi.org/10.1016/j.ydbio.2016.10.003.
Schneider RF, Li Y, Meyer A, Gunter HM. Regulatory gene networks that shape the development of adaptive phenotypic plasticity in a cichlid fish. Mol Ecol. 2014;23:4511–26. https://doi.org/10.1111/mec.12851.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:1–13. https://doi.org/10.1186/1471-2105-9-559.
Weirauch MT. Gene coexpression networks for the analysis of DNA microarray data. In: Dehmer M, Emmert-Streib F, Graber A, Salvador A, editors. Applied statistics for network biology: methods in systems biology. Weinheim: Wiley; 2011. p. 215–50.
Roy S, Bhattacharyya DK, Kalita JK. Reconstruction of gene co-expression network from microarray data using local expression patterns. BMC Bioinformatics. 2014;15(Suppl 7):1–14. https://doi.org/10.1186/1471-2105-15-S7-S10.
Oldham MC, Horvath S, Geschwind DH. Conservation and evolution of gene coexpression networks in human and chimpanzee brains. PNAS. 2006;103:17973–8. https://doi.org/10.1073/pnas.0605938103.
Filteau M, Pavey SA, St-Cyr J, Bernatchez L. Gene coexpression networks reveal key drivers of phenotypic divergence in lake whitefish. Mol Biol Evol. 2013;30:1384–96.
Johnston RA, Paxton KL, Moore FR, Wayne RK. Seasonal gene expression in a migratory songbird. Mol Ecol. 2016;25:5680–91.
Fruciano C, Meyer A, Franchini P. Divergent allometric trajectories in gene expression and coexpression produce species differences in sympatrically speciating Midas cichlid fish. Genome Biol Evol. 2019;11:1644–57.
Tian F, Liu S, Shi J, Qi H, Zhao K, Xie B. Transcriptomic profiling reveals molecular regulation of seasonal reproduction in Tibetan highland fish. Gymnocypris przewalskii BMC Genomics. 2019;20:1–13.
Konings A. Tanganyika cichlids in their natural habitat. 4th ed. El Paso: Cichlid Press; 2019.
Fujimura K, Okada N. Development of the embryo, larva and early juvenile of Nile tilapia Oreochromis niloticus (Pisces: Cichlidae) Developmental staging system. Dev Growth Differ. 2007;49:301–24.
Singh P, Börger C, More H, Sturmbauer C. The role of alternative splicing and differential gene expression in cichlid adaptive radiation. Genome Biol Evol. 2017;9:2764–81. https://doi.org/10.1093/gbe/evx204.
Brawand D, Wagner CE, Li YI, Malinsky M, Keller I, Fan S, et al. The genomic substrate for adaptive radiation in African cichlid fish. Nature. 2014;513:375–81. https://doi.org/10.1038/nature13726.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. https://doi.org/10.1186/gb-2013-14-4-r36.
Anders S, Pyl PT, Huber W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. https://doi.org/10.1186/s13059-014-0550-8.
Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? PLoS Comput Biol. 2011. https://doi.org/10.1371/journal.pcbi.1001057.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. https://doi.org/10.1186/s13059-014-0550-8.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300. https://doi.org/10.2307/2346101.
Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT analysis toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013;41:W77-83.
Schmitt T, Ogris C, Sonnhammer ELL. FunCoup 3.0: database of genome-wide functional coupling networks. Nucleic Acids Res. 2014. https://doi.org/10.1093/nar/gkt984.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015. https://doi.org/10.1093/nar/gku1003.
Bradford Y, Conlin T, Dunn N, Fashena D, Frazer K, Howe DG, et al. ZFIN: enhancements and updates to the Zebrafish Model Organism Database. Nucleic Acids Res. 2011. https://doi.org/10.1093/nar/gkq1077.
Smedley D, Haider S, Ballester B, Holland R, London D, Thorisson G, et al. BioMart – biological queries made easy. BMC Genomics. 2009;10:22.
