
Abstract
To assess the genetic diversity of the primary Qingke cultivars and establish their unique genetic profiles, 837 barley simple sequence repeat (SSR) primers were screened across 12 cultivars. The selection process involved the utilization of polyacrylamide gel electrophoresis and capillary electrophoresis technology, to identify primers exhibiting desirable characteristics, such as polymorphism, stability, and reproducibility. Subsequently, we analyzed the genetic diversity of the primary Qingke cultivars to for DNA fingerprints. A total of 18 pairs of SSR markers were selected as the optimal markers for constructing fingerprints of major Qingke cultivars. These included 83 observed alleles (Na), ranging from there to 11, with an average of 4.61 per pair. Notably, Bmag0496 and Scssr04163 exhibited higher allelic diversity, with 11 and 8 loci, respectively. The polymorphism information content (PIC) ranged from 0.36 to 0.74, with an average of 0.52. The expected heterozygosity (He) ranged from 0.4031 to 0.7682, with an average of 0.59, and the observed heterozygosity (Ho) varied between 0.13 and 0.67, with an average of 0.32. The outcomes obtained through phylogenetic tree analysis, population structure assessment and principal component analysis demonstrated that the primary Qingke cultivars could be classified into three distinct groups: group I primarily originated from Xizang and Qinghai provinces; group II mainly consisted of cultivars from Yunnan and Heilongjiang provinces; and group III predominantly comprised cultivars originating from Qinghai and Gansu provinces. Interestingly, the Sichuan cultivars were distributed across all three groups without any clear tendency toward a specific cluster or subgroup. These findings indicated that the genetic distance among Qingke cultivars was significantly correlated with geographic location but not exclusively determined by it. The construction of DNA fingerprints for the primary Qingke cultivars used these identified sets of SSR primers (18 pairs) laid a solid foundation for cultivar identification, conservation and utilization efforts related to this crop.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Qingke (Hordeum vulgare L. var. nudum Hook. f.), also known as naked barley or hulless barley and belongs to the barley genus in the family Gramineae, is a variety of barley in which the inner lemma of the grain is separated from the caryopsis at maturity, and the grain is exposed (Yao et al. 2021; Liang et al. 2012). Qingke is rich in β-glucan, dietary fiber, trace elements, and other nutrients and has health functions, such as chronic disease prevention, including diabetes, blood lipid reduction, diabetes prevention, and anti-tumor properties (Lyu et al. 2022). Owing to its short reproductive cycle, cold and drought tolerance is necessary to adapt to the extreme environment of the plateau (Lyu et al. 2022). Qingke is mainly distributed in five provinces and regions, namely Xizang, Qinghai, Gansu, Sichuan, and Yunnan (Chen et al. 2022), with a planting area of more than 270,000 hectares and an annual production of more than 1 million tons in China (Guo et al. 2020).
It is well known that the molecular identification of cultivars is a key step in a plant improvement strategy. Qingke is mainly planted on the Qinghai–Xizang Plateau (Chen et al. 2023). In the process of breeding new cultivars, cultivars within the region crossbreed with each other so that the parents and core germplasm containing excellent genes are repeatedly utilized. Thus, the genetic distance is becoming increasingly closer, reducing the genetic diversity. The similarity between new cultivars has increased, and the identification of genetic material in close proximity is sometimes difficult (Mériam et al. 2018). Cultivar identification is an effective tool for variety rights protection. With the development of molecular biology, the use of molecular marker technology to construct varietal DNA fingerprints to identify species has emerged as a more rapid, simple, economical, and reliable identification method than morphological identification (Mériam et al. 2018; Liang et al. 2023). To analyze the genetic diversity of existing primary Qingke cultivars, it is necessary to establish a Qingke DNA fingerprint library. Currently, simple sequence repeats or microsatellites (SSRs) method used in DNA fingerprints, has the advantages of high polymorphism, stability, good reproducibility, and mature technology (Sharan et al. 2024; Ilyas et al. 2023). The main methods for detecting SSR-labeled fragments are polyacrylamide gel electrophoresis (PAGE) and capillary electrophoresis (MCE) (Elumalai and Srinivasan 2023). They have been successfully applied to construct fingerprint profiles in sweet potato (Luo et al. 2023), Chimonanthus (Li et al. 2023), azuki bean (Vigna angularis), mung bean (Vigna radiata L.) (Zhao et al. 2022), wheat (Türkoğlu et al. 2023), and Amomum villosum (Li et al. 2022). However, this has not yet been reported for Qingke.
