
Abstract
Aminopeptidase N (APN) or CD13 is a conserved type II integral membrane zinc-dependent metalloprotease in the M1 family of ectoenzymes. APN is abundant in the kidneys and central nervous system. Identified substrates include Angiotensin III (Ang III); neuropeptides, including enkephalins and endorphins; and homones, including kallidan and somatostatin. It is developmentally expressed, a myelomonocytic marker for leukemias, and a receptor for coronovirus. There is evolving support for APN in the regulation of arterial blood pressure and the pathogenesis of hypertension. In rodent strains, intracerebraventricular (i.c.v.) infusions of APN reduces, while inhibitors of APN activity have a pressor effect on blood pressure. Dysregulation of central APN has been linked to the pathogenesis of hypertension in the spontaneously hypertensive rat. There is evidence that renal tubule APN inhibits Na flux and plays a mechanistic role in salt-adaptation. A functional polymorphism of the ANP gene has been identified in the Dahl salt-sensitive rat. Signaling by APN impacting on blood pressure is likely mediated by regulation of the metabolism of Ang III to Ang IV. Whether APN regulates arterial blood pressure in humans or is a therapeutic target for hypertension are subjects for future exploration.
Similar content being viewed by others

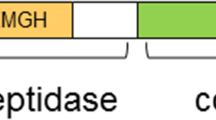
Aminopeptidase N (APN), which is also known as leucine aminopeptidase, alanyl aminopeptidase (AlaAP), aminopeptidase M, or CD13, is a conserved type II integral membrane zinc-dependent metalloprotease. This peptidase belongs to the M1 family of ectoenzymes [1–3]. Membrane-associated APN/CD13 is a non-covalently bonded homodimer with a mass of 160 kDa. Each subunit consists of 967 amino acids and contains a small (8–10 amino acid) N-terminal segment in the cytosol, a single transmembrane helical spanning domain, and an extracellular C-terminus (review [4]; Fig. 1). The APN protein is highly conserved in rabbit, rat, and human species with the differences occurring primarily in the stalk region immediately downstream from the transmembrane domain. APN contains a Zn2+-binding HELAH motif. The protein is highly glycosylated in vivo, and evidence suggests that unique asparagine-linked sugar chains contain a bisecting N-acetylglucosamine residue [5, 6]. Hemoporphyrins and Zn2+ inhibit APN activity [7, 8]. A soluble form has been detected in both plasma and urine [9–11].

Structure of aminopeptidase N. Type II homodimeric membrane with 160 kDa subunits, each with 10 glycosylation sites and 1 HELAH domain. The C-terminal domain is alleged to bind substrate and the N-terminal domain is the active center (reprinted from Ref. [31] and permission obtained from Elsevier)
APN preferentially cleaves neutral amino acids from the amino terminus of oligopeptides. In biological systems, primary peptide substrates identified include neuropeptides such as Met- and Leu-enkephalins [12], neurokinin A, Met-lys-bradykinin, and endorphins [13–15]; hormones, including somatostatin [16], kallidin [17, 18], and collagen type IV [19]. In addition, APN metabolizes the heptapeptide angiotensin 2–8 (Ang III) to the hexapeptide angiotensin 3–8 (Ang IV) via cleavage of the N-terminal arginine, suggesting that this protease can regulate both systemic and tissue renin-angiotensin-aldosterone (RAS) signaling [16, 20, 21].
APN is widely expressed. In the brain, APN is most abundant in the meninges, choroid plexus, pineal gland, paraventricular nucleus, and pituitary gland, but has also been detected in the cortex, caudate-putamen, subthalamic nucleus, central periaqueductal gray, and thalamus as well as in the dorsal and ventral horn of the spinal cord, hippocampus, nucleus accumbens, substantia nigra, hypothalamus (dorsomedial and ventromedial nuclei), raphe nucleus, pontine nucleus, inferior olive, and in the granular layer of the cerebellum [22–27]. In the kidney, APN is concentrated in renal epithelial cells along the apical side of the brush border membranes, mesangial cells, and glomeruli [5, 28, 29].
APN/CD13 has been established as a myelomonocytic marker in leukemia typing [2, 19, 30, 31], as a receptor for human coronavirus 229E and cytomegalovirus [32–34], as a mediator of both inflammation and cell invasion (review [31]), and as a regulator of analgesia via metabolism of endorphins and enkephalins [35–38].