Ahi EP, Singh P, Duenser A, Gessl W, Sturmbauer C. Divergence in larval jaw gene expression reflects differential trophic adaptation in haplochromine cichlids prior to foraging. BMC Evol Biol. 2019;19:150.
Medina M. Genomes, phylogeny, and evolutionary systems biology. PNAS. 2005;102:6630–5.
Loewe L. A framework for evolutionary systems biology. BMC Syst Biol. 2009;3:27.
Irisarri I, Singh P, Koblmüller S, Torres-Dowdall J, Henning F, Franchini P, et al. Phylogenomics uncovers early hybridization and adaptive loci shaping the radiation of Lake Tanganyika cichlid fishes. Nat Commun. 2018. https://doi.org/10.1038/s41467-018-05479-9.
Merkin J, Russell C, Chen P, Burge CB. Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science. 2012;338:1593–9.
Wanek AK, Sturmbauer C. Form, function and phylogeny: comparative morphometrics of Lake Tanganyika’s cichlid tribe Tropheini. Zool Scr. 2015;44:362–73.
Cerny R, Lwigale P, Ericsson R, Meulemans D, Epperlein HH, Bronner-Fraser M. Developmental origins and evolution of jaws: New interpretation of “maxillary” and “mandibular.” Dev Biol. 2004;276:225–36.
Kuratani S. Evolution of the vertebrate jaw from developmental perspectives. Evol Dev. 2012;14:76–92.
Gillis JA, Modrell MS, Baker CVH. Developmental evidence for serial homology of the vertebrate jaw and gill arch skeleton. Nat Commun. 2013;4:1–16.
Le Pabic P, Stellwag EJ, Scemama J, Carolina N. Embryonic development and skeletogenesis of the pharyngeal jaw apparatus in the cichlid Nile tilapia (Oreochromis niloticus). Anat Rec. 2009;292:1780–800.
Wainwright PC, Smith WL, Price SA, Tang KL, Sparks JS, Ferry LA, et al. The evolution of pharyngognathy: a phylogenetic and functional appraisal of the pharyngeal jaw key innovation in labroid fishes and beyond. Syst Biol. 2012;61:1001–27. https://doi.org/10.1093/sysbio/sys060.
Renz AJ, Gunter HM, Fischer J, Qiu H, Meyer A, Kuraku S. Ancestral and derived attributes of the dlx gene repertoires, cluster structure and expression patterns in an African cichlid fish. Science. 2009;308:2009–2009.
Gibert Y, Bernard L, Debiais-Thibaud M, Bourrat F, Joly J-S, Pottin K, et al. Formation of oral and pharyngeal dentition in teleosts depends on differential recruitment of retinoic acid signaling. FASEB J. 2010;24:3298–309. https://doi.org/10.1096/fj.09-147488.
Hulsey CD, Fraser GJ, Meyer A. Biting into the genome to phenome map: developmental genetic modularity of cichlid fish dentitions. Integr Comp Biol. 2016;56:373–88. https://doi.org/10.1093/icb/icw059.
Wray GA. The evolutionary significance of cis-regulatory mutations. Nat Rev Genet. 2007;8:206–16. https://doi.org/10.1038/nrg2063.
Meireles-Filho AC, Stark A. Comparative genomics of gene regulation-conservation and divergence of cis-regulatory information. Curr Opin Genet Dev. 2009;19:565–70.
Halfon MS. Perspectives on gene regulatory network evolution. Trends Genet. 2017;33:436–47. https://doi.org/10.1016/j.tig.2017.04.005.
Auman T, Chipman AD. The evolution of gene regulatory networks that define arthropod body plans. Integr Comp Biol. 2017;57:1–10.
Gao F, Davidson EH. Transfer of a large gene regulatory apparatus to a new developmental address in echinoid evolution. PNAS. 2008;105:6091–6.
Arduini BL, Bosse KM, Henion PD. Genetic ablation of neural crest cell diversification. Development. 2009;136:1987–94.
Jin Y-R, Han XH, Taketo MM, Yoon JK. Wnt9b-dependent FGF signaling is crucial for outgrowth of the nasal and maxillary processes during upper jaw and lip development. Development. 2012;139:1821–30.