This study utilized SSR molecular markers to distinguish 12 Qingke cultivars using the existing 837 pairs of barley primers. A total of 18 pairs of SSR markers were screened and used to analyze the genetic diversity of 63 Qingke primary cultivars. DNA fingerprinting of Qingke cultivars was constructed to provide technical support for rapid and accurate identification and protection of cultivars.
Materials and methods
Plant materials
The 63 Qingke primary cultivars used in this study were bred and preserved by Academy of Agricultural and Forestry Sciences, Qinghai University (Table 1). Nine cultivars of them were from Sichuan 20 cultivars from Qinghai, 17 cultivars from Xizang, six cultivars from Yunnan, nine cultivars from Gansu, and two cultivars from Heilongjiang. Three young leaves were collected from each cultivars, frozen in liquid nitrogen, and stored in a − 80 °C low-temperature freezer.
Methods
DNA extraction
The sample was placed in a 2.0-mL centrifuge tube and 700-μL CTAB extract preheated to 65 °C was added. The tubes were shaken well and incubated in a 65 °C thermostatic water bath for 45–60 min. Every 10 min, the centrifuge tube was removed and cooled to room temperature. After incubation, an equal volume of chloroform: isoamyl alcohol (24:1, V/V) solution was added, and the tubes were shaken gently up and down several times until the liquid was milky white. They were then centrifuged at 12,000 rpm for 10 min. The supernatant was transferred to a new 2.0-mL centrifuge tube, and 1.5 times the volume of precooled anhydrous ethanol was added. After gently mixing, the tubes were centrifuged at 12,000 rpm for 3 min to separate the phases. The tubes were removed from the centrifuge, and the supernatant was discarded. The samples were washed twice with 70% ethanol solution and dried under natural conditions. To dissolve the precipitate, 50 μL 1 × TE buffer (PH 8.0) was added. The quality of the DNA stock solution was tested using 0.8% agarose gel electrophoresis, and the concentration of the DNA stock solution was detected using a nucleic acid concentration meter. The stock solution was diluted to 50–150 ng/μL. The DNA was stored at − 20 °C.
SSR molecular marker screening
In this study, 12 primary Qingke cultivars (Table 1) and 837 pairs rom the barley genome and the literature were used. First, polyacrylamide gel electrophoresis was used to select consistency, stability, clear bands, polymorphism, etc., to co-screen candidate SSR molecular marker primers. Secondly, based on the principles of high efficiency, economy and uniform distribution, marker primers were selected as the preferred markers for authenticity identification of Qingke cultivars. The primers screened initially were examined for polymorphism by capillary electrophoresis. Primers were synthesized by Beijing Qingke Biotechnology Co (Beijing, China).
PCR amplification system
The PCR reaction system volume was 20 μL (Table 2). The PCR amplification program was as follows: 94 °C for 5 min; 94 °C for 30 s, 60–50 °C for 60 s (0.8 °C drop per cycle, total 12 cycles), and 72 °C for 30 s; 23 cycles of 94 °C for 30 s, 50 °C for 45 s, and 72 °C for 30 s; and final 72 °C for 10 min. The amplification products were stored at 4 °C.
Capillary electrophoresis fluorescence detection
Sample preparation
Fluorescence detection by capillary electrophoresis required the use of fluorescently labeled primers. Depending on the size range of the amplified fragments of the allelic variants, SSR primers were combined by labeling with four fluorescent dyes, which needed to be labeled at the 5' end of either the upstream or downstream primer to match the emission and absorption wavelengths of the capillary electrophoresis instrument.
According to the pre-determined combination of the primers, equal volumes of different fluorescent-labeled amplification products of the same combination of primers were obtained separately and mixed well. For genetic analysis, 1 μL of the mixture was added to a 96-well plate. To each well, 0.1 μL of molecular weight internal standard and 8.9 μL of deionized formamide were added. The samples were placed in the PCR instrument to denature at 95 °C for five min. They were removed, immediately placed on ice, cooled for more than 10 min, centrifuged for 10 s, and prepared for use.
Electrophoretic detection
The DNA analyzer (ABI 3730XL) was turned on to check the working conditions of the instrument. Buffer was added, and the glue was filled. A 96-well upper plate with samples was placed on the sample rack base. Data collection software was used following the genetic analyzer manual. The sample table was edited, and the run program was executed. The data were saved.