In this review, the role of APN in the regulation of blood pressure and the pathogenesis of hypertension is examined. Evidence for central and renal functions of APN is the focus.
Central APN in blood pressure regulation
A variety of studies in rats have provided evidence that central APN signaling regulates arterial blood pressure (review [39]). Intracerebraventricular (i.c.v.) infusions of the APN inhibitors bestatin and amistatin increase arterial blood pressure and are dipsogenic in normotensive (WKY and Sprague–Dawley) and hypertensive (SHR) rat strains [40–42]. In complementary studies, i.c.v. infusion of aminopeptidase M reduces arterial blood pressure in both the SHR and WKY rat strains [43, 44]. In microinfusion studies, APN placed explicitly into the paraventricular nucleus of the hypothalmous (PVN) decreases arterial blood pressure in both SHR and WKY rats, indicating that APN may specifically function to regulate blood pressure in the PVN [45, 46].
There is evidence that APN signaling in the brain is through the metabolism of Ang III to Ang IV. Ang III, versus Ang II, is the major active angiotensin metabolite in blood pressure control in the brain and i.c.v. infusion of Ang III causes a pressor response that is augmented by APN inhibitors bestatin and amistatin [47–52]. In further support of angiotensin metabolism mediating signaling by APN is the finding that (i) sarthran, a non-selective angiotensin antagonist blocks the pressor response to i.c.v. infusion of APN inhibitors [46, 51, 53] and (ii) the decrease in arterial blood pressure with i.c.v. infusion of aminopeptidase M in the SHR rats is prevented by an angiotensin type 1 receptor (AT1), as opposed to the angiotensin type 2 receptor (AT2) [54–56].
Other studies have indicated that the mechanism for central APN regulation of blood pressure may also involve secondary signaling independent of the metabolism of Ang III. Pretreatment with hexamethonium and/or an arginine vasopressin (AVP) receptor antagonist attenuates the hypotensive response to infusion of APN into the PVN in normotensive and hypertensive rat strains and i.v.c. administration of the APN inhibitor PC18 enhances vasopressin release, suggesting the involvement of signaling by vasopressin [45, 57]. The impact on arterial blood pressure of the metabolism of kinin (to bradykinin) and somatostatin by APN is not known.
A defect in receptor-mediated aminopeptidases has been postulated in the SHR rat [43, 55, 58]. Infusion of APN reduces arterial blood pressure in the SHR rat to a greater extent than in WKY rats [43]. There is reduced Ang II and Ang III metabolism, reflected in prolonged half-lives of Ang II and III and increased sensitivity to the i.c.v. injection of Ang II and Ang III, in the SHR versus WKY rat strains [59–62]. However, a reduction in metabolism of Ang III to Ang IV by APN does not fully account for the effect of APN infusion since similar decreases in central Ang II and Ang III levels have been observed in WKY and SHR rats following APN infusion even though blood pressure decreases to a greater extent in the SHR rat [43].
Renal APN in blood pressure regulation
There are several lines of evidence to suggest that renal APN regulates tubule salt handling, influences blood pressure, and plays a pathogenic role in hypertension.
First, renal tubule APN decreases basolateral Na/K ATPase, which is a preeminent Na+ transporter in renal tubule epithelial cells, and therefore most likely regulates Na+ flux [63]. We have linked regulation of Na/K ATPase to the production of Ang IV by APN [63]. Ang IV has been reported to promote natriuresis [64]. In the kidney, the primary Ang IV receptor (AT4) has been identified as an insulin-regulated membrane aminopeptidase (IRAP) [65, 66], and is present in human proximal and distal tubules, vascular smooth muscle, and endothelial cells [64, 67, 68]. Ang IV, via activation of AT4, stimulates mitogen-activated protein kinases (MAPKs), including p38 kinase and extracellular-signal-regulated-kinase (Erk)1/2; voltage-sensitive calcium channels; and tyrosine phosphorylation of focal adhesion kinase (FAK) and paxillin [67, 69–71]. Each of these regulates smooth muscle cell physiology and has been directly or indirectly related to blood pressure [72–75]. Infusion of Ang IV into the renal artery increases urinary Na+ excretion and cortical blood flow [64, 76]. Ang IV inhibits transcellular Na+ transport (measured by proximal tubule O2 consumption rates) in rat proximal tubules [77] and Na+ flux (reflected in increased Na+ uptake) [67] in human kidney (HK-2) cells. We linked Ang IV/AT4 to regulation of basolateral Na/K ATPase by APN by showing that siRNA to AT4 blocked the decrease in basolateral Na/K ATPase activity and abundance associated with over-expression of APN in LLCPK-1 cells [63]. We speculate that reducing Ang III levels may also be a mechanism by which APN reduces arterial blood pressure and induces a natriuresis since Ang III, like Ang II, is a vasoconstrictor and pressor agent [78–80]. In support of APN reducing Ang III signaling is the finding that inhibitors of APN increased AT2-mediated natriuresis and sensitivity to Ang III in AT1 receptor-blocked mice [56]. Thus, there is the indication that APN may promote natriuresis both by increasing Ang IV/AT4 and reducing Ang III signaling.