Xiong KM, Peterson RE, Heideman W. Aryl hydrocarbon receptor-mediated down-regulation of sox9b causes jaw malformation in zebrafish embryos. Mol Pharmacol. 2008;74:1544–53.
Mathew LK, Simonich MT, Tanguay RL. AHR-dependent misregulation of Wnt signaling disrupts tissue regeneration. Biochem Pharmacol. 2009;77:498–507.
Procházková J, Kabátková M, Bryja V, Umannová L, Bernatík O, Kozubík A, et al. The interplay of the aryl hydrocarbon receptor and β-catenin alters both AhR-dependent transcription and wnt/β-catenin signaling in liver progenitors. Toxicol Sci. 2011;122:349–60.
Zhang H, Yao Y, Chen Y, Yue C, Chen J, Tong J, et al. Crosstalk between AhR and wnt/β-catenin signal pathways in the cardiac developmental toxicity of PM2.5 in zebrafish embryos. Toxicology. 2016;355–356:31–8.
Planchart A, Mattingly CJ. 2,3,7,8-Tetrachlorodibenzo-p-dioxin upregulates FoxQ1b in zebrafish jaw primordium. Chem Res Toxicol. 2010;23:480–7.
Lin C-Y, Chen W-T, Lee H-C, Yang P-H, Yang H-J, Tsai H-J. The transcription factor Six1a plays an essential role in the craniofacial myogenesis of zebrafish. Dev Biol. 2009;331:152–66.
Nord H, Nygard Skalman L, von Hofsten J. Six1 regulates proliferation of Pax7-positive muscle progenitors in zebrafish. J Cell Sci. 2013;126:1868–80.
Yao Z, Farr GH, Tapscott SJ, Maves L, Maves L. Pbx and Prdm1a transcription factors differentially regulate subsets of the fast skeletal muscle program in zebrafish. Biol Open. 2013;2:546–55.
Birkholz DA, Olesnicky Killian EC, George KM, Artinger KB. Prdm1a is necessary for posterior pharyngeal arch development in zebrafish. Dev Dyn. 2009;238:2575–87.
Jackman WR, Draper BW, Stock DW. Fgf signaling is required for zebrafish tooth development. Dev Biol. 2004;274:139–57.
Barske L, Rataud P, Behizad K, Del Rio L, Cox SG, Crump JG. Essential role of Nr2f nuclear receptors in patterning the vertebrate upper jaw. Dev Cell. 2018;44(337–347):e5.
Britanova O, Depew MJ, Schwark M, Thomas BL, Miletich I, Sharpe P, et al. Satb2 haploinsufficiency phenocopies 2q32-q33 deletions, whereas loss suggests a fundamental role in the coordination of jaw development. Am J Hum Genet. 2006;79:668–78.
Sheehan-Rooney K, Pálinkášová B, Eberhart JK, Dixon MJ. A cross-species analysis of Satb2 expression suggests deep conservation across vertebrate lineages. Dev Dyn. 2010;239:3481–91.
Fish JL, Villmoare B, Köbernick K, Compagnucci C, Britanova O, Tarabykin V, et al. Satb2, modularity, and the evolvability of the vertebrate jaw. Evol Dev. 2011;13:549–64.
Bonnelye E, Aubin JE. Estrogen receptor-related receptor α: a mediator of estrogen response in bone. J Clin Endocrinol Metab. 2005;90:3115–21.
Kim Y-I, No Lee J, Bhandari S, Nam I-K, Yoo K-W, Kim S-J, et al. Cartilage development requires the function of Estrogen-related receptor alpha that directly regulates sox9 expression in zebrafish. Sci Rep. 2016;5:18011.
Ahi EP, Walker BS, Lassiter CS, Jónsson ZO. Investigation of the effects of estrogen on skeletal gene expression during zebrafish larval head development. PeerJ. 2016;4:e1878.