Data processing
GeneMaker V2.2.0 software was used to create the raw data according to the size of the molecular fragments of the amplification products. PowerMarker 3.25 and Popgen32 software were used to calculate Na, effective alleles (Ne), Shannon’s information index (I), PIC, major allele frequency (MAF), He, and Ho as indicators of the genetic diversity of the 18 pairs of primers. Genetic diversity indexes were calculated for 18 primer pairs using GenAIEx 6.51 and raw data on amplification product size. Nei’s genetic distance between the cultivars was assessed using PowerMarker 3.25, and a grouping tree was constructed using the neighbor-joining (NJ) method and visualized using iTOL (iTOL: Interactive Tree of Life (embl.de)). The population structure was analyzed using Bayesian modeling with Structure 2.3.4 and a Markov chain Monte Carlo (MCMC) approach with preset population groupings (K). Calculating, sampling, and grouping of individuals were based on allele frequencies. The parameter settings were as follows: K value range, 1–10; each K value for 10 independent runs; number of parameter iterations (length of burn in period), 100,000. The optimal K value was calculated on the STRUCTURE HARVESTER (http://taylor0.biology.ucla.edu/struct_harvest/) website. Finally, using the raw data on the size of amplification products, principal co-ordinates analysis (PCoA) was performed on 63 Qingke cultivars using GenAIEx 6.51.
DNA fingerprinting
Raw data (unit bp omitted) were created from the size of molecular fragments of the amplification products of 18 pairs of SSR primers in 63 primary Qingke cultivars using GeneMaker V2.2.0 software. The 18 pairs of SSR polymorphism primers were coded in a fixed order and recorded as 1–18 to read 63 primary Qingke cultivars in the form of allelic markers in the primer amplification of the product size. They were separated by “-” in the middle of the primer number, and site reads with “:” were connected to form a combination of allelic marker configurations. The time of the combination was separated by “/.” The tandem of 18 primers and allele configuration combinations constituted the DNA digital fingerprinting code for the cultivar.
Results
Primer screening
Utilizing the 12 primary Qingke cultivars (remarks in Table 1), we selected stable repeatability and excellent polymorphic primers from 837 SSR primer pairs. Based on the principles of efficiency, economy and simplicity, 18 pairs of SSR primers were selected as preferred markers for the identification of Qingke cultivars (Table 3). In addition, these 18 pairs of SSR primers were evenly distributed on Qingke chromosome 7, and their polymorphism contents varied. The SSR primers were combined into three sets of primers by labeling four fluorescent dyes according to the size range of the amplified fragments of the allelic variants (Table 4).
Analysis of genetic diversity
Fluorescence capillary electrophoresis was performed on 63 primary Qingke cultivars using 18 pairs of SSR primers. These included a total of 83 Na (Table 5). 83 Na uniquely genotype the 63 individuals, indicating that the 18 SSR markers had a high discriminatory ability. The number of Na ranged from three to 11, with an average of 4.61 per pair. Among them, Bmag0496 and GBM1516 had more bit points (11 and seven, respectively). The MAF ranged from 0.37 to 0.75, with a mean value of 0.54. The PIC ranged from 0.36 to 0.74, with a mean value of 0.52. The I value ranged from 0.71 to 1.80, with a mean value of 1.07. The He values ranged from 0.40 to 0.77, and the mean value was 0.59. The Ho values ranged from 0.13 to 0.67, with a mean value of 0.32. The data showed that the selected SSR markers were rich in polymorphism, and the 63 primary Qingke cultivars had high heterozygosity and rich genetic diversity.
Population structure analysis of 63 primary Qingke cultivars
Genetic distances and phylogenetic tree analysis
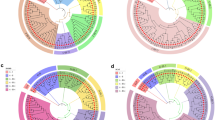
To understand the inter-individual genetic relationships Nei’s genetic distances among 63 primary Qingke cultivars were assessed using PowerMarker 3.25, and a phylogenetic tree was constructed based on the genetic distances using the NJ method (Fig. 1). The results of Nei’s genetic distance showed large genetic differences among 63 primary Qingke cultivars, with an average genetic distance of 0.55. The largest was between Kunlun 13 and Yunke 4, with a genetic distance of 0.94, which showed the most distant kinship. The smallest was between Zangqing 78 and Zangqing 80, with a genetic distance of 0.12, which showed the most closest kinship. The phylogenetic tree divided them into three groups. Group I contained 26 Qingke cultivars, 14 from Xizang, seven from Qinghai, three from Sichuan, and two from Gansu. Group II contained 16 Qingke cultivars, six from Yunnan, three from Sichuan, three from Xizang, two from Heilongjiang, one from Gansu, and one from Qinghai. Group III contained 21 Qingke cultivar, 12 from Qinghai, six from Gansu, and three from Sichuan. Among the 63 primary Qingke cultivars, Qingke from Xizang was mainly distributed in group I. Qingke from Qinghai was mainly distributed in groups III and I. Qingke from Yunnan and Heilongjiang were distributed in group II, and Qingke from Gansu was mainly distributed in group III. On the whole, most of the qualities from the same source were clustered in the same branch, showing close affinities. The results suggested that the geographic location of genetic cultivars had important relevance in cluster analysis. However, this correlation was not absolute due to environmental factors and artificial breeding choices, and the primary Qingke cultivars from Sichuan had three copies in each of the three groups.