Secondly, renal APN expression may be regulated by salt and reduced in hypertension. We have reported renal APN abundance and activity are increased by a high salt diet (8% vs. 0.8% NaCl) in salt-resistant rat strains, i.e., Dahl salt-resistant (SR) rats and Sprague–Dawley rats [81]. Consistent with increased APN activity is a reduction in renal Ang II/Ang III and increase in AT4 binding in SR rat strains in response to a salt-challenge [82, 83]. In contrast, renal APN does not increase in the Dahl salt-sensitive (SS) rat challenged with a high salt diet. Together, these results raise the possibility that renal APN regulates normal adaptation to high salt conditions and that dysregulation of APN contributes to salt sensitivity in the Dahl SS rat.
In a rodent nitric oxide inhibition model of hypertension, i.e., rats treated with N(omega)-nitro-l-arginine methyl ester (L-NAME), less renal, aortic, and serum APN activity than in controls has been reported [84, 85]. Although further studies are needed to determine the significance of APN in this model, a decrease in APN activity could be linked to the reported increase in the pressor response to Ang II by decreasing Ang III metabolism [86].
Thirdly, the APN gene maps to a reported quantitative trait locus (QTL) for hypertension in Dahl SS X Dahl SR rats on chromosome 1 and contains in the Dahl SS rat a functional single nucleotide polymorphism (SNP) four nucleotides upstream from a CCAAT/enhancer binding protein motif (CEBP-cis element; nucleotides −2256 to −2267) that is associated with CEBP binding and increased promotor activity of the 5′ flanking region [87]. Although this SNP is intriguing, transgenic and fine-mapping studies are needed to establish the relationship between the APN gene and hypertension in the Dahl rat.
Whether there are polymorphisms of the ANP gene in other models of genetic hypertension and, if so, whether they are linked to hypertension remains to be investigated. In the Milan Hypertensive (MHS) rat strain, greater renal cell membrane aminopeptidase M abundance than in the corresponding normotensive strain, i.e., Milan Normotensive (MNS), was identified in comparisons of 2D electrophoresis patterns of the two contrasting strains of rat [88]. However, Salardi et al. [88] showed that this is not genetically linked to hypertension in the MHS strain by generating recombinant strains in which the traits of hypertension and faster sodium transport do not cosegregate with the difference in expression or activity of APN.
APN and human arterial hypertension
Genetic association studies have raised the prospect that APN may be linked to human arterial hypertension. The human APN gene contains 20 exons and maps to chromosome 15q25–26 [89]. Thirty-three polymorphisms of adipocyte-derived APN have been identified from 48 unrelated Japanese individuals [90]. Two of the eight missense polymorphisms (Ile276Met and Lys528Arg) are reported to have a significant association with arterial blood pressure. Lys528Arg has been associated with essential and non-modulating hypertension [90, 91]. A reduction in the extent of regression of left ventricular hypertrophy following treatment with irbesartan, an AT1 receptor antagonist, has also been reported in patients with this polymorphism [92]. The Lys528Arg polymorphism has been shown to reduce enzymatic activity of APN by altering the tertiary structure and, in turn, reducing substrate binding Goto et al. [93]. Further analysis and examination of hypertensive subpopulations are needed to determine the significance of these mutations.