Fang X, Corrales J, Thornton C, Clerk T, Scheffler BE, Willett KL. Transcriptomic changes in zebrafish embryos and larvae following benzo[a]pyrene exposure. Toxicol Sci. 2015;146:395–411.
Auld KL, Berasi SP, Liu Y, Cain M, Zhang Y, Huard C, et al. Estrogen-related receptor α regulates osteoblast differentiation via Wnt/β-catenin signaling. J Mol Endocrinol. 2012;48:177–91.
Moens LN, van der Ven K, Van Remortel P, Del-Favero J, De Coen WM. Gene expression analysis of estrogenic compounds in the liver of common carp (Cyprinus carpio) using a custom cDNA microarray. J Biochem Mol Toxicol. 2007;21:299–311.
De WM, Keil D, van der Ven K, Vandamme S, Witters E, De CW. An integrated transcriptomic and proteomic approach characterizing estrogenic and metabolic effects of 17 α-ethinylestradiol in zebrafish (Danio rerio). Gen Comp Endocrinol. 2010;167:190–201.
Naya FJ, Olson E. MEF2: a transcriptional target for signaling pathways controlling skeletal muscle growth and differentiation. Curr Opin Cell Biol. 1999;11:683–8.
Youn H-D, Chatila TA, Liu JO, Nielsen S, Pines J, Kouzarides T, et al. Integration of calcineurin and MEF2 signals by the coactivator p300 during T-cell apoptosis. EMBO J. 2000;19:4323–31.
Wu H, Naya FJ, McKinsey TA, Mercer B, Shelton JM, Chin ER, et al. MEF2 responds to multiple calcium-regulated signals in the control of skeletal muscle fiber type. EMBO J. 2000;19:1963–73.
Abzhanov A, Kuo WP, Hartmann C, Grant BR, Grant PR, Tabin CJ. The calmodulin pathway and evolution of elongated beak morphology in Darwin’s finches. Nature. 2006;442:563–7.
Parsons KJ, Albertson RC. Roles for Bmp4 and CaM1 in shaping the jaw: evo-devo and beyond. Annu Rev Genet. 2009;43:369–88.
Gunter HM, Meyer A. Molecular investigation of mechanical strain-induced phenotypic plasticity in the ecologically important pharyngeal jaws of cichlid fish. J Appl Ichthyol. 2014;30:630–5. https://doi.org/10.1111/jai.12521.
Slusarski DC, Pelegri F. Calcium signaling in vertebrate embryonic patterning and morphogenesis. Dev Biol. 2007;307:1–13.
Monteiro P, Gilot D, Le Ferrec E, Rauch C, Lagadic-Gossmann D, Fardel O. Dioxin-mediated up-regulation of aryl hydrocarbon receptor target genes is dependent on the calcium/calmodulin/CaMKIalpha pathway. Mol Pharmacol. 2008;73:769–77.
Han EH, Kim HG, Im JH, Jeong TC, Jeong HG. Up-regulation of CYP1A1 by rutaecarpine is dependent on aryl hydrocarbon receptor and calcium. Toxicology. 2009;266:38–47.
Baumgarten L, Machado-Schiaffino G, Henning F, Meyer A. What big lips are good for: on the adaptive function of repeatedly evolved hypertrophied lips of cichlid fishes. Biol J Linn Soc. 2015;115:448–55. https://doi.org/10.1111/bij.12502.
Lecaudey LA, Sturmbauer C, Singh P, Ahi EP. Molecular mechanisms underlying nuchal hump formation in dolphin cichlid Cyrtocara moorii. Sci Rep. 2019;9:1–13.
Laugel-Haushalter V, Paschaki M, Marangoni P, Pilgram C, Langer A, Kuntz T, et al. RSK2 is a modulator of craniofacial development. PLoS ONE. 2014;9:e84343.
Langer HT, Afzal S, Kempa S, Spuler S. Nerve damage induced skeletal muscle atrophy is associated with increased accumulation of intramuscular glucose and polyol pathway intermediates. Sci Rep. 2020;10:1908. https://doi.org/10.1038/s41598-020-58213-1.