Phylogenetic tree of 63 primary Qingke cultivars. Orange, green, and blue-green dots correspond to Groups 1, 2, and 3, respectively. (Color figure online)
Structure analysis
To further assess the genetic characterization of primary Qingke cultivars, a population structure analysis was carried out in this experiment using STRUCTURE 2.3.4 and Evanno et al. (2005) of STRUCTURE HARVESTER (Fig. 2). The results showed that deltaK (ΔK) had a maximum value when K = three, indicating that the 63 primary Qingke cultivars could be categorized into three groups (Fig. 2A). As shown in Fig. 2B, when K = two, group II was divided into groups I and III, forming only two groups. When K = four, group I was divided into two groups, resulting in the formation of 4 groups. When K = three, in group I, the Q value was greater than 0.8 with 11 materials, accounting for 44% of such groups. Zangqing 1218, Zangqing 13, Zangqing 2000, Zangqing 25, Zangqing 28, Zangqing 320, Zangqing 78, Zangqing 80, Zangqing 811, Zangqing 83, and Ximala 22 belonged to group I in the phylogenetic tree and were all from Xizang. In group II, there were seven Qingke materials with a Q value greater than 0.8, accounting for 50% of such groups. Cultivars Diqing 3, DuanbaiQingke, Ganqing 1, Longke 4, Longke 5, Yunke 4, and ChangheiQingke belonged to group II in the phylogenetic tree. Except Ganqing 1 from Gansu, the other cultivars were from Yunnan and Heilongjiang. In group III, there were 15 Qingke materials with a Q value greater than 0.8, accounting for 62.5% of such groups. In group III, there were 15 Qingke materials with a Q value greater than 0.8, accounting for 62.5% of this group. Beiqing 1, Beiqing 3, Beiqing 4, Beiqing 6, Beiqing 8, Dulihuang, Ganqing 2, Ganqing 4, Ganqing 7, Huangqing 2, Kangqing 2, Kunlun12, Kunlun 13, Kunlun 14, and Kunlun 18 belonged to group III in the phylogenetic tree. Except for Kangqing 2, which was from Sichuan, the rest of the cultivars were from Qinghai and Gansu. In the three groups, only 50.7% of the Qingke cultivars were relatively homogeneous, and some of the cultivars were mixed types, indicating that there was also extensive hybridization among the three subgroups, especially in Sichuan, where the Qingke cultivars were distributed in all three groups (Fig. 2C).

Structure analysis of 63 primary Qingke cultivars. A Distribution of ΔK for different values of K. B Population structure analysis of 63 primary Qingke cultivars when K = 2–4; C Population structure analysis of 63 primary Qingke cultivars when K = 3. With K = 3, red, green, and blue bars correspond to Groups 1, 2, and 3, respectively. (Color figure online)
3.3.4 Principal component analysis (PCoA)
PCoA was performed on 63 primary Qingke cultivars using GenAIEx 6.51. The 63 primary Qingke cultivars were equally divided into 3 groups. The Qingke cultivars contained in each group were consistent with the results of the phylogenetic tree. They were consistent with the primary Qingke cultivars with a Q value greater than 0.8 in the STRUCTURE grouping groups and inconsistent with some primary Qingke cultivars with a Q value less than 0.8, namely Huangqing 1, Kangqing 3, Kangqing 6, Kunlun 17, and Kunlun 8, which all belonged to the STRUCTURE grouping analysis in the mixed type (Fig. 3).