The future
As with most basic studies and animal work, the ultimate goal is to transition from experimental results to human disease. In vivo measurements of APN and angiotensin metabolites in human hypertensive patients are paramount. Increased urinary levels of APN have been linked to human renal transplant rejection [94] and renal cell carcinoma [95]; however, the significance of this increase is not clear with respect to blood pressure. Investigation of the role of aminopeptidases in general and of APN in human hypertension is needed.
The anti-hypertensive potential of inhibitors of both brain aspartyl aminopeptidase, which converts Ang I to Angiotensin 2–10 Aminopeptidase A (GluAP) and metabolizes Ang II to Ang III, have been evaluated previously [39]. Significant development of APN inhibitors as therapeutic agents to target a variety of malignancies and inflammatory diseases has occurred [2]. Studies to evaluate APN as a therapeutic target for arterial hypertension, specifically focusing on defined subsets, e.g., salt-sensitive patients and these agents, may provide insight. Because APN signals through the angiotensin metabolites, Ang III, Ang IV, and AT4, these also may be therapeutic agents and targets.
References
Luan Y, Xu W (2007) The structure and main functions of aminopeptidase N. Curr Med Chem 14:639–647
Bauvois B, Dauzonne D (2005) Aminopeptidase-N/CD13 (EC 3.4.11.2) inhibitors: chemistry, biological evaluations, and therapeutic prospects. Med Res Rev 26:88–130
Tsujimoto M, Hattori A (2005) The oxytocinase subfamily of M1 aminopeptidases. Biochim Biophys Acta 1751:9–18
Sjostrom H, Noren O, Olsen J (2000) Structure and function of aminopeptidase N. Adv Exp Med Biol 477:25–34
Landry C, Santagata P, Bawab W et al (1993) Characterization of neutral endopeptidase 24.11 in dog glomeruli. Biochem J 291:773–779
Yamashita K, Tachibana Y, Matsuda Y, Katunuma N, Kochibe N, Kobata A (1988) Comparative studies of the sugar chains of aminopeptidase N and dipeptidylpeptidase IV purified from rat kidney brush-border membrane. Biochemistry 27:5565–5573
Garreau I, Chansel D, Vandermeersch S, Fruitier I, Piot JM, Ardaillou R (1998) Hemorphins inhibit angiotensin IV binding and interact with aminopeptidase N. Peptides 19:1339–1348
Ishii K, Usui S, Sugimura Y et al (2001) Aminopeptidase N regulated by zinc in human prostate participates in tumor cell invasion. Int J Cancer 92:49–54
Jung K, Pergande M, Wischke UW (1984) Characterization of particulate and soluble variants of the brush-border enzymes alanine aminopeptidase, alkaline phosphatase and gamma-glutamyltransferase in human urine. Biomed Biochim Acta 43:1357–1364
Kawai M, Otake Y, Hara Y (2003) High-molecular-mass isoform of aminopeptidase N/CD13 in serum from cholestatic patients. Clin Chim Acta 330:141–149
van Hensbergen Y, Broxterman HJ, Hanemaaijer R et al (2002) Soluble aminopeptidase N/CD13 in malignant and nonmalignant effusions and intratumoral fluid. Clin Cancer Res 8:3747–3754
Matsas R, Stephenson SL, Hryszko J, Kenny AJ, Turner AJ (1985) The metabolism of neuropeptides. Phase separation of synaptic membrane preparations with Triton X-114 reveals the presence of aminopeptidase N. Biochem J 231:445–449
Giros B, Gros C, Solhonne B, Schwartz JC (1986) Characterization of aminopeptidases responsible for inactivating endogenous (Met5) enkephalin in brain slices using peptidase inhibitors and anti-aminopeptidase M antibodies. Mol Pharmacol 29:281–287
Miller BC, Thiele DL, Hersh LB, Cottam GL (1994) Methionine enkephalin is hydrolyzed by aminopeptidase N on CD4+ and CD8+ spleen T cells. Arch Biochem Biophys 311:174–179
Lucius R, Sievers J, Mentlein R (1995) Enkephalin metabolism by microglial aminopeptidase N (CD13). J Neurochem 64:1841–1847