Moreno-Reyes R, Egrise D, Nève J, Pasteels J-L, Schoutens A. Selenium deficiency-induced growth retardation is associated with an impaired bone metabolism and osteopenia. J Bone Miner Res. 2001;16:1556–63. https://doi.org/10.1359/jbmr.2001.16.8.1556.
Parsons KJ, Wang J, Anderson G, Albertson RC. Nested levels of adaptive divergence: the genetic basis of craniofacial divergence and ecological sexual dimorphism. G3. 2015;5:1613–24. https://doi.org/10.1534/g3.115.018226.
Pakvasa M, Haravu P, Boachie-Mensah M, Jones A, Coalson E, Liao J, et al. Notch signaling: Its essential roles in bone and craniofacial development. Genes Dis. 2021;8:8–24. https://doi.org/10.1016/j.gendis.2020.04.006.
Ahi E, Kapralova K, Pálsson A, Maier V, Gudbrandsson J, Snorrason SS, et al. Transcriptional dynamics of a conserved gene expression network associated with craniofacial divergence in Arctic charr. EvoDevo. 2014;5:1–19. https://doi.org/10.1186/2041-9139-5-40.
Gunter HM, Schneider RF, Karner I, Sturmbauer C, Meyer A. Molecular investigation of genetic assimilation during the rapid adaptive radiations of East African cichlid fishes. Mol Ecol. 2017;26:6634–53.
Monroe DG, McGee-Lawrence ME, Oursler MJ, Westendorf JJ. Update on Wnt signaling in bone cell biology and bone disease. Gene. 2012;492:1–18.
Ahi EP, Steinhäuser SS, Pálsson A, Franzdóttir SR, Snorrason SS, Maier VH, et al. Differential expression of the Aryl hydrocarbon receptor pathway associates with craniofacial polymorphism in sympatric Arctic charr. EvoDevo. 2015;6:27.
Deregowski V, Gazzerro E, Priest L, Rydziel S, Canalis E. Notch 1 overexpression inhibits osteoblastogenesis by suppressing Wnt/beta-catenin but not bone morphogenetic protein signaling. J Biol Cchemistry. 2006;281:6203–10.
Stevens EA, Mezrich JD, Bradfield CA. The aryl hydrocarbon receptor: a perspective on potential roles in the immune system. Immunology. 2009;127:299–311.
Selleri L, Depew MJ, Jacobs Y, Chanda SK, Tsang KY, Cheah KS, et al. Requirement for Pbx1 in skeletal patterning and programming chondrocyte proliferation and differentiation. Development. 2001;128:3543–57.
Larbuisson A, Dalcq J, Martial JA, Muller M. Fgf receptors Fgfr1a and Fgfr2 control the function of pharyngeal endoderm in late cranial cartilage development. Differentiation. 2013;86:192–206.
Gunter HM, Fan S, Xiong F, Franchini P, Fruciano C, Meyer A. Shaping development through mechanical strain: the transcriptional basis of diet-induced phenotypic plasticity in a cichlid fish. Mol Ecol. 2013;22:4516–31. https://doi.org/10.1111/mec.12417.
Knight RD, Nair S, Nelson SS, Afshar A, Javidan Y, Geisler R, et al. lockjaw encodes a zebrafish tfap2a required for early neural crest development. Development. 2003;130:5755–68.
Liu W, Liu Y, Guo T, Hu C, Luo H, Zhang L, et al. TCF3, a novel positive regulator of osteogenesis, plays a crucial role in miR-17 modulating the diverse effect of canonical Wnt signaling in different microenvironments. Cell Death Dis. 2013;4:e539–e539.
Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–34.
Stegeman JJ, Goldstone JV, Hahn ME. Perspectives on zebrafish as a model in environmental toxicology. Fish Physiol. 2010;29:367–439.
Pfennig DW, Ehrenreich IM. Towards a gene regulatory network perspective on phenotypic plasticity, genetic accommodation and genetic assimilation. Mol Ecol. 2014;23:4438–40.