The 63 primary Qingke cultivars were subjected to principal coordinate analysis (PCoA) analysis based on genetic distance. Orange, green and blue dots correspond to Groups 1, 2, and 3, respectively. (Color figure online)
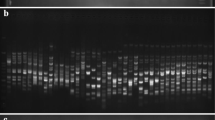
DNA fingerprinting of 63 primary Qingke cultivars
DNA fingerprinting could effectively identify the purity of Qingke quality, providing strong support for the identification, protection, and utilization of Qingke cultivars. Simplified fingerprints of 63 primary Qingke cultivars were constructed by combining the raw data of 63 primary Qingke cultivars established by the size of the molecular fragments amplified using 18 pairs of SSR polymorphic primers. The fingerprints constructed by this method intuitively reflected the unique bands of each cultivar and quickly and accurately identified 63 primary Qingke cultivars (Table 6).
Discussion
Genetic diversity of primary Qingke cultivars
Genetic diversity reflected the differences in genetic composition between populations or between individuals within populations, and was an important reference value for research on the origin and evolution of species, as well as their exploitation and utilization. SSR molecular markers can directly reflect the DNA level evolutionary relationships and genetic variation, providing technical security in the study of genetic polymorphism and population structure of species (Cheng et al. 2023). In this study, we analyzed the genetic diversity of 63 primary Qingke cultivars’ genotypes using 18 SSR molecular markers. 18 SSR molecular markers were distributed on seven chromosomes of the Qingke genome, which can be used as effective markers for the genetic diversity analysis of primary Qingke cultivars. Na, Ne, I, PIC, MAF, He and Ho indices were important indicators for assessing polymorphism of SSR markers and for evaluating the genetic diversity of organisms (Zeng et al. 2019). Different forms of Na were located in genes at the same location or genetic locus on chromosomes, and the level of Na reflected one of the indicators of population genetic variation (Ji et al. 2017). In this study, a total of 83 Na were identified, with the number of Na ranging from three to 11, with an average of 4.61per pair. These values were higher than those for the 70 Na connected to points ranging from two to 10, with an average of 3.68 achieved through 19 SSR primers for 60 Qingke cultivars from three counties in the Shangri-La region, Yunnan Province, which were reported by Guo et al.(2012). This indicated that the SSR markers applied in this study showed high polymorphism and excellent genetic quality in 63 primary Qingke cultivars. The results showed that the method could provide a good technical support for genetic diversity evaluation and kinship identification of 63 primary Qingke cultivars. The Na values obtained in this study were lower than those for the amplification of 562 large Qingke germplasm cultivars using 93 SSR primers (two–16, average 5.55) reported by Xu et al.(2017) and the amplification of 107 local Qingke cultivars from the Nepalese plateau (Pandey et al. 2006),utilizing 44 SSR primers (average 5.54). The low mean value of the number of Na was mainly attributed to the difference in the number of SSR marker primers screened. Whereas in this study, the SSR primers were screened based on stability, polymorphism, and high efficiency and were mainly used as preferred markers for the identification of Qingke cultivars. It is an index to estimate community biodiversity by using genetic information theory. The I values ranged from 0.71 to 1.80, with a mean value of 1.07, which was higher than 0.305 (Xu et al. 2017). PIC was a measure of the amount of information a genetic marker polymorphism could provide in a linkage analysis (Alaaeldin et al. 2024). PIC values were a measure of good and poor microsatellite marker polymorphisms. The larger the PIC, the greater the proportion of heterozygotes in a population and consequently in that seat, the higher the genetic information provided (Parada et al. 2012 ). Previously, the polymorphism of primers was categorized into three intervals based on PIC values as PIC < 0.25, 0.25 < PIC < 0.5, and PIC > 0.5 (Botstein 1980). The PIC values in this study ranged from 0.37 to 0.74, with a mean value of 0.52, which was significantly higher than 0.157 (Xu et al. 2017) and 0.3 (Guo et al. 2012). This indicated that the SSR markers selected in this study exhibited a high degree of polymorphism and were able to objectively reveal the level of genetic diversity of the 63 primary Qingke cultivars. Among them, 7 pairs of SSR markers had PIC values between 0.25 and 0.5, with medium polymorphism, and 11 pairs of SSR primers had PIC values > 0.5, which were highly polymorphic markers, accounting for 64.7% of all markers. The results showed that a large proportion of heterozygotes at these loci in 63 primary Qingke cultivars provided high genetic information and more genetic variation. Higher genetic diversity could be used to infer genetic distances between cultivars and for subsequent population structure analysis.