Ward PE, Benter IF, Dick L, Wilk S (1990) Metabolism of vasoactive peptides by plasma and purified renal aminopeptidase M. Biochem Pharmacol 40:1725–1732
Palmieri FE, Petrelli JJ, Ward PE (1985) Vascular, plasma membrane aminopeptidase M. Metabolism of vasoactive peptides. Biochem Pharmacol 34:2309–2317
Kokkonen JO, Kuoppala A, Saarinen J, Lindstedt KA, Kovanen PT (1999) Kallidin- and bradykinin-degrading pathways in human heart: degradation of kallidin by aminopeptidase M-like activity and bradykinin by neutral endopeptidase. Circulation 99:1984–1990
Saiki I, Fujii H, Yoneda J et al (1993) Role of aminopeptidase N (CD13) in tumor-cell invasion and extracellular matrix degradation. Int J Cancer 54:137–143
Chansel D, Czekalski S, Vandermeersch S, Ruffet E, Fournie-Zaluski MC, Ardaillou R (1998) Characterization of angiotensin IV-degrading enzymes and receptors on rat mesangial cells. Am J Physiol 275:F535–F542
Paul M, Poyan MA, Kreutz R (2006) Physiology of local renin-angiotensin systems. Physiol Rev 86:747–803
Hersh LB, Aboukhair N, Watson S (1987) Immunohistochemical localization of aminopeptidase M in rat brain and periphery: relationship of enzyme localization and enkephalin metabolism. Peptides 8:523–532
Gros C, Solhonne B, Giros B et al (1986) Immunohistochemical and subcellular studies of aminopeptidase M localization in rat brain: microvessels and synaptic membranes. NIDA Res Monogr 75:303–306
Noble F, Banisadr G, Jardinaud F et al (2001) First discrete autoradiographic distribution of aminopeptidase N in various structures of rat brain and spinal cord using the selective iodinated inhibitor [125I]RB 129. Neuroscience 105:479–488
Schnabel R, Bernstein HG, Luppa H, Lojda Z, Barth A (1992) Aminopeptidases in the circumventricular organs of the mouse brain: a histochemical study. Neuroscience 47:431–438
Barnes K, Bourne A, Cook PA, Turner AJ, Kenny AJ (1991) Membrane peptidases in the peripheral nervous system of the pig: their localization by immunohistochemistry at light and electron microscopic levels. Neuroscience 44:245–261
Solhonne B, Gros C, Pollard H, Schwartz JC (1987) Major localization of aminopeptidase M in rat brain microvessels. Neuroscience 22:225–232
Stefanovic V, Vlahovic P, Mitic-Zlatkovic M (1998) Epidermal growth factor upregulates aminopeptidase N and 5′-nucleotidase in human glomerular mesangial cells. Kidney Blood Press Res 21:310–316
Stange T, Kettmann U, Holzhausen HJ (1996) Immunoelectron microscopic single and double labelling of aminopeptidase N (CD 13) and dipeptidyl peptidase IV (CD 26). Acta Histochem 98:323–331
Pasqualini R, Koivunen E, Kain R et al (2000) Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res 60:722–727
Riemann D, Kehlen A, Langner J (1999) CD13—not just a marker in leukemia typing. Immunol Today 20:83–88
Lachance C, Arbour N, Cashman NR, Talbot PJ (1998) Involvement of aminopeptidase N (CD13) in infection of human neural cells by human coronavirus 229E. J Virol 72:6511–6519
Lassnig C, Sanchez CM, Egerbacher M et al (2005) Development of a transgenic mouse model susceptible to human coronavirus 229E. Proc Natl Acad Sci USA 102:8275–8280
Soderberg C, Sumitran-Karuppan S, Ljungman P, Moller E (1996) CD13-specific autoimmunity in cytomegalovirus-infected immunocompromised patients. Transplantation 61:594–600
Matsas R, Stephenson SL, Hryszko J, Kenny AJ, Turner AJ (1985) The metabolism of neuropeptides. Phase separation of synaptic membrane preparations with Triton X-114 reveals the presence of aminopeptidase N. Biochem J 231:445–449
Noble F, Roques BP (2007) Protection of endogenous enkephalin catabolism as natural approach to novel analgesic and antidepressant drugs. Expert Opin Ther Targets 11:145–159