Raff RA. The shape of life: genes, development and the evolution of animal form. Chicago: University of Chicago Press; 1996.
Raff RA. Larval homologies and radical evolutionary changes in early development. In: Bock GK, Cardew G, editors. Novartis foundation symposium 222–homology: homology: novartis foundation symposium 222. Chichester: Wiley; 1999. p. 110–21.
Acknowledgements
We acknowledge C. Börger for assistance in data generation, W. Gessl for fish husbandry and photographs, and M. Koller for image editing.
Funding
Austrian Science Fund (FWF) P29838 (awarded to CS) funded the research (fish purchase and husbandry, and sequencing costs). A PhD scholarship from the Austrian Centre of Limnology (University of Graz), funded the salary of PS during the project. The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript. Open Access Funding by the FWF.
Author information
Authors and Affiliations
Contributions
CS, PS conceived the study. PS conducted the bioinformatics analyses. EPA conducted the gene-ontology and pathway analysis. PS, EPA drafted the manuscript. CS supervised the study and contributed to the writing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approval is not applicable as no experiments were conducted on the fish prior to sampling. Fish sacrifice was approved by the University of Graz, Institute of Biology.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1
. Schematic depiction of the dissection strategy utilised in this study. Red dotted lines mark the cuts made. RNA from oral jaws (upper + lower) and the lower pharyngeal jaw were separately extracted. The dissection included the following tissues: bone, cartilage, teeth, muscle, tendons, fat, and blood vessels. Figure S2. Conditional coexpression analysis: fitting scale free topology to establish softpower for constructing the separate OJA and LPJA adjacency matrices. Softpower of 18 was chosen for both. Figure S3. Global coexpression analysis: fitting scale free topology to establish softpower for constructing jaw adjacency matrices with OJA and PJA data together. Softpower of 6 was chosen. Figure S4. Conditional coexpression analysis: Preservation of modules in the GCNs underlying oral (OJA) and lower pharyngeal jaws (LPJA). a Preservation of genes found in LPJA modules in the OJA coexpression network calculated by a Zsummary statistic based on a permutation test that takes into account the connectivity and density of genes in a module. Zsummary < 2 represents lack of preservation (dotted blue line). Zsummary between 2 and 10 implies moderate preservation. Zsummary > 10 supports strong preservation of module. b Visual representation of module preservation. Top: LPJA modules in the LPJA GCN. Bottom: LPJA modules in the OJA GCN. Figure S5. Global coexpression analysis: Gene co-expression network of the oral and pharyngeal jaws. Dendrograms produced by average linkage hierarchical clustering of 16,669 genes based on topological overlap matrix (TOM). Modules within the network were assigned colours based on the horizontal bar underneath the dendrogram. Figure S6. Global coexpression analysis: Barplot of mean trait-based gene significance across modules in the oral and pharyngeal jaw co-expression network. Figure S7. Global coexpression analysis: Per module gene significance and connectivity in the oral and pharyngeal jaw co-expression network and connectivity in the oral and pharyngeal jaw coexpression network. Figure S8. Global coexpression analysis: enriched pathways in Black and Purple species-specific gene expression modules.
Additional file 2.
File S1. RNAseq mapping read statistics across samples and results from differential gene expression between PJA and PJA.
Additional file 3.
File S2. a Gene ontology enrichment analysis of differentially expressed genes between LPJA and OJA. The enriched genes in GOs related to skeletogenesis, muscle formation and biogenesis of organic compounds were used to predict their potential upstream transcriptional regulators.
Additional file 4.
File S2. b Gene names of genes in jaw- and species-specific modules. Gene ontology enrichment analyses of identified gene networks showing species or jaw specific coexpression. Subsets of related GOs were used to predict upstream transcription factors and downstream sub-module(s) for each network.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Singh, P., Ahi, E.P. & Sturmbauer, C. Gene coexpression networks reveal molecular interactions underlying cichlid jaw modularity. BMC Ecol Evo 21, 62 (2021). https://doi.org/10.1186/s12862-021-01787-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-021-01787-9