Phylogenetic tree, population structure, and principal component analysis of primary Qingke cultivars
Custer analysis can reflect the relationship between individuals (Amritha et al. 2024). Population structure analysis better reflects the genetic background and gene exchange among the 63 Qingke primary cultivars than cluster analysis (Zhao et al. 2021). Principal component analysis can not only visualize and three-dimensionally indicate the genetic distance and kinship between the 63 Qingke primary cultivars, but also reflect the degree of dispersion between different categories (Zhang et al. 2022). The results obtained based on group analysis, population structure, and principal component analysis of 63 Qingke primary cultivars were highly consistent and corroborated each other. The results showed that they were divided into three groups, which were analyzed in relation to their geographic origins. Group I mainly contained the primary cultivars of Xizang and Qinghai. Group II mainly included the primary cultivars of Qinghai and Gansu, and group III mainly included the primary cultivars of Yunnan and Heilongjiang, indicating that the grouping results of these primary cultivars were closely related to their geographic origins. Chen et al. (2023) and Wang et al. (2014) similarly indicated that the population structure of Qingke in the Qinghai–Xizang Plateau region is closely related to its geographic origin. Population structure analysis showed that 52.38% of Qingke cultivars had a Q value greater than 0.8. This germplasm was relatively singular, and the genetic structure was relatively simple, which could provide a research basis for subsequent Qingke genetic breeding. However, the genotypes of some cultivars in the three groups were mixed; for example, the genotypes of nine primary Qingke cultivars from Sichuan were mixed, accounting for three in each of the three groups, suggesting that the genotypes of these Qingke cultivars showed high heterozygosity and that the correlation with geographic location was not noticeable. Similarly, geography had less influence on the genetic structure of Ethiopian barley local cultivars, with climatic factors accounting for 37.4% of the explained variation (Abebe et al. 2015). In summary, most Qingke cultivars had a certain correlation between taxon division and geographic location, while the correlation of some cultivars with mixed genotypes was not clear. This might be the r were divided into three actors, such as environmental factors, gene exchange, and artificial selection. The analyzed data told us that the results of cluster analysis, population structure and principal component licensing could be used as a reference when selecting breeding parents. Priority is given to the more distantly related cultivars in preference to those with a simple genetic structure in the population structure analysis, so that the genetic differences in the progeny material obtained are richer.
DNA fingerprinting of primary Qingke cultivars
The DNA fingerprinting technique analyzes the genetic material itself, avoiding the influence of factors such as the environment, sample morphology, and the source of the material, and identifies the germplasm genotype as an indicator at the molecular level, which is regarded as the baseline method for cultivar identification (Zhang et al. 2023). Currently, DNA fingerprinting is widely reported in plants, such as sugar beet (Liang et al. 2023), cotton (Santosh et al. 2022), Atractylodes chinensis (fam. Asteraceae) (Ma et al. 2022), and sunflower (Ibrar et al. 2022). Šurlan-Momirović et al. 2013 used SSR markers for DNA fingerprinting of 145 barley from Serbia, and Jaiswal et al. (Jaiswal et al. 2007) used randomly amplified polymorphic DNA markers for DNA fingerprinting of 26 barley species. However, research related to the construction of the DNA fingerprints of Qingke has not been reported. In this study, we successfully constructed 63 digital DNA fingerprints of Qingke cultivars to provide fast and accurate technical support for the detection of Qingke cultivars, to ensure the seed quality of cultivars from the source, and to play the role of cultivar on yield.
Conclusion
Genetic diversity and population structure of markers for SSR analysis and their fingerprints were constructed using 18 pairs of polymorphic primers for 63 primary Qingke cultivars from Sichuan, Qinghai, Xizang, Yunnan, Gansu, and Heilongjiang. The average values of Na, PIC, I, He, and Ho were 4.61, 0.52, 1.07, 0.59, and 0.32, which were within acceptable limits. According to the genetic distance construction of the evolutionary tree, population structure and PCoA showed that the three grouping results were basically the same. There was rich and high genetic diversity among the genotypes of 63 primary Qingke cultivars, which were divided into three groups. In this study, efficient SSR marker primers of Qingke were screened to more accurately assess the genetic characteristics. Based on the results of DNA fingerprints of 63 primary Qingke cultivars, we can be applied to the identification of new cultivars of Qingke and the authenticity of cultivars. Based on the results of genetic diversity and population structure analyses of 63 primary Qingke cultivars, we can select cultivars that are genetically far apart when selecting parents for breeding, thus maintaining rich genetic differences in offspring. This study lays the foundation for guiding the selection of breeding parents and new cultivars of Qingke in Sichuan, Qinghai, Tibet, Yunnan, Gansu and Heilongjiang, and promoting the innovation of germplasm resources in the future.