Fischer HS, Zernig G, Hauser KF, Gerard C, Hersh LB, Saria A (2002) Neutral endopeptidase knockout induces hyperalgesia in a model of visceral pain, an effect related to bradykinin and nitric oxide. J Mol Neurosci 18:129–134
Chen H, Noble F, Coric P, Fournie-Zaluski MC, Roques BP (1998) Aminophosphinic inhibitors as transition state analogues of enkephalin-degrading enzymes: a class of central analgesics. Proc Natl Acad Sci USA 95:12028–12033
Banegas I, Prieto I, Vives F et al (2006) Brain aminopeptidases and hypertension. J Renin Angiotensin Aldosterone Syst 7:129–134
Quirk WS, Harding JW, Wright JW (1987) Amastatin and bestatin-induced dipsogenicity in the Sprague-Dawley rat. Brain Res Bull 19:145–147
Harding JW, Felix D (1987) The effects of the aminopeptidase inhibitors amastatin and bestatin on angiotensin-evoked neuronal activity in rat brain. Brain Res 424:299–304
Wilson WL, Roques BP, Llorens-Cortes C, Speth RC, Harding JW, Wright JW (2005) Roles of brain angiotensins II and III in thirst and sodium appetite. Brain Res 1060:108–117
Wright JW, Jensen LL, Cushing LL, Harding JW (1989) Leucine aminopeptidase M-induced reductions in blood pressure in spontaneously hypertensive rats. Hypertension 13:910–915
Wright JW, Amir HZ, Murray CE et al (1991) Use of aminopeptidase M as a hypotensive agent in spontaneously hypertensive rats. Brain Res Bull 27:545–551
Batt CM, Jensen LL, Harding JW, Wright JW (1996) Microinfusion of aminopeptidase M into the paraventricular nucleus of the hypothalamus in normotensive and hypertensive rats. Brain Res Bull 39:235–240
Jensen LL, Harding JW, Wright JW (1989) Increased blood pressure induced by central application of aminopeptidase inhibitors is angiotensinergic-dependent in normotensive and hypertensive rat strains. Brain Res 490:48–55
Dewey AL, Wright JW, Hanesworth JM, Harding JW (1988) Effects of aminopeptidase inhibition on the half-lives of [125I]angiotensins in the cerebroventricles of the rat. Brain Res 448:369–372
Wright JW, Quirk WS, Hanesworth JM, Harding JW (1988) Influence of aminopeptidase inhibitors on brain angiotensin metabolism and drinking in rats. Brain Res 441:215–220
Sullivan MJ, Harding JW, Wright JW (1988) Differential effects of aminopeptidase inhibitors on angiotensin-induced pressor responses. Brain Res 456:249–253
Abhold RH, Sullivan MJ, Wright JW, Harding JW (1987) Binding, degradation and pressor activity of angiotensins II and III after aminopeptidase inhibition with amastatin and bestatin. J Pharmacol Exp Ther 242:957–962
Wright JW, Roberts KA, Cook VI, Murray CE, Sardinia MF, Harding JW (1990) Intracerebroventricularly infused [D-Arg1]angiotensin III, is superior to [D-Asp1]angiotensin II, as a pressor agent in rats. Brain Res 514:5–10
Wright JW, Tamura-Myers E, Wilson WL et al (2003) Conversion of brain angiotensin II to angiotensin III is critical for pressor response in rats. Am J Physiol Regul Integr Comp Physiol 284:R725–R733
Batt CM, Klein EW, Harding JW, Wright JW (1988) Pressor responses to amastatin, bestatin and Plummer’s inhibitors are suppressed by pretreatment with the angiotensin receptor antagonist sarthran. Brain Res Bull 21:731–735
Wright JW, Krebs LT, Stobb JW, Harding JW (1995) The angiotensin IV system: functional implications. Front Neuroendocrinol 16:23–52
Wright JW, Hamilton TA, Harding JW (1995) Anomalous effects of losartan on aminopeptidase-induced reductions of blood pressure in SHR. Brain Res Bull 36:169–174
Padia SH, Kemp BA, Howell NL et al (2007) Intrarenal aminopeptidase N inhibition augments natriuretic responses to angiotensin III in angiotensin type 1 receptor-blocked rats. Hypertension 49:625–630
Reaux A, de Mota N, Zini S et al (1999) PC18, a specific aminopeptidase N inhibitor, induces vasopressin release by increasing the half-life of brain angiotensin III. Neuroendocrinology 69:370–376
Wright JW, Sullivan MJ, Quirk WS, Batt CM, Harding JW (1987) Heightened blood pressure and drinking responsiveness to intracerebroventricularly applied angiotensins in the spontaneously hypertensive rat. Brain Res 420:289–294
Wright JW, Sullivan MJ, Bredl CR, Hanesworth JM, Cushing LL, Harding JW (1987) Delayed cerebroventricular metabolism of [125I]angiotensins in the spontaneously hypertensive rat. J Neurochem 49:651–654
Harding JW, Yoshida MS, Dilts RP, Woods TM, Wright JW (1986) Cerebroventricular and intravascular metabolism of [125I]angiotensins in rat. J Neurochem 46:1292–1297
Wright JW, Sullivan MJ, Harding JW (1985) Dysfunction of central angiotensinergic aminopeptidase activity in spontaneously hypertensive rats. Neurosci Lett 61:351–356
Batt CM, Jensen LL, Hanesworth JM, Harding JW, Wright JW (1990) Intracerebroventricularly applied peptidase inhibitors increase endogenous angiotensin levels. Brain Res 529:126–130
Kotlo K, Shukla S, Tawar U, Skidgel RA, Danziger RS (2007) Aminopeptidase N reduces basolateral Na/K ATPase in proximal tubule cells. Amer J Physiol Renal Physiol 293(4):F1047–F1053
Hamilton TA, Handa RK, Harding JW, Wright JW (2001) A role for the angiotensin IV/AT4 system in mediating natriuresis in the rat. Peptides 22:935–944
Albiston AL, McDowall SG, Matsacos D et al (2001) Evidence that the angiotensin IV (AT (4)) receptor is the enzyme insulin-regulated aminopeptidase. J Biol Chem 276:48623–48626
Chai SY, Fernando R, Peck G et al (2004) The angiotensin IV/AT4 receptor. Cell Mol Life Sci 61:2728–2737
Handa RK (2001) Characterization and signaling of the AT (4) receptor in human proximal tubule epithelial (HK-2) cells. J Am Soc Nephrol 12:440–449
Czekalski S, Chansel D, Vandermeersch S, Ronco P, Ardaillou R (1996) Evidence for angiotensin IV receptors in human collecting duct cells. Kidney Int 50:1125–1131
Chen JK, Zimpelmann J, Harris RC, Burns KD (2001) Angiotensin IV induces tyrosine phosphorylation of focal adhesion kinase and paxillin in proximal tubule cells. Am J Physiol Renal Physiol 280:F980–F988
Dulin N, Madhun ZT, Chang CH, Berti-Mattera L, Dickens D, Douglas JG (1995) Angiotensin IV receptors and signaling in opossum kidney cells. Am J Physiol 269:F644–F652
Li YD, Block ER, Patel JM (2002) Activation of multiple signaling modules is critical in angiotensin IV-induced lung endothelial cell proliferation. Am J Physiol Lung Cell Mol Physiol 283:L707–L716
Lee CK, Kim J, Won KJ et al (2006) Phorbol ester-induced contraction through p38 mitogen-activated protein kinase is diminished in aortas from DOCA-salt hypertensive rats. Arch Pharm Res 29:1024–1031
Kim J, Lee YR, Lee CH et al (2005) Mitogen-activated protein kinase contributes to elevated basal tone in aortic smooth muscle from hypertensive rats. Eur J Pharmacol 514:209–215
Xu Q, Liu Y, Gorospe M, Udelsman R, Holbrook NJ (1996) Acute hypertension activates mitogen-activated protein kinases in arterial wall. J Clin Invest 97:508–514
Blanc A, Pandey NR, Srivastava AK (2003) Synchronous activation of ERK 1/2, p38mapk and PKB/Akt signaling by H2O2 in vascular smooth muscle cells: potential involvement in vascular disease (review). Int J Mol Med 11:229–234
Coleman JK, Krebs LT, Hamilton TA et al (1998) Autoradiographic identification of kidney angiotensin IV binding sites and angiotensin IV-induced renal cortical blood flow changes in rats. Peptides 19:269–277
Handa RK, Krebs LT, Harding JW, Handa SE (1998) Angiotensin IV AT4-receptor system in the rat kidney. Am J Physiol 274:F290–F299