References
Abebe TD, Naz AA, LéON J (2015) Landscape genomics reveal signatures of local adaptation in barley (Hordeum vulgare L.). Front Plant Sci [j] 6:813
Alaaeldin MSE, Abedlaziz RE, Salah KLA et al (2024) Genetic diversity assessment of Trichoderma spp. isolated from various Egyptian locations using its gene sequencing marker, rep-PCR, and their cellulolytic activity [J]. Egypt J Biol Pest Control. https://doi.org/10.1186/s41938-024-00784-6
Amritha M, Dhanya O, Nair GA et al (2024) Genetic diversity studies between Ipomoea wild species and cultivated sweet potato Ipomoea batatas using SSR markers. South Afr J Botany 169:452–463
Botstein D (1980) A theory of modular evolution for bacteriophages. Ann NY Acad Sci [j] 354:484–490
Chen X, Ma M, Liu X et al (2022) Multi-scale structure of A-and B-type granules of normal and waxy hull-less barley starch. Int J Biol Macromol [j] 200:42–49
Chen T, Xu J, Wang L et al (2023) Landscape genomics reveals adaptive genetic differentiation driven by multiple environmental variables in naked barley on the Qinghai-Tibetan Plateau. Heredity [j] 131:316–326
Cheng L, Ning Z, Jiang ZC et al (2023) Analysis of genetic diversity and population structure in sweetpotato using SSR markers. J Integr Agric [j] 22:3408–3415
Elumalai K, Srinivasan A (2023) Genetic diversity and population structure of sugarcane introgressed hybrids by SSR markers. 3 Biotech [j] 13:399
Guo Y, Li Y, Huang Y et al (2012) Genetic diversity analysis of hulless barley from Shangri-la region revealed by SSR and AFLP markers. Genetic Resour Crop Evol [j] 59:1543–1552
Guo T, Horvath C, Chen L et al (2020) Understanding the nutrient composition and nutritional functions of highland barley (Qingke): a review. Trends Food Sci Technol [j] 103:109–117
Ibrar D, Khan S, Mahmood T et al (2022) Molecular markers-based DNA fingerprinting coupled with morphological diversity analysis for prediction of heterotic grouping in sunflower (Helianthus annuus L.). Front Plant Sci [j] 13:916845
Ilyas MZ, Park H, Jang SJ et al (2023) Association mapping for evaluation of population structure, genetic diversity, and physiochemical traits in drought-stressed maize germplasm using SSR markers. Plants [j] 12:4092
Jaiswal S, Prasad L, Kumar U et al (2007) Characterization of genetic diversity in barley (Hordeum vulgare L.) through DNA fingerprinting. Indian J Genet Plant Breed [j] 67:169–170
Ji H, Huang J, Li H et al (2017) Genetic diversity analysis of microsatellite markers in Xingguo gray geese [J]. Anim Husb Vet Med 49(08):6–9
Li WX, Li JL, He JJ et al (2022) Genetic diversity and population structure of germplasm resources of Amomum villosum based on SSR markers. Zhongguo Zhong Yao Za Zhi = Zhongguo Zhongyao Zazhi = China J Chinese Mater Medica [j] 47:4618–4626
Li Z, He L, Wang X et al (2023) Genetic diversity analysis and fingerprints of Chrysanthemum × morifolium based on SSR molecular markers. Sheng Wu Gong Cheng Xue Bao = Chin J Biotechnol [j] 39:2839–2860
Liang J, Deng G, Long H et al (2012) Virus-induced silencing of genes encoding LEA protein in Tibetan hulless barley (Hordeum vulgare ssp. vulgare) and their relationship to drought tolerance. Molecular Breeding [j] 30:441–451
Liang XM, Pi Z, Wu ZD et al (2023) Constructing DNA fingerprinting and evaluating genetic diversity among sugar beet (Beta vulgaris L.) varieties based on four molecular markers. Sugar Tech [j] 25:1361–1373
Luo Z, Yao Z, Yang Y et al (2023) Genetic fingerprint construction and genetic diversity analysis of sweet potato (Ipomoea batatas) germplasm resources. BMC Plant Biol [j] 23:355
Lyu Y, Ma S, Liu J et al (2022) A systematic review of highland barley: ingredients, health functions and applications. Grain Oil Sci Technol [j] 5:35–43
Ma S, Zhao J, Su W et al (2022) Transcriptome-derived SSR markers for DNA fingerprinting and inter-populations genetic diversity assessment of Atractylodes chinensis. Nucleus [j] 65:321–329
Mériam BR, Leila R, Rahma J et al (2018) Fingerprinting and genetic purity assessment of F1 barley hybrids and their salt-tolerant parental lines using nSSR molecular markers. 3 Biotech [j] 8:57
Pandey M, Wagner C, Friedt W et al (2006) Genetic relatedness and population differentiation of Himalayan hulless barley (Hordeum vulgare L.) landraces inferred with SSRs. Theor Appl Genet [j] 113:715–729
Parada R, Książkiewicz J, Jaszczak KK (2012) Studies on resources of genetic diversity in conservative flocks of geese using microsatellite DNA polymorphic markers[J]. Mol Biol Rep 39:5219–5297
Santosh H, Bargat A, Santhy V et al (2022) Microsatellite marker based DNA fingerprinting of cotton (Gossypium spp.) hybrids and their parents. Electron J Plant Breed [j] 13:780–789
Sharan H, Kumar A, Singh S (2024) Newly developed SSRs based genetic diversity patterns in trait specific populations of German chamomile (Matricaria chamomilla L.). Ind Crops Prod [j] 210:118152
Šurlan-Momirović G, Krämer I, Bratković K et al (2013) Molecular characterization of barley (Hordeum vulgare L.) accessions of the Serbian Genebank by SSR fingerprinting. Genetika [j] 45:167–180
Türkoğlu A, Haliloğlu K, Mohammadi SA et al (2023) Genetic diversity and population structure in türkiye bread wheat genotypes revealed by simple sequence repeats (SSR) markers. Genes [j] 14:1182
Wang L, Xu J, Xia T et al (2014) Population structure and linkage disequilibrium in six-rowed barley landraces from the Qinghai-Tibetan Plateau. Crop Sci [j] 54:2011–2022
Xu Q, Zeng X, Lin B et al (2017) A microsatellite diversity analysis and the development of core-set germplasm in a large hulless barley (Hordeum vulgare L.) collection. BMC Genet [j] 18:1–10
Yao X, Wang Y, Yao Y et al (2021) Identification microRNAs and target genes in Tibetan hulless barley to BLS infection. Agron J [j] 113:2273–2292
Zeng K, Song W, Wang Q et al (2019) Analysis of genetic diversity and population genetic structure of Siniperca chuatsi germplasm based on microsatellite markers [J]. J Huazhong Agric Univ 38(06):104–115
Zhang Y, Huang G, Mo Y, Lu S, Zhao Y, Tang Y, Lu Z, San B, Rong T (2022) CDDP combined with SRAP markers to analyze the genetic diversity of sportive fruit varieties[J]. South China Fruit Tree 51(02):57–63
Zhang M, Zheng C, Li J et al (2023) Genetic diversity, population structure, and DNA fingerprinting of Ailanthus altissima var. erythrocarpa based on EST-SSR markers. Sci Rep [j] 13:19315
Zhao Y, Wang R, Liu Q et al (2021) Genetic diversity of ancient Camellia sinensis (L.) O. Kuntze in Sandu County of Guizhou Province in China[J]. Diversity 13(6):276–276
Zhao Q, Chen Z, Khalifa MA et al (2022) DNA fingerprinting construction and genetic diversity analysis for azuki bean (Vigna angularis) and mung bean (Vigna radiata L.) germplasm resources. Legum Res Int J [j] 45:135–142
Acknowledgements
We thank LetPub (www.letpub.com) for linguistic assistance and pre-submis sion expert review.
Funding
This research was supported by the National Key Research and Development Project (2022YFD2301300), the Qinghai Key Laboratory of Hulless Barley Genetics and Breeding (2023-1_5), the Agricultural product quality supervision project (111821301092362041), and the Agriculture Research System of China (CARS-05).
Author information
Authors and Affiliations
Contributions
Qian Hu contributed in experimental design, methodology, software, investigation, data analysis, and writing original draft. Youhua Yao and Yongmei Cui contributed in software, data analysis, and writing and editing. Xin Li contributed in software, investigation, and writing. Likun An contributed in investigation, review and editing. Yixiong Bai contributed in software, data analysis, and writing. Baojun Ding contributed in software, supervision, data analysis, and writing. Xiaohua Yao and Kunlun Wu contributed in supervision, funding acquisition, writing, review and editing. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hu, Q., Yao, Y., Cui, Y. et al. Genetic diversity analysis and DNA fingerprinting of primary Qingke (Hordeum vulgare L. var. nudum Hook. f.) cultivars. Genet Resour Crop Evol (2024). https://doi.org/10.1007/s10722-024-02054-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10722-024-02054-8