Johnson JA, Dostal DE, Elsberry-Gonder A (1990) Angiotensin III and pressor responsiveness in 3-day renal artery stenosis rabbits. Am J Physiol 258:H540–H548
Suzuki S, Doi Y, Aoi W, Kuramochi M, Hashiba K (1984) Effect of angiotensin III on blood pressure, renin-angiotensin-aldosterone system in normal and hypertensive subjects. Jpn Heart J 25:75–85
Ardaillou R, Chansel D (1997) Synthesis and effects of active fragments of angiotensin II. Kidney Int 52:1458–1468
Farjah M, Washington TL, Roxas BP, Geenen DL, Danziger RS (2004) Dietary NaCl regulates renal aminopeptidase N: relevance to hypertension in the Dahl rat. Hypertension 43:282–285
Meng QC, Durand J, Chen YF, Oparil S (1995) Effects of dietary salt on angiotensin peptides in kidney. J Am Soc Nephrol 6:1209–1215
Grove KL, Deschepper CF (1999) High salt intake differentially regulates kidney angiotensin IV AT4 receptors in Wistar-Kyoto and spontaneously hypertensive rats. Life Sci 64:1811–1818
Vlahovic P, Cvetkovic T, Nikolic J, Sokolovic D (2007) Ethanol inhibitory effect on rat kidney brush border aminopeptidases. Nephron Exp Nephrol 106:e73–e76
Linardi A, Panunto PC, Ferro ES, Hyslop S (2004) Peptidase activities in rats treated chronically with N (omega)-nitro-L-arginine methyl ester (L-NAME). Biochem Pharmacol 68:205–214
Hu L, Sealey JE, Chen R et al (2004) Nitric oxide synthase inhibition accelerates the pressor response to low-dose angiotensin II, exacerbates target organ damage, and induces renin escape. Am J Hypertens 17:395–403
Kotlo K, Hughes DE, Herrera VL et al (2007) Functional polymorphism of the Anpep gene increases promoter activity in the Dahl salt-resistant rat promoter. Hypertension 49:1–6
Salardi S, Falchetto R, Troffa C et al (1993) Relationships among alterations in renal membrane sodium transport, renin and aminopeptidase M activities in genetic hypertension. Biochim Biophys Acta 1182(1):22–29
Lerche C, Vogel LK, Shapiro LH, Noren O, Sjostrom H (1996) Human aminopeptidase N is encoded by 20 exons. Mamm Genome 7:712–713
Yamamoto N, Nakayama J, Yamakawa-Kobayashi K, Hamaguchi H, Miyazaki R, Arinami T (2002) Identification of 33 polymorphisms in the adipocyte-derived leucine aminopeptidase (ALAP) gene and possible association with hypertension. Hum Mutat JID - 9215429 19:251–257
Williams JS, Raji A, Williams GH, Conlin PR (2006) Non-modulating hypertension is associated with insulin resistance and the Lys528Arg variant human adipocyte-derived leucine aminopeptidase. Hypertension 48(4):e33
Hallberg P, Lind L, Michaelsson K et al (2003) Adipocyte-derived leucine aminopeptidase genotype and response to antihypertensive therapy. BMC Cardiovasc Disord 3:11. Epub: 2003 Sep 18: 11
Goto Y, Hattori A, Ishii Y, Tsujimoto M (2006) Reduced activity of the hypertension-associated Lys528Arg mutant of human adipocyte-derived leucine aminopeptidase (A-LAP)/ER-aminopeptidase-1. FEBS Lett 580:1833–1838
Sri KK, Kirubakaran MG, Pandey AP, Kanagasabapathy AS (1985) Urinary N-acetyl-beta-D-glucosaminidase and aminopeptidase N in the diagnosis of graft rejection after live donor renal transplantation. Clin Chim Acta 150:69–85
Kitamura Y, Watanabe M, Komatsubara S, Sakata Y (1990) Urinary excretion of glycine.prolile dipeptidile aminopeptidase, N-acetyl-beta-D-glucosaminidase, alanine aminopeptidase and low molecular protein in patients with renal cell carcinoma. Hinyokika Kiyo 36:535–539
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Danziger, R.S. Aminopeptidase N in arterial hypertension. Heart Fail Rev 13, 293–298 (2008). https://doi.org/10.1007/s10741-007-9061-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-007-9061-y